

Ion-Triggered In Situ Gelling Intranasal Spray of Dronedarone Hydrochloride Nanocarriers: In Vitro Optimization and In Vivo Pharmacokinetic Appraisal

 , , ,
, , ,
Abstract
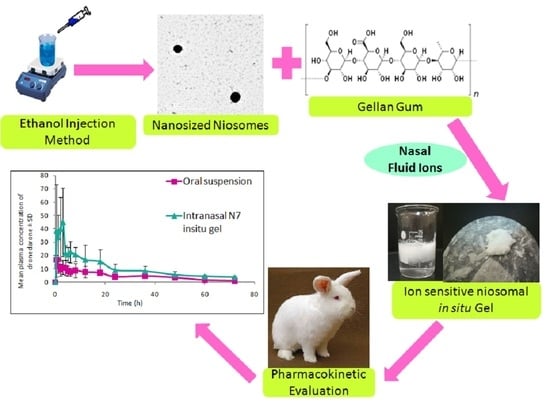
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of DRN Loaded Niosomes
2.3. Characterization of the Prepared Niosomes
2.3.1. Vesicle Size, Size Distribution, and Zeta Potential
2.3.2. Entrapment Efficiency (EE%)
2.4. Factorial Experimental Design of the Study
2.5. Selection and Validation of the Optimal DRN Loaded Niosomes Formula
2.5.1. Transmission Electron Microscopy (TEM)
2.5.2. Lyophilization of the Optimized DRN Loaded Niosomes
2.5.3. Differential Scanning Calorimetry (DSC)
2.6. Stability Study of the Optimal DRN Loaded Niosomes Formula
2.7. Preparation of Niosomal Ion Sensitive In Situ Gelling Intranasal Spray
2.8. Evaluation of DRN Niosomal In Situ Gelling Intranasal Spray
2.8.1. Assessment of In Situ Gelation Capability
2.8.2. Measurement of pH
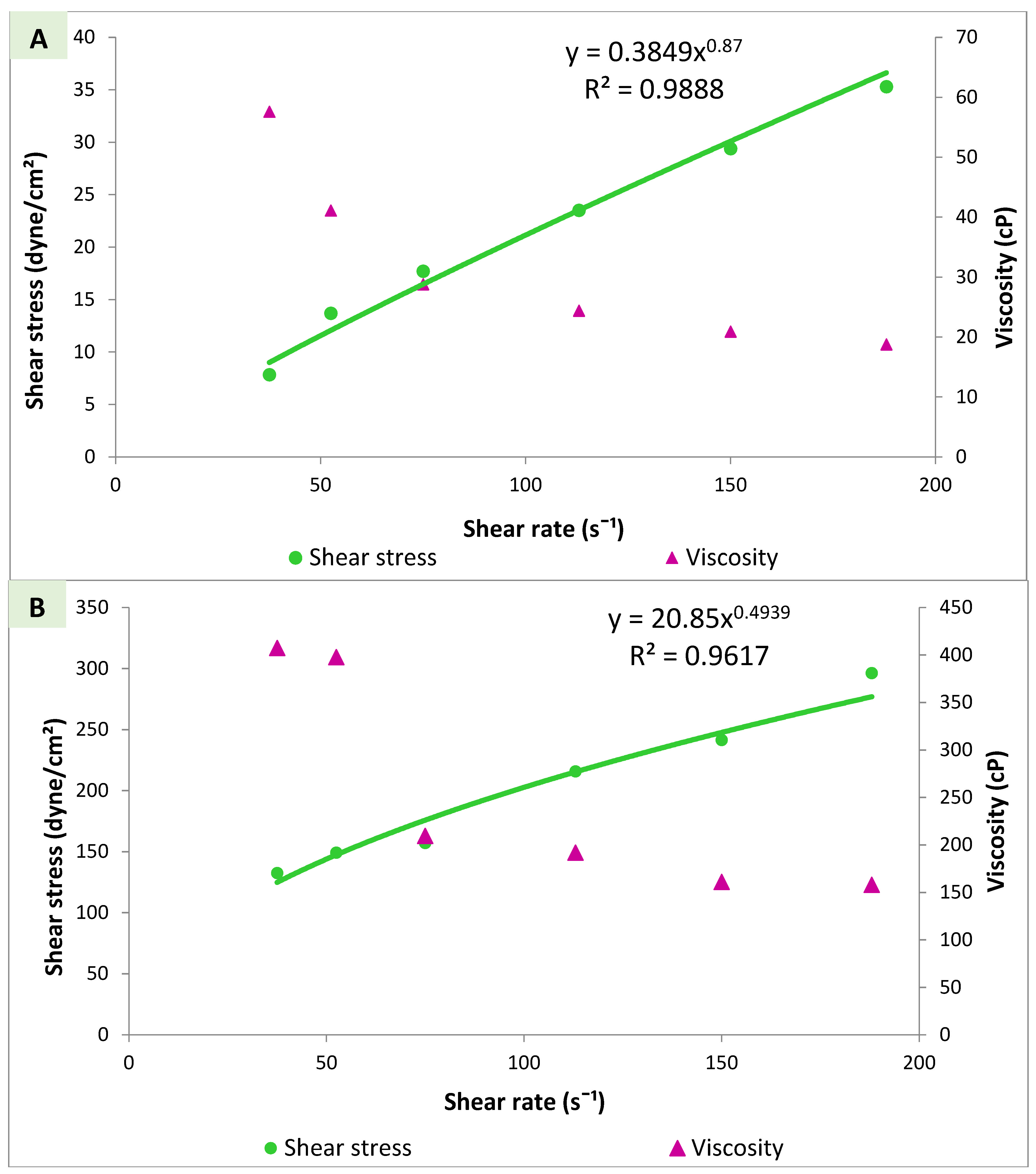
2.8.3. Assessment of Rheological Properties
2.8.4. Evaluation of Gel Strength
2.8.5. Evaluation of Mucoadhesion Force
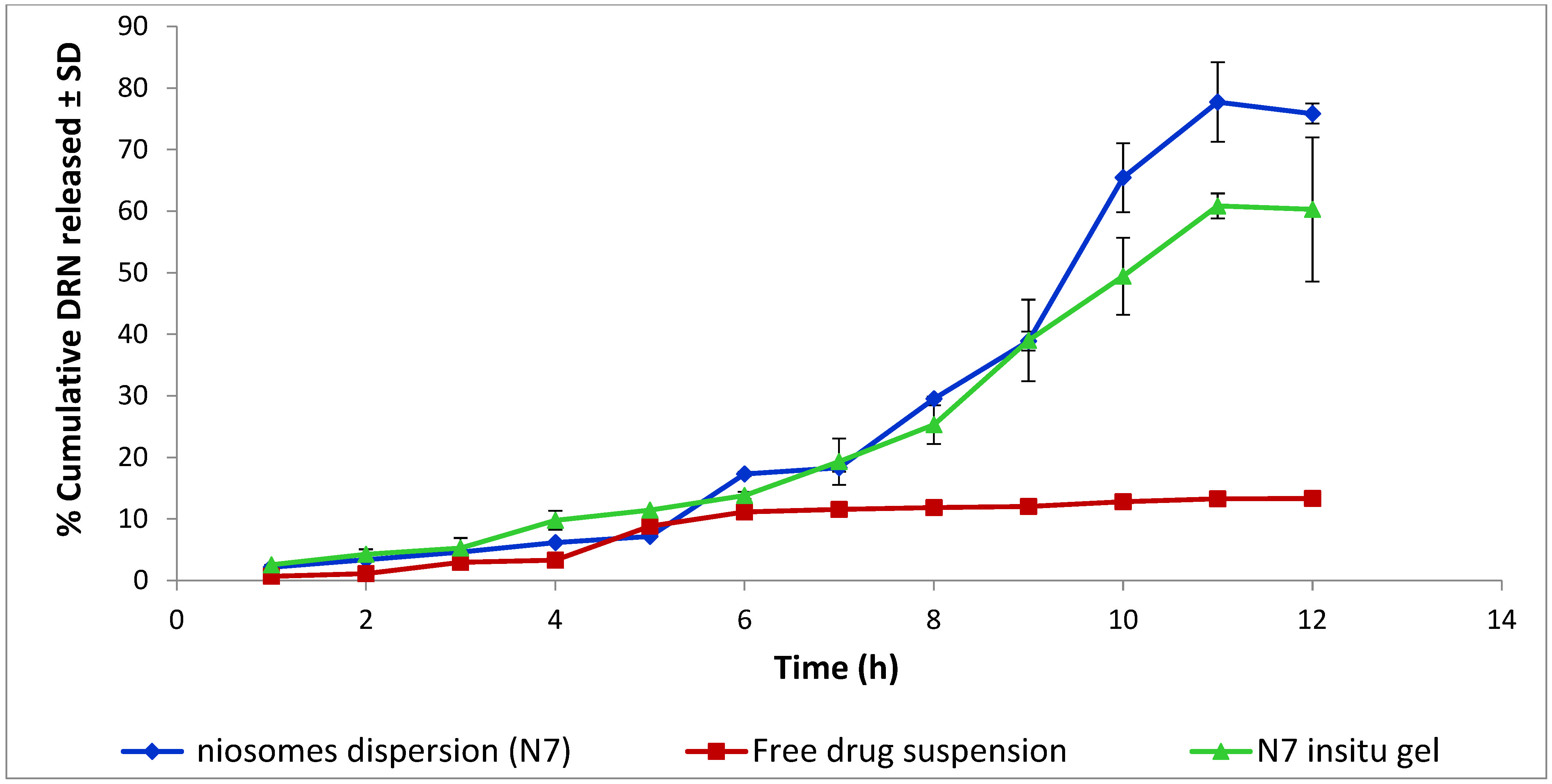
2.8.6. In Vitro Release Study of Niosomal Dispersion and Niosomal In Situ Gel
2.8.7. Release Mechanism of Optimal DRN Niosomal In Situ Gel
- n < 0.5 (0.45)—quasi-Fickian diffusion.
- n = 0.5 (0.45)—diffusion mechanism.
- 0.5 (0.45) < n < 1—non-Fickian diffusion.
- n = 1 (0.89)—case II transport (zero-order release).
- n > 1 (0.89)—super case II transport.
2.9. In Vivo Pharmacokinetic Assessment
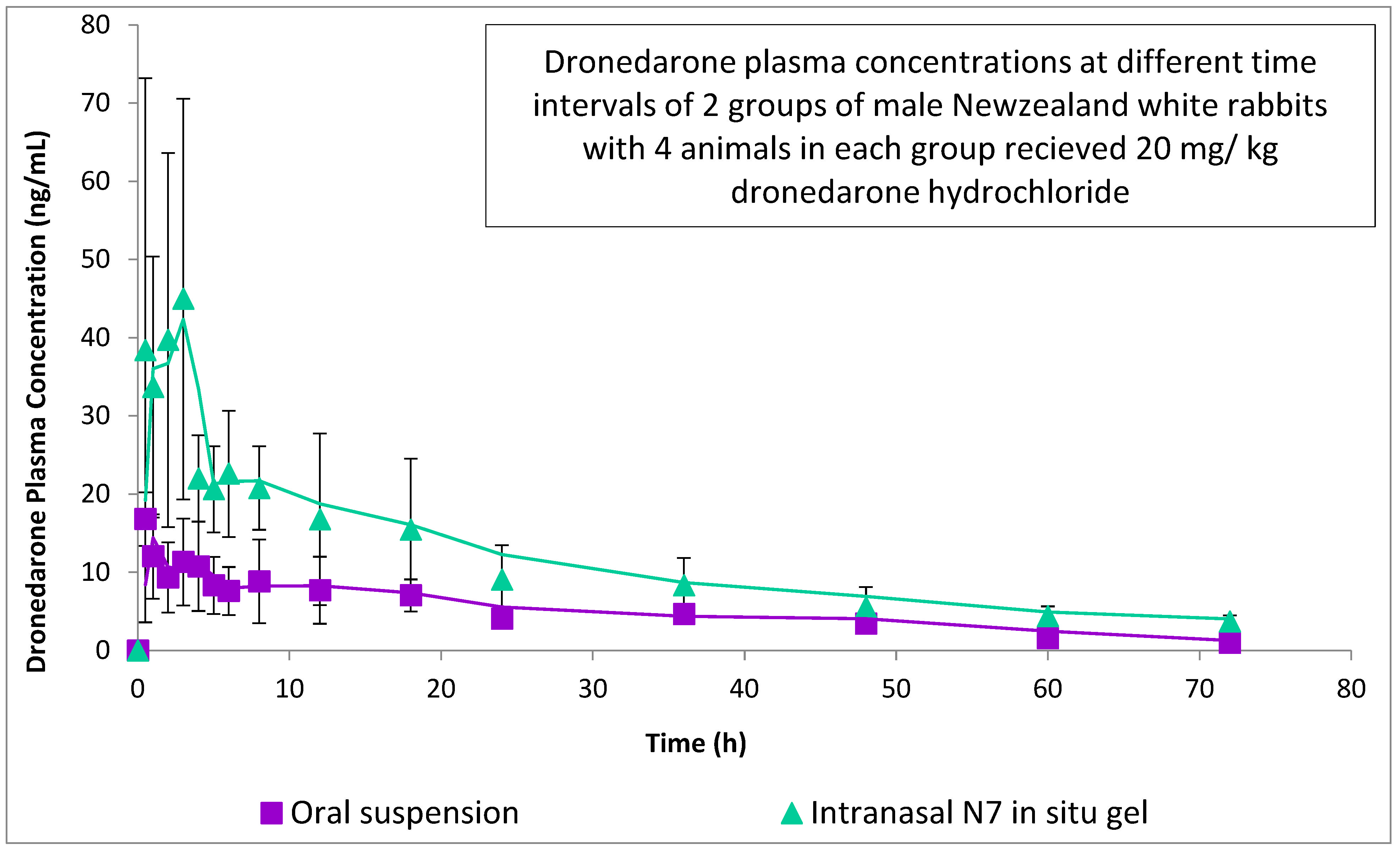
2.9.1. Animal Study Design and Samples Collection
2.9.2. Bioanalytical Method Description and Chromatographic Conditions
2.9.3. Plasma Samples Preparation
3. Results and Discussion
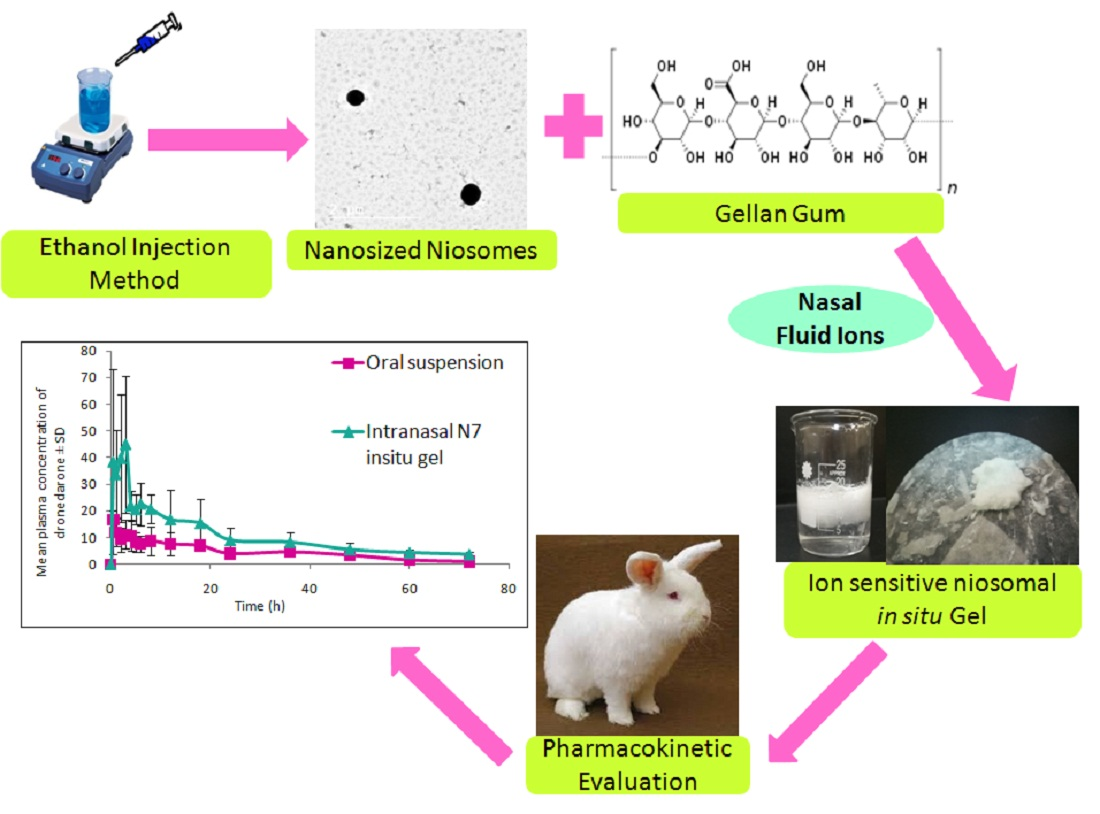
3.1. Analysis of 32 Full Factorial Design
3.1.1. Effect of Variables on VS
3.1.2. Effect of Variables on PDI
3.1.3. Effect of Variables on ZP
3.1.4. Effect of Variables on EE%
3.2. Selection and Validation of the Optimal DRN Loaded Niosomes
3.2.1. Transmission Electron Microscopy (TEM)
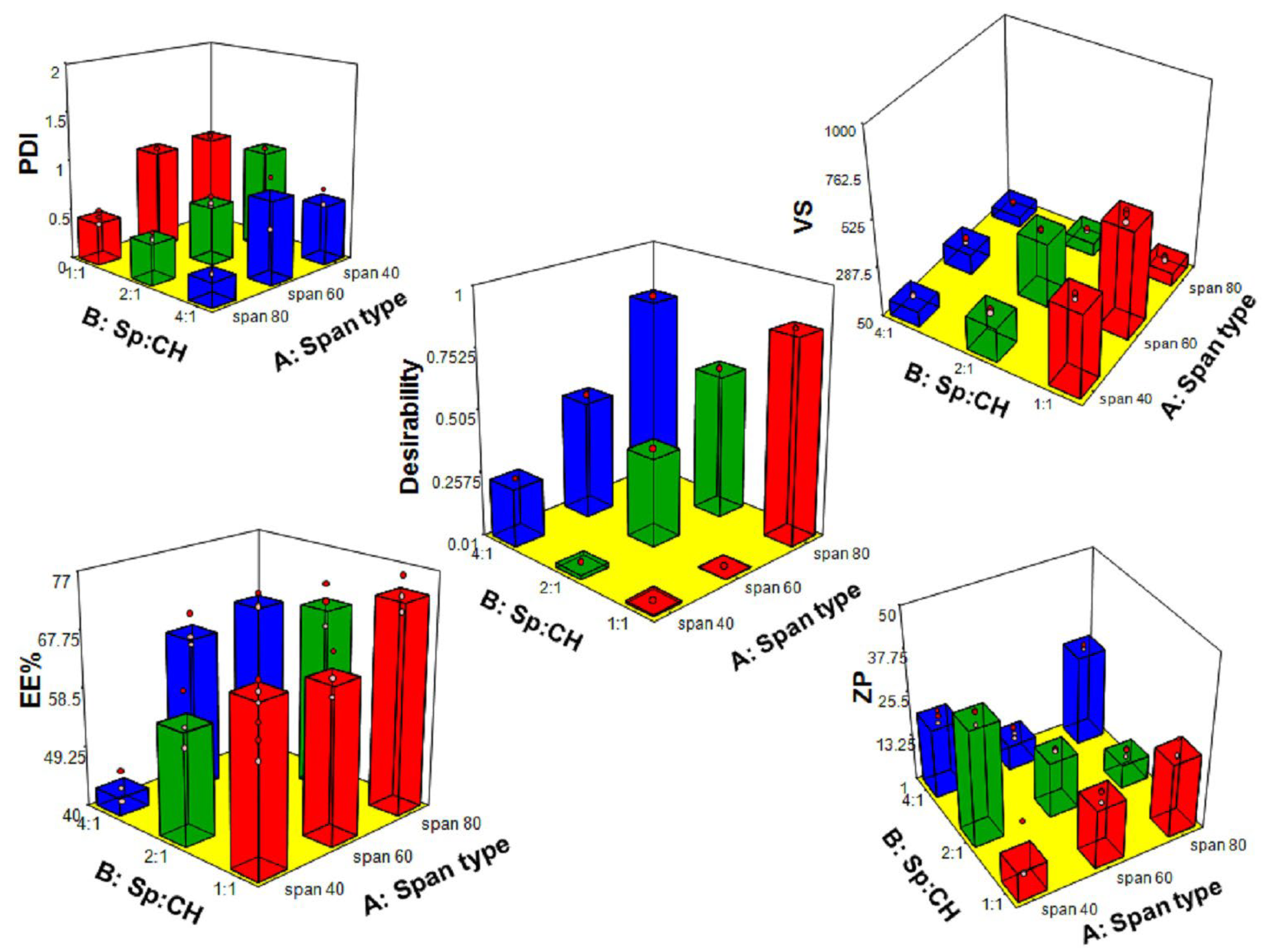
3.2.2. Differential Scanning Calorimetry (DSC)
3.3. Stability Study of the Optimal DRN Loaded Niosomes Formula
3.4. Evaluation of DRN Niosomal In Situ Gel
3.4.1. Assessment of In Situ Gelation Capability
3.4.2. Measurement of pH
3.4.3. Viscosity Measurements and Rheological Properties
3.4.4. Evaluation of Gel Strength
3.4.5. Evaluation of Mucoadhesion Force
3.5. In Vitro Release Study of Niosomes Dispersion and Niosomal In Situ Gel
Release Mechanism of Optimal DRN Niosomal In Situ Gel
3.6. In Vivo Pharmacokinetic Study
3.6.1. Liquid Chromatography/Mass Spectrometry (LC-MS/MS) Bioanalytical Method for Determination of Dronedarone in Rabbits’ Plasma
3.6.2. Experimental Observation and Estimated Pharmacokinetic Parameters
3.6.3. Statistical Analysis of Pharmacokinetic Parameters
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schweizer, P.A.; Becker, R.; Katus, H.A.; Thomas, D. Dronedarone: Current evidence for its safety and efficacy in the management of atrial fibrillation. Drug Des. Dev. Ther. 2011, 5, 27–39. [Google Scholar]
- Kathofer, S.; Thomas, D.; Karle, C.A. The novel antiarrhythmic drug dronedarone: Comparison with amiodarone. Cardiovasc. Drug Rev. 2005, 23, 217–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.N.; Connolly, S.J.; Crijns, H.J.; Roy, D.; Kowey, P.R.; Capucci, A.; Radzik, D.; Aliot, E.M.; Hohnloser, S.H. Dronedarone for maintenance of sinus rhythm in atrial fibrillation or flutter. N. Engl. J. Med. 2007, 357, 987–999. [Google Scholar] [CrossRef] [Green Version]
- Jardan, Y.A.; Brocks, D.R. The pharmacokinetics of dronedarone in normolipidemic and hyperlipidemic rats. Biopharm. Drug Dispos. 2016, 37, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Han, S.D.; Jung, S.W.; Jang, S.W.; Son, M.; Kim, B.M.; Kang, M.J. Reduced food-effect on intestinal absorption of dronedarone by self-microemulsifying drug delivery system (SMEDDS). Biol. Pharm. Bull. 2015, 38, 1026–1032. [Google Scholar] [CrossRef] [Green Version]
- Iram, F.; Ali, S.; Ahmad, A.; Khan, S.A.; Husain, A. A review on dronedarone: Pharmacological, pharmacodynamic and pharmacokinetic profile. J. Acute Dis. 2016, 5, 102–108. [Google Scholar] [CrossRef] [Green Version]
- Moghassemi, S.; Hadjizadeh, A. Nano-niosomes as nanoscale drug delivery systems: An illustrated review. J. Control. Release 2014, 185, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Kazi, K.M.; Mandal, A.S.; Biswas, N.; Guha, A.; Chatterjee, S.; Behera, M.; Kuotsu, K. Niosome: A future of targeted drug delivery systems. J. Adv. Pharm. Technol. Res. 2010, 1, 374–380. [Google Scholar]
- Osanloo, M.; Assadpour, S.; Mehravaran, A.; Abastabar, M.; Akhtari, J. Niosome-loaded antifungal drugs as an effective nanocarrier system: A mini review. Curr. Med. Mycol. 2018, 4, 31–36. [Google Scholar] [CrossRef]
- Mahmoud, K.; Swidan, S.; El-Nabarawi, M.; Teaima, M. Lipid based nanoparticles as a novel treatment modality for hepatocellular carcinoma: A comprehensive review on targeting and recent advances. J. Nanobiotechnol. 2022, 20, 1–42. [Google Scholar] [CrossRef]
- Sultan, A.A.; El-Gizawy, S.A.; Osman, M.A.; El Maghraby, G.M. Niosomes for oral delivery of nateglinide: In situ-in vivo correlation. J. Liposome Res. 2018, 28, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi-Ghadi, Z.; Ebrahimnejad, P.; Talebpour Amiri, F.; Nokhodchi, A. Improved oral delivery of quercetin with hyaluronic acid containing niosomes as a promising formulation. J. Drug Target. 2021, 29, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Eid, R.K.; Essa, E.A.; El Maghraby, G.M. Essential oils in niosomes for enhanced transdermal delivery of felodipine. Pharm. Dev. Technol. 2019, 24, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Zidan, A.S.; Hosny, K.M.; Ahmed, O.A.; Fahmy, U.A. Assessment of simvastatin niosomes for pediatric transdermal drug delivery. Drug Deliv. 2016, 23, 1536–1549. [Google Scholar] [CrossRef]
- Abou-Taleb, H.A.; Khallaf, R.A.; Abdel-Aleem, J.A. Intranasal niosomes of nefopam with improved bioavailability: Preparation, optimization, and in-vivo evaluation. Drug Des. Dev. Ther. 2018, 12, 3501–3516. [Google Scholar] [CrossRef] [Green Version]
- Pando, D.; Matos, M.; Gutiérrez, G.; Pazos, C. Formulation of resveratrol entrapped niosomes for topical use. Colloids Surf. B Biointerfaces 2015, 128, 398–404. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, L.; Xiang, S.; Jiang, C.; Wu, W.; Ruan, S.; Du, Q.; Chen, T.; Xue, Y.; Chen, H.; et al. Formulation and characterization of a 3D-printed cryptotanshinone-loaded niosomal hydrogel for topical therapy of acne. AAPS PharmSciTech 2020, 21, 159. [Google Scholar] [CrossRef]
- Mali, N.; Darandale, S.; Vavia, P. Niosomes as a vesicular carrier for topical administration of minoxidil: Formulation and in vitro assessment. Drug Deliv. Transl. Res. 2013, 3, 587–592. [Google Scholar] [CrossRef]
- Ag Seleci, D.; Maurer, V.; Stahl, F.; Scheper, T.; Garnweitner, G. Rapid microfluidic preparation of niosomes for targeted drug delivery. Int. J. Mol. Sci. 2019, 20, 4696. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.R.; Jin, H.; Wang, Y.; Chen, C.; Li, M.; Mao, S.J.; Wang, Q.; Li, H. Anti-CD123 antibody-modified niosomes for targeted delivery of daunorubicin against acute myeloid leukemia. Drug Deliv. 2017, 24, 882–890. [Google Scholar] [CrossRef] [Green Version]
- Ghori, M.U.; Mahdi, M.H.; Smith, A.M.; Conway, B.R. Nasal drug delivery systems: An overview. Am. J. Pharmacol. Sci. 2015, 3, 110–119. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, A.; Madhv, N.S. Nasal cavity, a promising transmucosal platform for drug delivery and research approaches from nasal to brain targetting. J. Drug Deliv. Ther. 2012, 2. [Google Scholar] [CrossRef]
- Behl, C.; Pimplaskar, H.; Sileno, A.; Demeireles, J.; Romeo, V. Effects of physicochemical properties and other factors on systemic nasal drug delivery. Adv. Drug Deliv. Rev. 1998, 29, 89–116. [Google Scholar] [CrossRef]
- Sworn, G.; Sanderson, G.; Gibson, W. Gellan gum fluid gels. Food Hydrocoll. 1995, 9, 265–271. [Google Scholar] [CrossRef]
- Mahdi, M.H.; Conway, B.R.; Smith, A.M. Evaluation of gellan gum fluid gels as modified release oral liquids. Int. J. Pharm. 2014, 475, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Teaima, M.H.; Alsofany, J.M.; El-Nabarawi, M.A. Clove oil endorsed transdermal flux of dronedarone hydrochloride loaded bilosomal nanogel: Factorial design, in vitro evaluation and ex vivo permeation. AAPS PharmSciTech 2022, 23, 1–14. [Google Scholar] [CrossRef]
- Kovvasu, S.P.; Kunamaneni, P.; Yeung, S.; Rueda, J.; Betageri, G.V. Formulation of dronedarone hydrochloride-loaded proliposomes: In vitro and in vivo evaluation using Caco-2 and Rat Model. AAPS PharmSciTech 2019, 20, 226. [Google Scholar] [CrossRef]
- Gambhire, V.M.; Gambhire, M.S.; Ranpise, N.S. Solid lipid nanoparticles of dronedarone hydrochloride for oral delivery: Optimization, in vivo pharmacokinetics and uptake studies. Pharm. Nanotechnol. 2019, 7, 375–388. [Google Scholar] [CrossRef]
- Kakkar, S.; Kaur, I.P. Spanlastics—A novel nanovesicular carrier system for ocular delivery. Int. J. Pharm. 2011, 413, 202–210. [Google Scholar] [CrossRef]
- Teaima, M.H.; Eltabeeb, M.A.; El-Nabarawi, M.A.; Abdellatif, M.M. Utilization of propranolol hydrochloride mucoadhesive invasomes as a locally acting contraceptive: In-vitro, ex-vivo, and in-vivo evaluation. Drug Deliv. 2022, 29, 2549–2560. [Google Scholar] [CrossRef]
- Pravalika, K.; Induri, M.; Sudhakar, M.; Fathima, A. Spectrophotometric Estimation of Dronedarone Hydrochloride in Pharmaceutical Dosage Forms by using Multivariate Technique. World J. Pharm. Sci. 2013, 1, 15–18. [Google Scholar]
- Abdelbary, A.A.; Abd-Elsalam, W.H.; Al-Mahallawi, A.M. Fabrication of novel ultradeformable bilosomes for enhanced ocular delivery of terconazole: In vitro characterization, ex vivo permeation and in vivo safety assessment. Int. J. Pharm. 2016, 513, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Teaima, M.; Yasser, M.; Elfar, N.; Shoueir, K.; El-Nabarawi, M.; Helal, D. Construction of sublingual trilaminated Eszopiclone fast dissolving film for the treatment of Insomnia: Formulation, characterization and In vivo clinical comparative pharmacokinetic study in healthy human subjects. PLoS ONE 2022, 17, e0266019. [Google Scholar] [CrossRef] [PubMed]
- Zeb, A.; Qureshi, O.S.; Kim, H.S.; Cha, J.H.; Kim, J.K. Improved skin permeation of methotrexate via nanosized ultradeformable liposomes. Int. J. Nanomed. 2016, 11, 3813–3824. [Google Scholar]
- Teaima, M.H.; Badawi, N.M.; Attia, D.A.; El-Nabarawi, M.A.; Elmazar, M.M.; Mousa, S.A. Efficacy of pomegranate extract loaded solid lipid nanoparticles transdermal emulgel against Ehrlich ascites carcinoma. Nanomed. Nanotechnol. Biol. Med. 2022, 39, 102466. [Google Scholar] [CrossRef]
- Garhy, D.M.; Ismail, S.; Ibrahim, H.K.; Ghorab, M.M. Buccoadhesive gel of carvedilol nanoparticles for enhanced dissolution and bioavailability. J. Drug Deliv. Sci. Technol. 2018, 47, 151–158. [Google Scholar] [CrossRef]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar]
- Olejnik, A.; Kapuscinska, A.; Schroeder, G.; Nowak, I. Physico-chemical characterization of formulations containing endomorphin-2 derivatives. Amino Acids 2017, 49, 1719–1731. [Google Scholar] [CrossRef] [Green Version]
- Clark, J.D.; Gebhart, G.F.; Gonder, J.C.; Keeling, M.E.; Kohn, D.F. Special report: The 1996 guide for the care and use of laboratory animals. ILAR J. 1997, 38, 41–48. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [Green Version]
- Alsofany, J.M.; Hamza, M.Y.; Abdelbary, A.A. Fabrication of nanosuspension directly loaded fast-dissolving films for enhanced oral bioavailability of olmesartan medoxomil: In vitro characterization and pharmacokinetic evaluation in healthy human volunteers. AAPS PharmSciTech 2018, 19, 2118–2132. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, T.; Sternberg, B.; Florence, A.T. Preparation and properties of vesicles (niosomes) of sorbitan monoesters (Span 20, 40, 60 and 80) and a sorbitan triester (Span 85). Int. J. Pharm. 1994, 105, 1–6. [Google Scholar] [CrossRef]
- Khazaeli, P.; Pardakhty, A.; Shoorabi, H. Caffeine-loaded niosomes: Characterization and in vitro release studies. Drug Deliv. 2007, 14, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Ruckmani, K.; Jayakar, B.; Ghosal, S.K. Nonionic surfactant vesicles (niosomes) of cytarabine hydrochloride for effective treatment of leukemias: Encapsulation, storage, and in vitro release. Drug Dev. Ind. Pharm. 2000, 26, 217–222. [Google Scholar] [CrossRef]
- Abdelbary, G.A.; Aburahma, M.H. Oro-dental mucoadhesive proniosomal gel formulation loaded with lornoxicam for management of dental pain. J. Liposome Res. 2015, 25, 107–121. [Google Scholar] [CrossRef]
- Gupta, M.; Vaidya, B.; Mishra, N.; Vyas, S.P. Effect of surfactants on the characteristics of fluconazole niosomes for enhanced cutaneous delivery. Artif. Cells Blood Substit. Biotechnol. 2011, 39, 376–384. [Google Scholar] [CrossRef]
- Coderch, L.; Fonollosa, J.; De Pera, M.; Estelrich, J.; De La Maza, A.; Parra, J.L. Influence of cholesterol on liposome fluidity by EPR. Relationship with percutaneous absorption. J. Control. Release 2000, 68, 85–95. [Google Scholar] [CrossRef]
- Ramana, L.N.; Sethuraman, S.; Ranga, U.; Krishnan, U.M. Development of a liposomal nanodelivery system for nevirapine. J. Biomed. Sci. 2010, 17, 57. [Google Scholar] [CrossRef] [Green Version]
- Hathout, R.M.; Mansour, S.; Mortada, N.D.; Guinedi, A.S. Liposomes as an ocular delivery system for acetazolamide: In vitro and in vivo studies. AAPS PharmSciTech 2007, 8, E1–E12. [Google Scholar] [CrossRef]
- Taylor, P. Ostwald ripening in emulsions: Estimation of solution thermodynamics of the disperse phase. Adv. Colloid Interface Sci. 2003, 106, 261–285. [Google Scholar] [CrossRef]
- Teaima, M.; Hababeh, S.; Khanfar, M.; Alanazi, F.; Alshora, D.; El-Nabarawi, M. Design and optimization of pioglitazone hydrochloride Self-Nanoemulsifying Drug Delivery System (SNEDDS) incorporated into an orally disintegrating tablet. Pharmaceutics 2022, 14, 425. [Google Scholar] [CrossRef] [PubMed]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M. Impact of particle size and polydispersity index on the clinical applications of lipidic nanocarrier systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mahallawi, A.M.; Abdelbary, A.A.; Aburahma, M.H. Investigating the potential of employing bilosomes as a novel vesicular carrier for transdermal delivery of tenoxicam. Int. J. Pharm. 2015, 485, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Aithal, B.K.; Sunil Kumar, M.R.; Rao, B.N.; Upadhya, R.; Prabhu, V.; Shavi, G.; Arumugam, K.; Sajankila, S.P.; Udupa, N.; Satyamoorthy, K.; et al. Evaluation of pharmacokinetic, biodistribution, pharmacodynamic, and toxicity profile of free juglone and its sterically stabilized liposomes. J. Pharm. Sci. 2011, 100, 3517–3528. [Google Scholar] [CrossRef] [PubMed]
- Badr-Eldin, S.M.; Aldawsari, H.M.; Alhakamy, N.A.; Fahmy, U.A.; Ahmed, O.A.A.; Neamatallah, T.; Tima, S.; Almaghrabi, R.H.; Alkudsi, F.M.; Alamoudi, A.A.; et al. Merging experimental design and nanotechnology for the development of optimized simvastatin spanlastics: A promising combined strategy for augmenting the suppression of various human cancer cells. Pharmaceutics 2022, 14, 1024. [Google Scholar] [CrossRef]
- Park, J.; Ngo, H.V.; Jin, H.E.; Lee, K.W.; Lee, B.J. Hydroxyl group-targeted conjugate and its self-assembled nanoparticle of peptide drug: Effect of degree of saturation of fatty acids and modification of physicochemical properties. Int. J. Nanomed. 2022, 17, 2243–2260. [Google Scholar] [CrossRef]
- Hao, Y.; Zhao, F.; Li, N.; Yang, Y.; Li, K. Studies on a high encapsulation of colchicine by a niosome system. Int. J. Pharm. 2002, 244, 73–80. [Google Scholar] [CrossRef]
- Tatykhanova, G.n.; Aseyev, V.; Kudaibergenov, S.E. Mucoadhesive properties of gellan and its modified derivatives. Rev. Adv. Chem. 2020, 10, 140–157. [Google Scholar] [CrossRef]
- Hao, J.; Zhao, J.; Zhang, S.; Tong, T.; Zhuang, Q.; Jin, K.; Chen, W.; Tang, H. Fabrication of an ionic-sensitive in situ gel loaded with resveratrol nanosuspensions intended for direct nose-to-brain delivery. Colloids Surf. B Biointerfaces 2016, 147, 376–386. [Google Scholar] [CrossRef]
- Owen, D.H.; Peters, J.J.; Katz, D.F. Rheological properties of contraceptive gels. Contraception 2000, 62, 321–326. [Google Scholar] [CrossRef]
- Belgamwar, V.S.; Chauk, D.S.; Mahajan, H.S.; Jain, S.A.; Gattani, S.G.; Surana, S.J. Formulation and evaluation of in situ gelling system of dimenhydrinate for nasal administration. Pharm. Dev. Technol. 2009, 14, 240–248. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Cardoso, V.M.; Gremião, M.P.D.; Cury, B.S.F. Mucin-polysaccharide interactions: A rheological approach to evaluate the effect of pH on the mucoadhesive properties. Int. J. Biol. Macromol. 2020, 149, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Tanaka, Y.; Sakurai, M. Dynamic mechanical properties of aqueous gellan solutions in the sol-gel transition region. Carbohydr. Polym. 1996, 30, 101–108. [Google Scholar] [CrossRef]
- Paul, A.; Fathima, K.M.; Nair, S.C. Intra nasal in situ gelling system of lamotrigine using ion activated mucoadhesive polymer. Open Med. Chem. J. 2017, 11, 222–244. [Google Scholar] [CrossRef]
- Heredia, N.S.; Vizuete, K.; Flores-Calero, M.; Pazmiño, V.K.; Pilaquinga, F.; Kumar, B.; Debut, A. Comparative statistical analysis of the release kinetics models for nanoprecipitated drug delivery systems based on poly(lactic-co-glycolic acid). PLoS ONE 2022, 17, e0264825. [Google Scholar] [CrossRef] [PubMed]
- Guerra-Ponce, W.L.; Gracia-Vásquez, S.L.; González-Barranco, P.; Camacho-Mora, I.A.; Gracia-Vásquez, Y.A.; Orozco-Beltrán, E.; Felton, L.A. In vitro evaluation of sustained released matrix tablets containing ibuprofen: A model poorly water-soluble drug. Braz. J. Pharm. Sci. 2016, 52, 751–759. [Google Scholar] [CrossRef]
- Karthika, J.; Vishalakshi, B. pH responsive gels of gellan gum and carboxymethyl cellulose: Matrix for ketoprofen delivery. Int. J. ChemTech Res. 2014, 6, 1055–1069. [Google Scholar]
- Peppas, N.A.; Narasimhan, B. Mathematical models in drug delivery: How modeling has shaped the way we design new drug delivery systems. J. Control. Release 2014, 190, 75–81. [Google Scholar] [CrossRef]
- Klech, C.M.; Simonelli, A.P. Examination of the moving boundaries associated with non-fickian water swelling of glassy gelatin beads: Effect of solution pH. J. Membr. Sci. 1989, 43, 87–101. [Google Scholar] [CrossRef]
- Saifi, Z.; Rizwanullah, M.; Mir, S.R.; Amin, S. Bilosomes nanocarriers for improved oral bioavailability of acyclovir: A complete characterization through in vitro, ex-vivo and in vivo assessment. J. Drug Deliv. Sci. Technol. 2020, 57, 101634. [Google Scholar] [CrossRef]
- Kumar, A.; Pathak, K.; Bali, V. Ultra-adaptable nanovesicular systems: A carrier for systemic delivery of therapeutic agents. Drug Discov. Today 2012, 17, 1233–1241. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Independent Variables (Factors) | Constrains | |||
|---|---|---|---|---|
| X1: Span® type | Sp40 | Sp60 | Sp80 | |
| X2: Sp: CH ratio (w:w) | 1:1 | 2:1 | 4:1 | |
| Dependent variables (Responses) | Constrains | |||
| Y1:VS (nm) | Minimize | |||
| Y2:PDI | Minimize | |||
| Y3:ZP (mV) | Maximize | |||
| Y4:EE% | Maximize | |||
| Responses | Y1: VS (nm) | Y2: PDI | Y3: ZP (mV) | Y4: EE% |
| Minimum | 100.90 | 0.23 | 4.24 | 40.52 |
| Maximum | 633.20 | 1.00 | 35.70 | 76.34 |
| Ratio | 6.28 | 4.27 | 8.42 | 1.8 |
| Model | 2FI | 2FI | 2FI | 2FI |
| Adequate precision | 62.44 | 11.97 | 13.75 | 17.86 |
| R2 | 0.996 | 0.90 | 0.91 | 0.93 |
| Adjusted R2 | 0.994 | 0.86 | 0.86 | 0.905 |
| Predicted R2 | 0.991 | 0.78 | 0.79 | 0.85 |
| Model p-value | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| Significant variables | X1, X2 | X1, X2 | X1 | X1, X2 |
| Ion Source Settings | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ion Source | Gas Temp. (°C) | Gas Flow (L/min) | Sheath Gas Temp (°C) | Sheath Gas Flow (L/min) | Nebulizer (psi) | Capillary (V) | Nozzle Voltage | ||||
| ESI | 300 | 10 | 300 | 10 | 50 | 4500 | 2000 | ||||
| Scan Settings | |||||||||||
| Compound name | Scan Type | Polarity | Precursor ion | MS1 Res | Product Ion | MS2 Res | Dwell Time | Fragmentor | Collision Energy | Cell Accelerator Voltage | |
| Dronedarone | MRM | +ve | 557.2 | Wide | 100 | Wide | 200 | 120 | 25 | 3 | |
| Nitrofurantoin | MRM | −ve | 237 | Wide | 151.9 | Wide | 300 | 100 | 18 | 3 | |
| Run No | Formula | Variables | Responses ± SD | ||||
|---|---|---|---|---|---|---|---|
| X1: Span® Type | X2: Sp:CH | Y1:VS (nm) | Y2: PDI | Y3: ZP (mV) | Y4: EE% | ||
| 1 | N1 | Sp40 | 1:1 | 490.74 ± 16.43 | 1.00 ± 0.00 | −9.62 ± 8.74 | 66.17 ± 1.63 |
| 2 | N2 | Sp40 | 2:1 | 227.77 ± 10.66 | 1.00 ± 0.00 | −34.33 ± 0.03 | 57.66 ± 4.44 |
| 3 | N3 | Sp40 | 4:1 | 130.83 ± 5.50 | 0.61 ± 0.09 | −21.00 ± 1.85 | 42.96 ± 2.58 |
| 4 | N4 | Sp60 | 1:1 | 607.64 ± 29.50 | 1.00 ± 0.00 | −18.10 ± 1.91 | 64.63 ± 3.42 |
| 5 | N5 | Sp60 | 2:1 | 378.62 ± 8.30 | 0.59 ± 0.05 | −16.70 ± 0.46 | 50.51 ± 3.20 |
| 6 | N6 | Sp60 | 4:1 | 154.64 ± 11.52 | 0.83 ± 0.30 | −8.00 ± 1.54 | 64.55 ± 2.63 |
| 7 | N7 | Sp80 | 1:1 | 121.27 ± 13.31 | 0.43 ± 0.07 | −22.23 ± 2.84 | 73.44 ± 2.80 |
| 8 | N8 | Sp80 | 2:1 | 113.65 ± 6.16 | 0.41 ± 0.04 | −7.80 ± 1.20 | 68.80 ± 3.40 |
| 9 | N9 | Sp80 | 4:1 | 105.87 ± 6.71 | 0.25 ± 0.02 | −26.70 ± 0.78 | 66.32 ± 1.30 |
| Parameters | Freshly Prepared | After 90 Days at 4 °C | After 90 Days at 25 °C | p-Value |
|---|---|---|---|---|
| VS (nm) | 121.27 ± 13.31 | 121.58 ± 12.89 | 125.38 ± 13.43 | 0.915 |
| PDI | 0.432 ± 0.07 | 0.42 ± 0.10 | 0.44 ± 0.05 | 0.919 |
| ZP (mV) | −22.23 ± 2.80 | −21.56± 2.09 | −20.44 ± 1.89 | 0.652 |
| EE% | 73.44 ± 2.78 | 71.70± 2.79 | 70.99 ± 2.44 | 0.548 |
| Model | N7 In Situ Gel Regression Data | |
|---|---|---|
| R² | n | |
| Zero order | 0.8985 | |
| First order | 0.8368 | |
| Korsmeyer–Peppas | 0.9515 | 1.45 |
| Higuchi | 0.7185 | |
| Hixson Crowell | 0.8591 | |
| Parameters | Intranasal In Situ gel (Treatment N) | Oral Suspension (Treatment O) | p-Value |
|---|---|---|---|
| Cmax (ng/mL) | 57.99 ± 29.66 | 16.79 ± 3.42 | 0.033 |
| Tmax (h) | 2.5 | 0.5 | 0.174 |
| AUC0–72 (ng h/mL) | 766.93 ± 251.74 | 320.76 ± 93.48 | 0.016 |
| AUC0–∞ (ng h/mL) | 919.73 ± 250.03 | 469.92 ± 107.25 | 0.016 |
| t1/2 (h) | 28.28 ± 11.03 | 31.25 ± 6.64 | |
| Kel (h−1) | 0.027 ± 0.008 | 0.023 ± 0.006 | |
| % Relative bioavailability | 195.72 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teaima, M.H.; Helal, D.A.; Alsofany, J.M.; El-Nabarawi, M.A.; Yasser, M. Ion-Triggered In Situ Gelling Intranasal Spray of Dronedarone Hydrochloride Nanocarriers: In Vitro Optimization and In Vivo Pharmacokinetic Appraisal. Pharmaceutics 2022, 14, 2405. https://doi.org/10.3390/pharmaceutics14112405
Teaima MH, Helal DA, Alsofany JM, El-Nabarawi MA, Yasser M. Ion-Triggered In Situ Gelling Intranasal Spray of Dronedarone Hydrochloride Nanocarriers: In Vitro Optimization and In Vivo Pharmacokinetic Appraisal. Pharmaceutics. 2022; 14(11):2405. https://doi.org/10.3390/pharmaceutics14112405
Chicago/Turabian StyleTeaima, Mahmoud H., Doaa A. Helal, Jihad M. Alsofany, Mohamed A. El-Nabarawi, and Mohamed Yasser. 2022. "Ion-Triggered In Situ Gelling Intranasal Spray of Dronedarone Hydrochloride Nanocarriers: In Vitro Optimization and In Vivo Pharmacokinetic Appraisal" Pharmaceutics 14, no. 11: 2405. https://doi.org/10.3390/pharmaceutics14112405
APA StyleTeaima, M. H., Helal, D. A., Alsofany, J. M., El-Nabarawi, M. A., & Yasser, M. (2022). Ion-Triggered In Situ Gelling Intranasal Spray of Dronedarone Hydrochloride Nanocarriers: In Vitro Optimization and In Vivo Pharmacokinetic Appraisal. Pharmaceutics, 14(11), 2405. https://doi.org/10.3390/pharmaceutics14112405