
Peptides vs. Polymers: Searching for the Most Efficient Delivery System for Mitochondrial Gene Therapy

 , ,
, ,  ,
,  and
and 
Abstract
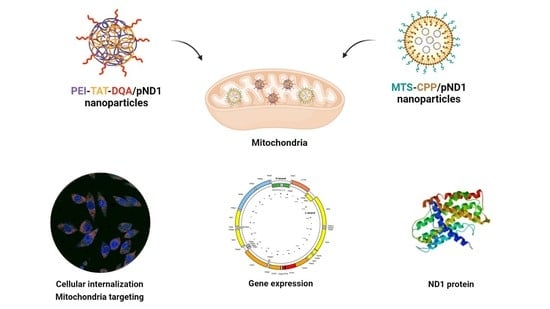
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Synthesis of Peptides and PEI–DQA
2.2.2. Nuclear Magnetic Resonance Spectroscopy (NMR)
2.2.3. Formulation of Peptide/pDNA and PEI–DQA/TAT/pDNA Complexes
2.2.4. Determination of Size and Surface Charge
2.2.5. Cell Culture
2.2.6. Cytotoxicity Evaluation
2.2.7. Fluorescence Confocal Microscopy
FITC Plasmid Labeling
Live Cell Imaging
2.2.8. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
2.2.9. Mitochondrial Isolation
2.2.10. Protein Quantification
2.2.11. Statistical Analysis
3. Results and Discussion
3.1. Synthesis and Characterization of PEI–SA, and PEI–DQA
3.2. pDNA Complexation Capacity
3.3. Characterization of PEI–DQA/TAT/pND1 Complexes
3.4. Cytotoxic Profile
3.5. Mitochondrial Targeting Ability
3.6. Evaluation of Gene Expression
3.7. Quantification of Protein
3.8. Integrity of Mitochondria
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Herst, P.M.; Rowe, M.R.; Carson, G.M.; Berridge, M.V. Functional Mitochondria in Health and Disease. Front. Endocrinol. 2017, 8, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filograna, R.; Mennuni, M.; Alsina, D.; Larsson, N. Mitochondrial DNA copy number in human disease: The more the better? FEBS Lett. 2021, 595, 976–1002. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Hong, D.; Zhang, W.; Yao, S.; Qi, X.; Lv, H.; Zheng, R.; Feng, L.; Huang, Y.; Yuan, Y.; et al. Mutations in mitochondrially encoded complex I enzyme as the second common cause in a cohort of Chinese patients with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes. J. Hum. Genet. 2011, 56, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Hertweck, K.; Dasgupta, S. The Landscape of mtDNA Modifications in Cancer: A Tale of Two Cities. Front. Oncol. 2017, 7, 262. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, D.; Blaza, J.; Larsson, N.-G.; Hirst, J. The Enigma of the Respiratory Chain Supercomplex. Cell Metab. 2017, 25, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Filippi, M.-D.; Ghaffari, S. Mitochondria in the maintenance of hematopoietic stem cells: New perspectives and opportunities. Blood 2019, 133, 1943–1952. [Google Scholar] [CrossRef]
- Mehta, M.; Weinberg, S.; Chandel, M.M.M.S.E.W.N.S. Mitochondrial control of immunity: Beyond ATP. Nat. Rev. Immunol. 2017, 17, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Kozlov, A.V.; Camara, A.K.S. Mitochondria in Health and Diseases. Cells 2020, 9, 1177. [Google Scholar] [CrossRef] [PubMed]
- Popov, L. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, M.; Prasad, S.; Tripathi, A.; Pandey, A.N.; Ali, I.; Singh, A.K.; Shrivastav, T.G.; Chaube, S.K. Apoptosis in mammalian oocytes: A review. Apoptosis 2015, 20, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 2018, 34, 879–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Zarante, A.M.; Almannai, M.; Scaglia, F. Therapies for mitochondrial diseases and current clinical trials. Mol. Genet. Metab. 2017, 122, 1–9. [Google Scholar] [CrossRef]
- Pfeffer, G.; Majamaa, K.; Turnbull, D.M.; Thorburn, D.; Chinnery, P.F. Treatment for mitochondrial disorders. Cochrane Database Syst. Rev. 2012, 2012, CD004426. [Google Scholar] [CrossRef] [PubMed]
- Siva, M.A.; Mahalakshmi, R.; Bhakta-Guha, D.; Guha, G. Gene therapy for the mitochondrial genome: Purging mutations, pacifying ailments. Mitochondrion 2019, 46, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, E.; Batista, C.; Sousa, F.; Queiroz, J.; Costa, D. Mitochondrial Gene Therapy: Advances in Mitochondrial Gene Cloning, Plasmid Production, and Nanosystems Targeted to Mitochondria. Mol. Pharm. 2017, 14, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Faria, R.; Vivés, E.; Boisguerin, P.; Sousa, A.; Costa, D. Development of Peptide-Based Nanoparticles for Mitochondrial Plasmid DNA Delivery. Polymers 2021, 13, 1836. [Google Scholar] [CrossRef] [PubMed]
- Yasuzaki, Y.; Yamada, Y.; Ishikawa, T.; Harashima, H. Validation of Mitochondrial Gene Delivery in Liver and Skeletal Muscle via Hydrodynamic Injection Using an Artificial Mitochondrial Reporter DNA Vector. Mol. Pharm. 2015, 12, 4311–4320. [Google Scholar] [CrossRef] [PubMed]
- Vercauteren, D.; Rejman, J.; Martens, T.F.; Demeester, J.; De Smedt, S.C.; Braeckmans, K. On the cellular processing of non-viral nanomedicines for nucleic acid delivery: Mechanisms and methods. J. Control. Release 2012, 161, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Emmanuele, V.; Quinzii, C.M. Emerging therapies for mitochondrial diseases. Essays Biochem. 2018, 62, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Neves, A.R.; Sousa, A.; Faria, R.; Albuquerque, T.; Queiroz, J.; Costa, D. Cancer gene therapy mediated by RALA/plasmid DNA vectors: Nitrogen to phosphate groups ratio (N/P) as a tool for tunable transfection efficiency and apoptosis. Colloids Surf. B Biointerfaces 2020, 185, 110610. [Google Scholar] [CrossRef]
- Gómez-Aguado, I.; Rodríguez-Castejón, J.; Vicente-Pascual, M.; Rodríguez-Gascón, A.; Del Pozo-Rodríguez, A.; Aspiazu, M. Ángeles, S. Nucleic Acid Delivery by Solid Lipid Nanoparticles Containing Switchable Lipids: Plasmid DNA vs. Messenger RNA. Molecules 2020, 25, 5995. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, K.; Zhao, J.; Gao, B.; Feng, Y. Polymeric nano-carriers for on-demand delivery of genes via specific responses to stimuli. J. Mater. Chem. B 2020, 8, 9621–9641. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ding, S.; Zhang, Z.; Wang, L.; You, Y. Cationic micelle: A promising nanocarrier for gene delivery with high transfection efficiency. J. Gene Med. 2019, 21, e3101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, D.; Albuqerque, T.; Queiroz, J.; Valente, A. A co-delivery platform based on plasmid DNA peptide-surfactant complexes: Formation, characterization and release behavior. Colloids Surf. B Biointerfaces 2019, 178, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, T.; Faria, R.; Sousa, Â.; Neves, A.R.; Queiroz, J.A.; Costa, D. Polymer-peptide ternary systems as a tool to improve the properties of plasmid DNA vectors in gene delivery. J. Mol. Liq. 2020, 309, 113157. [Google Scholar] [CrossRef]
- Neves, A.R.; Albuquerque, T.; Faria, R.; Paul, M.; Biswas, S.; Sousa, Â.; Costa, D. Development of Tailor-Made Dendrimer Ternary Complexes for Drug/Gene Co-Delivery in Cancer. Pharmaceutics 2021, 13, 1256. [Google Scholar] [CrossRef]
- Falanga, A.; Lombardi, L.; Galdiero, E.; Del Genio, V.; Galdiero, S. The world of cell penetrating: The future of medical applications. Future Med. Chem. 2020, 12, 1431–1446. [Google Scholar] [CrossRef]
- Deshayes, S.; Konate, K.; Dussot, M.; Chavey, B.; Vaissière, A.; Van, T.N.N.; Aldrian, G.; Padari, K.; Pooga, M.; Vivès, E.; et al. Deciphering the internalization mechanism of WRAP:siRNA nanoparticles. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183252. [Google Scholar] [CrossRef]
- Sousa, Â.; Almeida, A.M.; Faria, R.; Konate, K.; Boisguerin, P.; Queiroz, J.; Costa, D. Optimization of peptide-plasmid DNA vectors formulation for gene delivery in cancer therapy exploring design of experiments. Colloids Surf. B Biointerfaces 2019, 183, 110417. [Google Scholar] [CrossRef] [PubMed]
- Konate, K.; Dussot, M.; Aldrian, G.; Vaissière, A.; Viguier, V.; Neira, I.F.; Couillaud, F.; Vivès, E.; Boisguerin, P.; Deshayes, S. Peptide-Based Nanoparticles to Rapidly and Efficiently “Wrap ’n Roll” siRNA into Cells. Bioconjug. Chem. 2019, 30, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.M.; Filipczak, N.; Torchilin, V.P. Cell penetrating peptides: A versatile vector for co-delivery of drug and genes in cancer. J. Control. Release 2021, 330, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Gessner, I.; Neundorf, I. Nanoparticles Modified with Cell-Penetrating Peptides: Conjugation Mechanisms, Physicochemical Properties, and Application in Cancer Diagnosis and Therapy. Int. J. Mol. Sci. 2020, 21, 2536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derakhshankhah, H.; Jafari, S. Cell penetrating peptides: A concise review with emphasis on biomedical applications. Biomed. Pharmacother. 2018, 108, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Ruseska, I.; Zimmer, A. Internalization mechanisms of cell-penetrating peptides. Beilstein J. Nanotechnol. 2020, 11, 101–123. [Google Scholar] [CrossRef] [PubMed]
- Bakar, L.M.; Abdullah, M.Z.; Doolaanea, A.A.; Ichwan, S.J.A. PLGA-Chitosan nanoparticle-mediated gene delivery for oral cancer treatment: A brief review. J. Phys. Conf. Ser. 2017, 884, 012117. [Google Scholar] [CrossRef]
- Lellouche, E.; Locatelli, E.; Israel, L.L.; Naddaka, M.; Kurlander, E.; Michaeli, S.; Lellouche, J.-P.; Franchini, M.C. Maghemite-containing PLGA-PEG-based polymeric nanoparticles for siRNA delivery: Toxicity and silencing evaluation. RSC Adv. 2017, 7, 26912–26920. [Google Scholar] [CrossRef] [Green Version]
- Piperno, A.; Sciortino, M.T.; Giusto, E.; Montesi, M.; Panseri, S.; Scala, A. Recent Advances and Challenges in Gene Delivery Mediated by Polyester-Based Nanoparticles. Int. J. Nanomed. 2021, 16, 5981–6002. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liao, X.; Zhang, G.; Mu, C. Dynamical Analysis of the Generalized Lorenz Systems. J. Dyn. Control Syst. 2017, 23, 349–362. [Google Scholar] [CrossRef]
- Costa, D.; Briscoe, W.H.; Queiroz, J. Polyethylenimine coated plasmid DNA-surfactant complexes as potential gene delivery systems. Colloids Surf. B Biointerfaces 2015, 133, 156–163. [Google Scholar] [CrossRef]
- Faria, R.; Sousa, Â.; Neves, A.R.; Queiroz, J.A.; Costa, D. Methotrexate-plasmid DNA polyplexes for cancer therapy: Characterization, cancer cell targeting ability and tuned in vitro transfection. J. Mol. Liq. 2019, 292, 292. [Google Scholar] [CrossRef]
- Cheraghi, R.; Alipour, M.; Nazari, M.; Hosseinkhani, S. Optimization of conditions for gene delivery system based on PEI. Nanomedicine 2017, 4, 8–16. [Google Scholar]
- Benjaminsen, R.V.; Mattebjerg, M.A.; Henriksen, J.R.; Moghimi, S.M.; Andresen, T.L. The Possible “Proton Sponge” Effect of Polyethylenimine (PEI) Does Not Include Change in Lysosomal pH. Mol. Ther. 2013, 21, 149–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaltimbacher, V.; Bonnet, C.; Lecoeuvre, G.; Forster, V.; Sahel, J.-A.; Corral-Debrinski, M. mRNA localization to the mitochondrial surface allows the efficient translocation inside the organelle of a nuclear recoded ATP6 protein. RNA 2006, 12, 1408–1417. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, C.; Kaltimbacher, V.; Ellouze, S.; Augustin, S.; Bénit, P.; Forster, V.; Rustin, P.; Sahel, J.-A.; Corral-Debrinski, M. Allotopic mRNA Localization to the Mitochondrial Surface Rescues Respiratory Chain Defects in Fibroblasts Harboring Mitochondrial DNA Mutations Affecting Complex I or V Subunits. Rejuvenation Res. 2007, 10, 127–144. [Google Scholar] [CrossRef]
- Bonnet, C.; Augustin, S.; Ellouze, S.; Bénit, P.; Bouaita, A.; Rustin, P.; Sahel, J.-A.; Corral-Debrinski, M. The optimized allotopic expression of ND1 or ND4 genes restores respiratory chain complex I activity in fibroblasts harboring mutations in these genes. Biochim. Biophys. Acta 2008, 1783, 1707–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellouze, S.; Augustin, S.; Bouaita, A.; Bonnet, C.; Simonutti, M.; Forster, V.; Picaud, S.; Sahel, J.-A.; Corral-Debrinski, M. Optimized Allotopic Expression of the Human Mitochondrial ND4 Prevents Blindness in a Rat Model of Mitochondrial Dysfunction. Am. J. Hum. Genet. 2008, 83, 373–387. [Google Scholar] [CrossRef] [Green Version]
- Korake, S.; Gajbhiye, K.R. Dequalinium-Derived Nanoconstructs: A Promising Vehicle for Mitochondrial Targeting. Curr. Drug Deliv. 2021, 18, 1056–1063. [Google Scholar] [CrossRef]
- Chuah, J.-A.; Matsugami, A.; Hayashi, F.; Numata, K. Self-Assembled Peptide-Based System for Mitochondrial-Targeted Gene Delivery: Functional and Structural Insights. Biomacromolecules 2016, 17, 3547–3557. [Google Scholar] [CrossRef] [PubMed]
- Lyrawati, D.; Trounson, A.; Cram, D. Expression of GFP in the Mitochondrial Compartment Using DQAsome-Mediated Delivery of an Artificial Mini-mitochondrial Genome. Pharm. Res. 2011, 28, 2848–2862. [Google Scholar] [CrossRef] [PubMed]
- Weissig, V. DQAsomes as the Prototype of Mitochondria-Targeted Pharmaceutical Nanocarriers: Preparation, Characterization, and Use. Methods Mol. Biol. 2015, 1265, 1–11. [Google Scholar] [CrossRef]
- Mallick, S.; Song, S.J.; Bae, Y.; Choi, J.S. Self-assembled nanoparticles composed of glycol chitosan-dequalinium for mitochondria-targeted drug delivery. Int. J. Biol. Macromol. 2019, 132, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Weissig, V.; Torchilin, V.P. Towards Mitochondrial Gene Therapy: DQAsomes as a Strategy. J. Drug Target. 2001, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zupančič, Š.; Kocbek, P.; Zariwala, M.G.; Renshaw, D.; Gul, M.O.; Elsaid, Z.; Taylor, K.M.G.; Somavarapu, S. Design and Development of Novel Mitochondrial Targeted Nanocarriers, DQAsomes for Curcumin Inhalation. Mol. Pharm. 2014, 11, 2334–2345. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yin, H.; Yan, F.; Sun, M.; Du, L.; Peng, W.; Li, Q.; Feng, Y.; Zhou, Y. Folate-mediated mitochondrial targeting with doxorubicin-polyrotaxane nanoparticles overcomes multidrug resistance. Oncotarget 2014, 6, 2827–2842. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Khan, A.; Liu, Y.; Feng, J.; Dai, L.; Wang, G.; Alam, N.; Tong, L.; Ni, Y. Chitosan oligosaccharide-based dual pH responsive nano-micelles for targeted delivery of hydrophobic drugs. Carbohydr. Polym. 2019, 223, 115061. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, Q.; Xia, X.; Li, X.; Ruan, W.; Zheng, M.; Zou, Y.; Shi, B. Polymeric Nanoparticles for Mitochondria Targeting Mediated Robust Cancer Therapy. Front. Bioeng. Biotechnol. 2021, 9, 755727. [Google Scholar] [CrossRef] [PubMed]
- Faria, R.; Albuquerque, T.; Neves, A.R.; Bhatt, H.; Biswas, S.; Cardoso, A.M.; de Lima, M.C.P.; Jurado, A.S.; Costa, D. Physicochemical characterization and targeting performance of triphenylphosphonium nano-polyplexes. J. Mol. Liq. 2020, 316, 113873. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faria, R.; Paul, M.; Biswas, S.; Vivès, E.; Boisguérin, P.; Sousa, Â.; Costa, D. Peptides vs. Polymers: Searching for the Most Efficient Delivery System for Mitochondrial Gene Therapy. Pharmaceutics 2022, 14, 757. https://doi.org/10.3390/pharmaceutics14040757
Faria R, Paul M, Biswas S, Vivès E, Boisguérin P, Sousa Â, Costa D. Peptides vs. Polymers: Searching for the Most Efficient Delivery System for Mitochondrial Gene Therapy. Pharmaceutics. 2022; 14(4):757. https://doi.org/10.3390/pharmaceutics14040757
Chicago/Turabian StyleFaria, Rúben, Milan Paul, Swati Biswas, Eric Vivès, Prisca Boisguérin, Ângela Sousa, and Diana Costa. 2022. "Peptides vs. Polymers: Searching for the Most Efficient Delivery System for Mitochondrial Gene Therapy" Pharmaceutics 14, no. 4: 757. https://doi.org/10.3390/pharmaceutics14040757
APA StyleFaria, R., Paul, M., Biswas, S., Vivès, E., Boisguérin, P., Sousa, Â., & Costa, D. (2022). Peptides vs. Polymers: Searching for the Most Efficient Delivery System for Mitochondrial Gene Therapy. Pharmaceutics, 14(4), 757. https://doi.org/10.3390/pharmaceutics14040757