
The Current State of the Art in PARP Inhibitor-Based Delivery Nanosystems

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
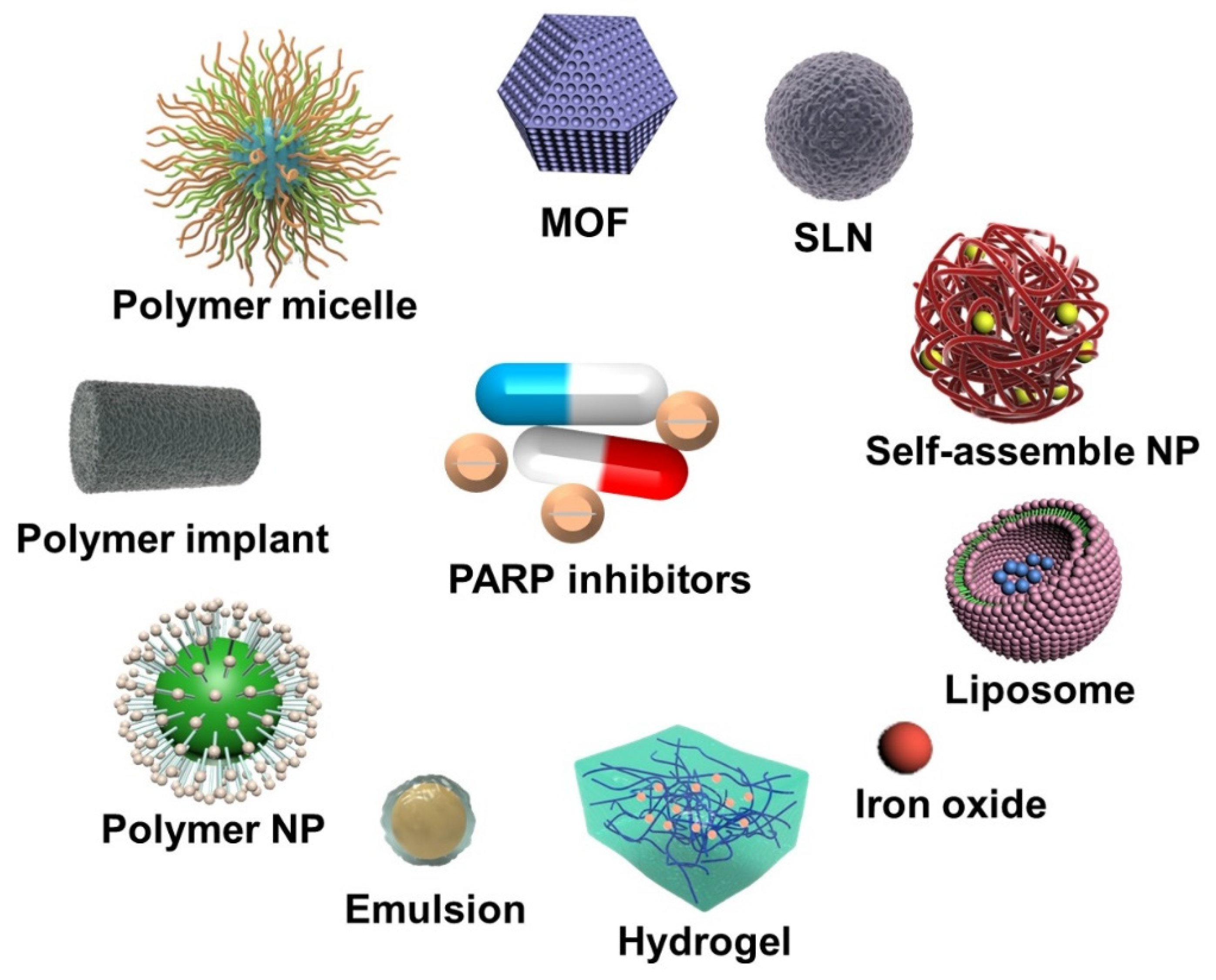
2. Nanoparticle-Based PARP Inhibitor Delivery Systems
2.1. PARPi Monotherapy
2.1.1. Olaparib-Based Nanosystems
2.1.2. Talazoparib-Based Nanosystems
2.1.3. Others
2.2. Combined Therapy
2.2.1. Chemotherapeutic Drugs
Hybrid Nanosystems
Self-Assembled NPs
Active Targeted NPs
2.2.2. Radiotherapy
Olaparib-Based Nanosystems
Talazoparib-Based Nanosystems
2.2.3. Photodynamic Therapy
2.3. Others
3. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chi, J.; Su, Y.C.; Parakrama, R.; Fayyaz, F.; Saif, M.W. The role of PARP inhibitors in BRCA mutated pancreatic cancer. Ther. Adv. Gastroenterol. 2021, 14, 17562848211014818. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Wu, Z.; Cui, P.; Tao, H.; Zhang, S.; Hu, Y. The Synergistic Effect of PARP Inhibitors and Immune Checkpoint Inhibitors. Clin. Med. Insights Oncol. 2021, 15, 117955492199628. [Google Scholar] [CrossRef] [PubMed]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair 2018, 71, 177–182. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Bono, J. Inhibition of Poly(ADP-Ribose)Polymerase in Tumors from BRCA Mutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knelson, E.H.; Patel, S.A.; Sands, J.M. PARP Inhibitors in Small-Cell Lung Cancer: Rational Combinations to Improve Responses. Cancers 2021, 13, 727. [Google Scholar] [CrossRef]
- Boussios, S.; Karihtala, P.; Moschetta, M.; Karathanasi, A.; Pavlidis, N. Combined Strategies with Poly(ADP-Ribose)Polymerase (PARP) Inhibitors for the Treatment of Ovarian Cancer: A Literature Review. Diagnostics 2019, 9, 87. [Google Scholar] [CrossRef] [Green Version]
- Murai, J.; Huang, S.-Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Bonadio, R.C.; Estevez-Diz, M.D.P. Perspectives on PARP Inhibitor Combinations for Ovarian Cancer. Front. Oncol. 2021, 11, 754524. [Google Scholar] [CrossRef]
- Turk, A.A.; Wisinski, K.B. PARP inhibitors in breast cancer: Bringing synthetic lethality to the bedside. Cancer 2018, 124, 2498–2506. [Google Scholar] [CrossRef]
- Cortesi, L.; Rugo, H.S.; Jackisch, C. An Overview of PARP Inhibitors for the Treatment of Breast Cancer. Target Oncol. 2021, 16, 255–282. [Google Scholar] [CrossRef] [PubMed]
- Layman, R.M.; Arun, B. PARP Inhibitors in Triple-Negative Breast Cancer Including Those with BRCA Mutations. Cancer J. 2021, 27, 67–75. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, V.; Paunu, K.; Ahlskog, J.K.; Varnai, R.; Sipeky, C.; Sundvall, M. PARP Inhibitors in Prostate Cancer—The Preclinical Rationale and Current Clinical Development. Genes 2019, 10, 565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathade, A.D.; Kommineni, N.; Bulbake, U.; Thummar, M.M.; Samanthula, G.; Khan, W. Preparation and Comparison of Oral Bioavailability for Different Nano-formulations of Olaparib. AAPS PharmSciTech 2019, 20, 276. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Herman, T.F.; Santos, C. First Pass Effect; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Wang, X.; Zhang, C.; Han, N.; Luo, J.; Zhang, S.; Wang, C.; Jia, Z.; Du, S. Triglyceride-mimetic prodrugs of scutellarin enhance oral bioavailability by promoting intestinal lymphatic transport and avoiding first-pass metabolism. Drug Deliv. 2021, 28, 1664–1672. [Google Scholar] [CrossRef]
- Fang, J. Bioavailability of anthocyanins. Drug Metab. Rev. 2014, 46, 508–520. [Google Scholar] [CrossRef]
- Heo, Y.A.; Dhillon, S. Olaparib Tablet: A Review in Ovarian Cancer Maintenance Therapy. Target Oncol. 2018, 13, 801–808. [Google Scholar] [CrossRef]
- Moore, K.N.; Birrer, M.J. Administration of the Tablet Formulation of Olaparib in Patients with Ovarian Cancer: Practical Guidance and Expectations. Oncologist 2018, 23, 697–703. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Moreno, V.; Dean, E.J.; Drew, Y.; Nicum, S.; Ranson, M.; Plummer, R.; Swaisland, H.; Burke, W.; Mccormack, P. Phase I study to determine the bioavailability and tolerability of a tablet formulation of the PARP inhibitor olaparib in patients with advanced solid tumors: Dose-escalation phase. J. Clin. Oncol. 2012, 30, 3051-3051. [Google Scholar] [CrossRef]
- Singh, B.; Yang, S.; Krishna, A.; Sridhar, S. Nanoparticle Formulations of Poly(ADP-Ribose)Polymerase Inhibitors for Cancer Therapy. Front. Chem. 2020, 8, 594619. [Google Scholar] [CrossRef] [PubMed]
- Yordanova, M.; Hubert, A.; Hassan, S. Expanding the Use of PARP Inhibitors as Monotherapy and in Combination in Triple-Negative Breast Cancer. Pharmaceuticals 2021, 14, 1270. [Google Scholar] [CrossRef] [PubMed]
- Farokhzad, O.C.; Langer, R. Impact of nanotechnology on drug delivery. ACS Nano 2009, 3, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, B.S.; Buabeid, M.; Ibrahim, N.A.; Kharaba, Z.J.; Ijaz, M.; Noreen, S.; Murtaza, G. Potential of Nanocarrier-Based Drug Delivery Systems for Brain Targeting: A Current Review of Literature. Int. J. Nanomed. 2021, 16, 7517–7533. [Google Scholar] [CrossRef]
- Zhao, C.Y.; Cheng, R.; Yang, Z.; Tian, Z.M. Nanotechnology for Cancer Therapy Based on Chemotherapy. Molecules 2018, 23, 826. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Zhang, Y.; Zhang, Q.; Zhang, L. Nanoparticle-Hydrogel: A Hybrid Biomaterial System for Localized Drug Delivery. Ann. Biomed. Eng. 2016, 44, 2049–2061. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Li, J.; Zhao, Q.; Pan, T.; Zhong, H.; Wang, W. Advanced and Innovative Nano-Systems for Anticancer Targeted Drug Delivery. Pharmaceutics 2021, 13, 1151. [Google Scholar] [CrossRef]
- Ge, Z.; Wang, Q.; Zhu, Q.; Yusif, M.; Yu, J.; Xu, X. Improved oral bioavailability, cellular uptake, and cytotoxic activity of zingerone via nano-micelles drug delivery system. J. Microencapsul. 2021, 38, 394–404. [Google Scholar] [CrossRef]
- Ashfaq, U.A.; Riaz, M.; Yasmeen, E.; Yousaf, M.Z. Recent Advances in Nanoparticle-Based Targeted Drug-Delivery Systems against Cancer and Role of Tumor Microenvironment. Crit. Rev. Ther. Drug Carr. Syst. 2017, 34, 317–353. [Google Scholar] [CrossRef] [PubMed]
- Amreddy, N.; Babu, A.; Muralidharan, R.; Panneerselvam, J.; Srivastava, A.; Ahmed, R.; Mehta, M.; Munshi, A.; Ramesh, R. Recent Advances in Nanoparticle-Based Cancer Drug and Gene Delivery. Adv. Cancer Res. 2018, 137, 115–170. [Google Scholar] [PubMed]
- Feng, S.; Ren, Y.; Li, H.; Tang, Y.; Yan, J.; Shen, Z.; Zhang, H.; Chen, F. Cancer Cell-Membrane Biomimetic Boron Nitride Nanospheres for Targeted Cancer Therapy. Int. J. Nanomed. 2021, 16, 2123–2136. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-Based Drug Delivery in Cancer Therapy and Its Role in Overcoming Drug Resistance. Front. Mol. Biosci. 2020, 7, 193. [Google Scholar] [CrossRef]
- Zhou, L.; Li, Y.; Liang, Q.; Liu, J.; Liu, Y. Combination therapy based on targeted nano drug co-delivery systems for liver fibrosis treatment: A review. J. Drug Target. 2022, 30, 577–588. [Google Scholar] [CrossRef]
- Liang, Y.; Liu, Z.Y.; Wang, P.Y.; Li, Y.J.; Wang, R.R.; Xie, S.Y. Nanoplatform-based natural products co-delivery system to surmount cancer multidrug-resistant. J. Control. Release 2021, 336, 396–409. [Google Scholar] [CrossRef]
- Sargazi, S.; Mukhtar, M.; Rahdar, A.; Barani, M.; Pandey, S.; Díez-Pascual, A.M. Active Targeted Nanoparticles for Delivery of Poly(ADP-Ribose)Polymerase (PARP) Inhibitors: A Preliminary Review. Int. J. Mol. Sci. 2021, 22, 10319. [Google Scholar] [CrossRef]
- Dréan, A.; Lord, C.J.; Ashworth, A. PARP inhibitor combination therapy. Crit. Rev. Oncol. Hematol. 2016, 108, 73–85. [Google Scholar] [CrossRef]
- Hennes, E.R.; Dow-Hillgartner, E.N.; Bergsbaken, J.J.; Piccolo, J.K. PARP-inhibitor potpourri: A comparative review of class safety, efficacy and cost. J. Oncol. Pharm. Pract. Off. Publ. Int. Soc. Oncol. Pharm. Pract. 2020, 26, 718–729. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Rodrigues, D.N.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Van de Ven, A.L.; Tangutoori, S.; Baldwin, P.; Qiao, J.; Gharagouzloo, C.; Seitzer, N.; Clohessy, J.G.; Makrigiorgos, G.M.; Cormack, R.; Pandolfi, P.P.; et al. Nanoformulation of Olaparib Amplifies PARP Inhibition and Sensitizes PTEN/TP53-Deficient Prostate Cancer to Radiation. Mol. Cancer Ther. 2017, 16, 1279–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frampton, J.E. Olaparib: A review of its use as maintenance therapy in patients with ovarian cancer. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2015, 29, 143–150. [Google Scholar] [CrossRef]
- Baldwin, P.; Ohman, A.W.; Tangutoori, S.; Dinulescu, D.M.; Sridhar, S. Intraperitoneal delivery of NanoOlaparib for disseminated late-stage cancer treatment. Int. J. Nanomed. 2018, 13, 8063–8074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, B.; Barick, K.C.; Hassan, P.A. Recent advances in active targeting of nanomaterials for anticancer drug delivery. Adv. Colloid Interface Sci. 2021, 296, 102509. [Google Scholar] [CrossRef] [PubMed]
- Mazzucchelli, S.; Truffi, M.; Baccarini, F.; Beretta, M.; Corsi, F. H-Ferritin-nanocaged olaparib: A promising choice for both BRCA-mutated and sporadic triple negative breast cancer. Sci. Rep. 2017, 7, 7505. [Google Scholar] [CrossRef] [Green Version]
- Guido, C.; Maiorano, G.; Cortese, B.; D’Amone, S.; Palamà, I.E. Biomimetic Nanocarriers for Cancer Target Therapy. Bioengineering 2020, 7, 111. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.; He, R.; Xu, D.; Zang, J.; Weeranoppanant, N.; Dong, H.; Li, Y. Cell membrane biomimetic nanoparticles for inflammation and cancer targeting in drug delivery. Biomater. Sci. 2020, 8, 552–568. [Google Scholar] [CrossRef]
- Xia, Q.; Zhang, Y.; Li, Z.; Hou, X.; Feng, N. Red blood cell membrane-camouflaged nanoparticles: A novel drug delivery system for antitumor application. Acta Pharm. Sin. B 2019, 9, 675–689. [Google Scholar] [CrossRef]
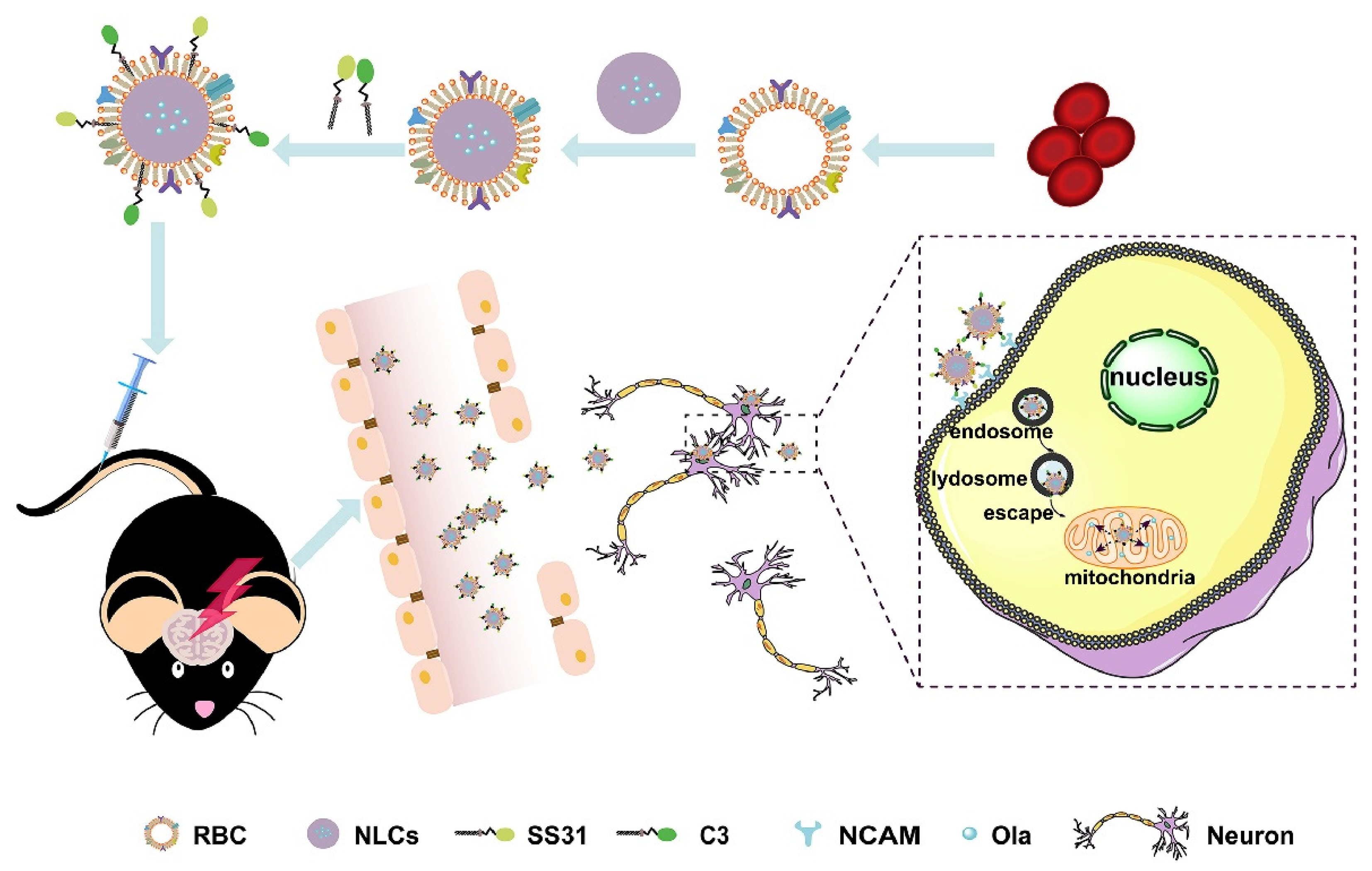
- Sun, J.; Liu, J.; Gao, C.; Zheng, J.; Zhang, J.; Ding, Y.; Gong, W.; Yang, M.; Li, Z.; Wang, Y.; et al. Targeted delivery of PARP inhibitors to neuronal mitochondria via biomimetic engineered nanosystems in a mouse model of traumatic brain injury. Acta Biomater. 2022, 140, 573–585. [Google Scholar] [CrossRef]
- Hoy, S.M. Talazoparib: First Global Approval. Drugs 2018, 78, 1939–1946. [Google Scholar] [CrossRef]
- Shen, Y.; Rehman, F.L.; Feng, Y.; Boshuizen, J.; Bajrami, I.; Elliott, R.; Wang, B.; Lord, C.J.; Post, L.E.; Ashworth, A. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin. Cancer Res. 2013, 19, 5003–5015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskiler, G.G. Talazoparib to treat BRCA-positive breast cancer. Drugs Today 1998, 55, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, P.; Tangutoori, S.; Sridhar, S. In vitro analysis of PARP inhibitor nanoformulations. Int. J. Nanomed. 2018, 13, 59–61. [Google Scholar] [CrossRef] [Green Version]
- Ahlawat, J.; Henriquez, G.; Narayan, M. Enhancing the Delivery of Chemotherapeutics: Role of Biodegradable Polymeric Nanoparticles. Molecules 2018, 23, 2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belz, J.E.; Kumar, R.; Baldwin, P.; Ojo, N.C.; Leal, A.S.; Royce, D.B.; Zhang, D.; van de Ven, A.L.; Liby, K.T.; Sridhar, S. Sustained Release Talazoparib Implants for Localized Treatment of BRCA1-deficient Breast Cancer. Theranostics 2017, 7, 4340–4349. [Google Scholar] [CrossRef]
- Mu, H.; Holm, R. Solid lipid nanocarriers in drug delivery: Characterization and design. Expert Opin. Drug Deliv. 2018, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Selvamuthukumar, S.; Velmurugan, R. Nanostructured lipid carriers: A potential drug carrier for cancer chemotherapy. Lipids Health Dis. 2012, 11, 159. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Du, C.; Guo, N.; Teng, Y.; Meng, X.; Sun, H.; Li, S.; Yu, P.; Galons, H. Composition design and medical application of liposomes. Eur. J. Med. Chem. 2019, 164, 640–653. [Google Scholar] [CrossRef]
- Guimarães, D.; Cavaco-Paulo, A.; Nogueira, E. Design of liposomes as drug delivery system for therapeutic applications. Int. J. Pharm. 2021, 601, 120571. [Google Scholar] [CrossRef]
- Zhang, D.; Baldwin, P.; Leal, A.S.; Carapellucci, S.; Sridhar, S.; Liby, K.T. A nano-liposome formulation of the PARP inhibitor Talazoparib enhances treatment efficacy and modulates immune cell populations in mammary tumors of BRCA-deficient mice. Theranostics 2019, 9, 6224–6238. [Google Scholar] [CrossRef]
- Mehra, N.K.; Tekmal, R.R.; Palakurthi, S. Development and Evaluation of Talazoparib Nanoemulsion for Systemic Therapy of BRCA1-mutant Cancer. Anticancer Res. 2018, 38, 4493–4503. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Z.Y.; Wu, N.; Chen, Y.C.; Cheng, Q.; Wang, J. PARP inhibitor resistance: The underlying mechanisms and clinical implications. Mol. Cancer 2020, 19, 107. [Google Scholar] [CrossRef] [PubMed]
- Eskiler, G.G.; Cecener, G.; Egeli, U.; Tunca, B. Synthetically Lethal BMN 673 (Talazoparib) Loaded Solid Lipid Nanoparticles for BRCA1 Mutant Triple Negative Breast Cancer. Pharm. Res. 2018, 35, 218. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.M.; El-Karim, S.S.A.; Mahmoud, A.H.; Amr, A.E.E.; Al-Omar, M.A. A Comparative Study of the Anticancer Activity and PARP-1 Inhibiting Effect of Benzofuran-Pyrazole Scaffold and Its Nano-Sized Particles in Human Breast Cancer Cells. Molecules 2019, 24, 2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasa, S.S.K.; Diakova, G.; Suzuki, R.; Mills, A.M.; Gutknecht, M.F.; Klibanov, A.L.; Slack-Davis, J.K.; Kelly, K.A. Plectin-targeted liposomes enhance the therapeutic efficacy of a PARP inhibitor in the treatment of ovarian cancer. Theranostics 2018, 8, 2782–2798. [Google Scholar] [CrossRef] [PubMed]
- Yeung, Y.T.; Aziz, F.; Guerrero-Castilla, A.; Arguelles, S. Signaling Pathways in Inflammation and Anti-inflammatory Therapies. Curr. Pharm. Des. 2018, 24, 1449–1484. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [Green Version]
- Correia, A.S.; Gärtner, F.; Vale, N. Drug combination and repurposing for cancer therapy: The example of breast cancer. Heliyon 2021, 7, e05948. [Google Scholar] [CrossRef]
- Pezaro, C. PARP inhibitor combinations in prostate cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835919897537. [Google Scholar] [CrossRef] [Green Version]
- Min, A.; Im, S.A. PARP Inhibitors as Therapeutics: Beyond Modulation of PARylation. Cancers 2020, 12, 394. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- McCrorie, P.; Mistry, J.; Taresco, V.; Lovato, T.; Fay, M.; Ward, I.; Ritchie, A.A.; Clarke, P.A.; Smith, S.J.; Marlow, M.; et al. Etoposide and olaparib polymer-coated nanoparticles within a bioadhesive sprayable hydrogel for post-surgical localised delivery to brain tumours. Eur. J. Pharm. Biopharm. 2020, 157, 108–120. [Google Scholar] [CrossRef]
- Novohradsky, V.; Zajac, J.; Vrana, O.; Kasparkova, J.; Brabec, V. Simultaneous delivery of olaparib and carboplatin in PEGylated liposomes imparts this drug combination hypersensitivity and selectivity for breast tumor cells. Oncotarget 2018, 9, 28456–28473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, R.; Patra, B.; Varadharaj, S.; Verma, R.S. Establishing the promising role of novel combination of triple therapeutics delivery using polymeric nanoparticles for Triple negative breast cancer therapy. BioImpacts 2021, 11, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.A.; Shanker, R. Toxicity of Nanomaterials. Biomed. Res. Int. 2015, 2015, 521014. [Google Scholar] [CrossRef]
- Allen, C. The question of toxicity of nanomaterials and nanoparticles. J. Control. Release 2019, 304, 288. [Google Scholar] [CrossRef] [PubMed]
- Ao, M.; Yu, F.; Li, Y.; Zhong, M.; Tang, Y.; Yang, H.; Wu, X.; Zhuang, Y.; Wang, H.; Sun, X.; et al. Carrier-free nanoparticles of camptothecin prodrug for chemo-photothermal therapy: The making, in vitro and in vivo testing. J. Nanobiotechnol. 2021, 19, 350. [Google Scholar] [CrossRef]
- Zhou, M.; Han, S.; Aras, O.; An, F. Recent Advances in Paclitaxel-based Self-Delivery Nanomedicine for Cancer Therapy. Curr. Med. Chem. 2021, 28, 6358–6374. [Google Scholar] [CrossRef]
- Zhu, L.; Guo, Y.; Qian, Q.; Yan, D.; Li, Y.; Zhu, X.; Zhang, C. Carrier-Free Delivery of Precise Drug-Chemogene Conjugates for Synergistic Treatment of Drug-Resistant Cancer. Angew. Chem. 2020, 59, 17944–17950. [Google Scholar] [CrossRef]
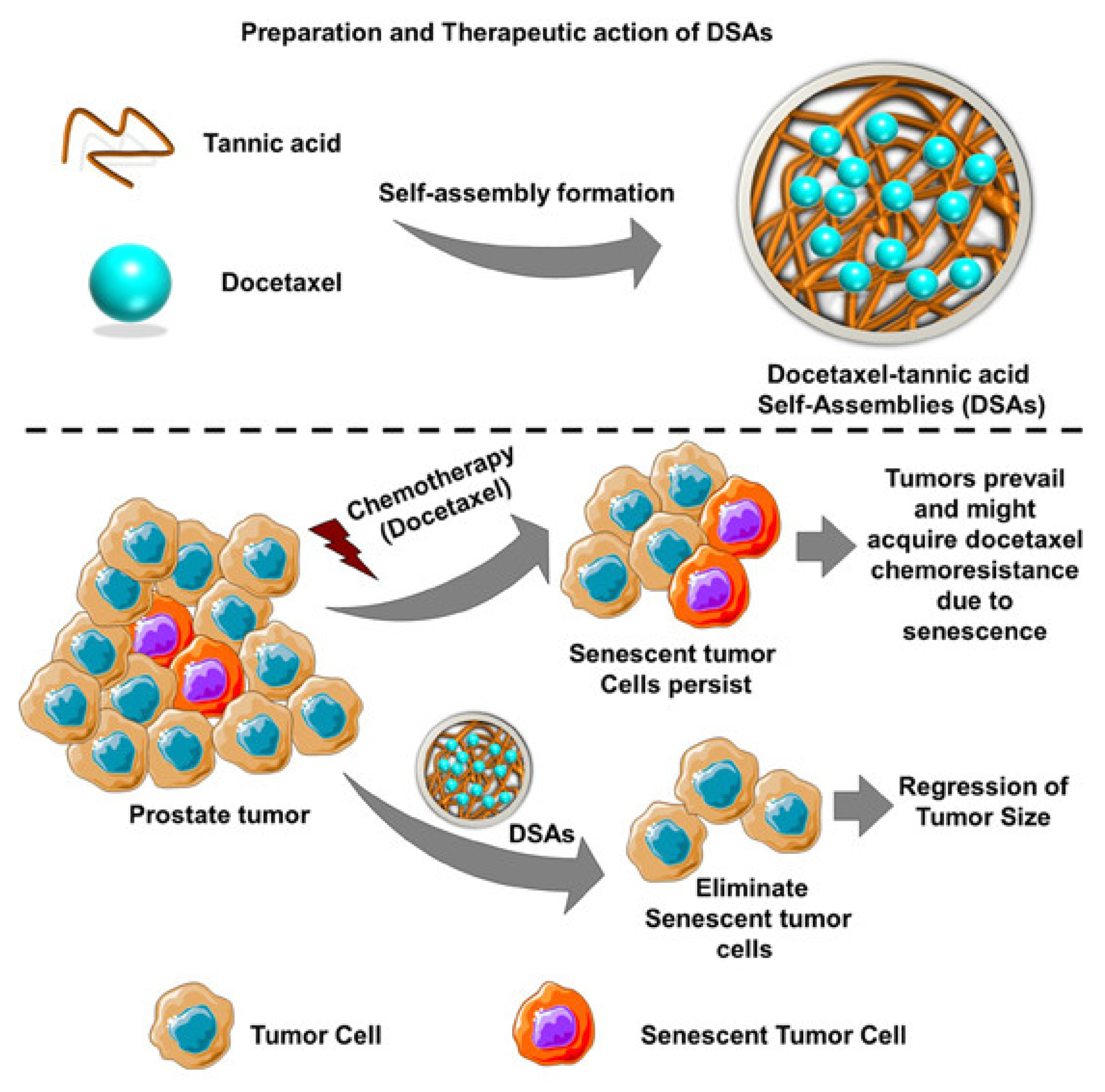
- Nagesh, P.K.B.; Chowdhury, P.; Hatami, E.; Kumari, S.; Kashyap, V.K.; Tripathi, M.K.; Wagh, S.; Meibohm, B.; Chauhan, S.C.; Jaggi, M.; et al. Cross-Linked Polyphenol-Based Drug Nano-Self-Assemblies Engineered to Blockade Prostate Cancer Senescence. ACS Appl. Mater. Interfaces 2019, 11, 38537–38554. [Google Scholar] [CrossRef]
- Chowdhury, P.; Nagesh, P.K.B.; Hatami, E.; Wagh, S.; Dan, N.; Tripathi, M.K.; Khan, S.; Hafeez, B.B.; Meibohm, B.; Chauhan, S.C.; et al. Tannic acid-inspired paclitaxel nanoparticles for enhanced anticancer effects in breast cancer cells. J. Colloid Interface Sci. 2019, 535, 133–148. [Google Scholar] [CrossRef] [PubMed]
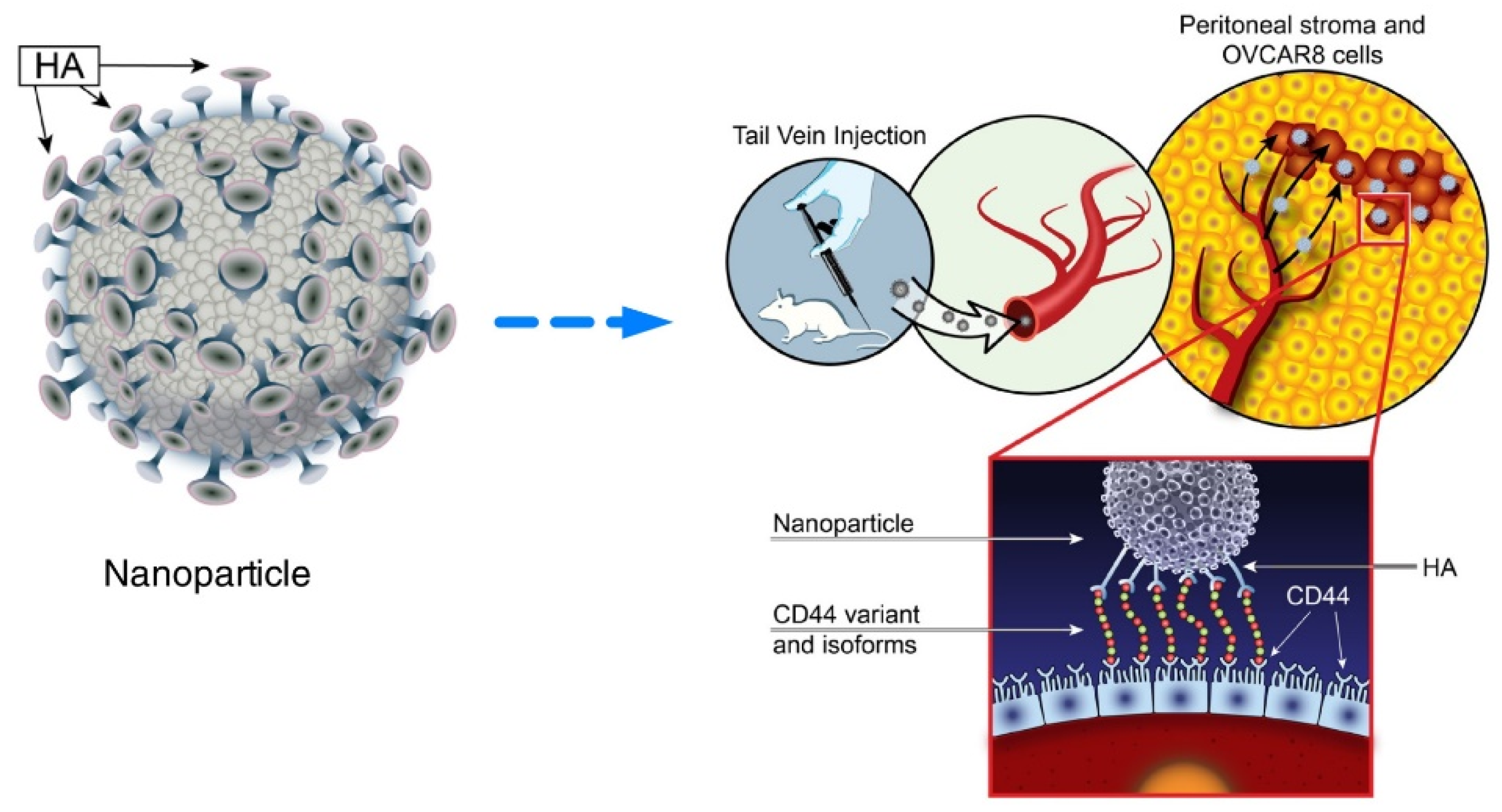
- Sacks, J.D.; Barbolina, M.V. Expression and Function of CD44 in Epithelial Ovarian Carcinoma. Biomolecules 2015, 5, 3051–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mensah, L.B.; Morton, S.W.; Li, J.; Xiao, H.; Quadir, M.A.; Elias, K.M.; Penn, E.; Richson, A.K.; Ghoroghchian, P.P.; Liu, J.; et al. Layer-by-layer nanoparticles for novel delivery of cisplatin and PARP inhibitors for platinum-based drug resistance therapy in ovarian cancer. Bioeng. Transl. Med. 2019, 4, e10131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, C.; Qi, Y.; Zhang, Y.; Wang, Y.; Zhao, X.; Min, H.; Han, X.; Lang, J.; Qin, H.; Shi, Q.; et al. Epidermal Growth Factor Receptor-Targeting Peptide Nanoparticles Simultaneously Deliver Gemcitabine and Olaparib to Treat Pancreatic Cancer with Breast Cancer 2 (BRCA2) Mutation. ACS Nano 2018, 12, 10785–10796. [Google Scholar] [CrossRef] [PubMed]
- Citrin, D.E. Recent Developments in Radiotherapy. N. Engl. J. Med. 2017, 377, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Barcellini, A.; Loap, P.; Murata, K.; Villa, R.; Kirova, Y.; Okonogi, N.; Orlandi, E. PARP Inhibitors in Combination with Radiotherapy: To Do or Not to Do? Cancers 2021, 13, 5380. [Google Scholar] [CrossRef]
- Jannetti, S.A.; Zeglis, B.M.; Zalutsky, M.R.; Reiner, T. Poly(ADP-Ribose)Polymerase (PARP) Inhibitors and Radiation Therapy. Front. Pharmacol. 2020, 11, 170. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Liu, J.; Hu, C.; Li, D.; Yang, J.; Wu, Z.; Yang, L.; Chen, Y.; Fu, S.; Wu, J. Olaparib nanoparticles potentiated radiosensitization effects on lung cancer. Int. J. Nanomed. 2018, 13, 8461–8472. [Google Scholar] [CrossRef] [Green Version]
- Farran, B.; Montenegro, R.C.; Kasa, P.; Pavitra, E.; Huh, Y.S.; Han, Y.K.; Kamal, M.A.; Nagaraju, G.P.; Raju, G.S.R. Folate-conjugated nanovehicles: Strategies for cancer therapy. Mater. Sci. Eng. C Mater. Biol. Appl. 2020, 107, 110341. [Google Scholar] [CrossRef]
- Li, D.; Hu, C.; Yang, J.; Liao, Y.; Chen, Y.; Fu, S.Z.; Wu, J.B. Enhanced Anti-Cancer Effect of Folate-Conjugated Olaparib Nanoparticles Combined with Radiotherapy in Cervical Carcinoma. Int. J. Nanomed. 2020, 15, 10045–10058. [Google Scholar] [CrossRef]
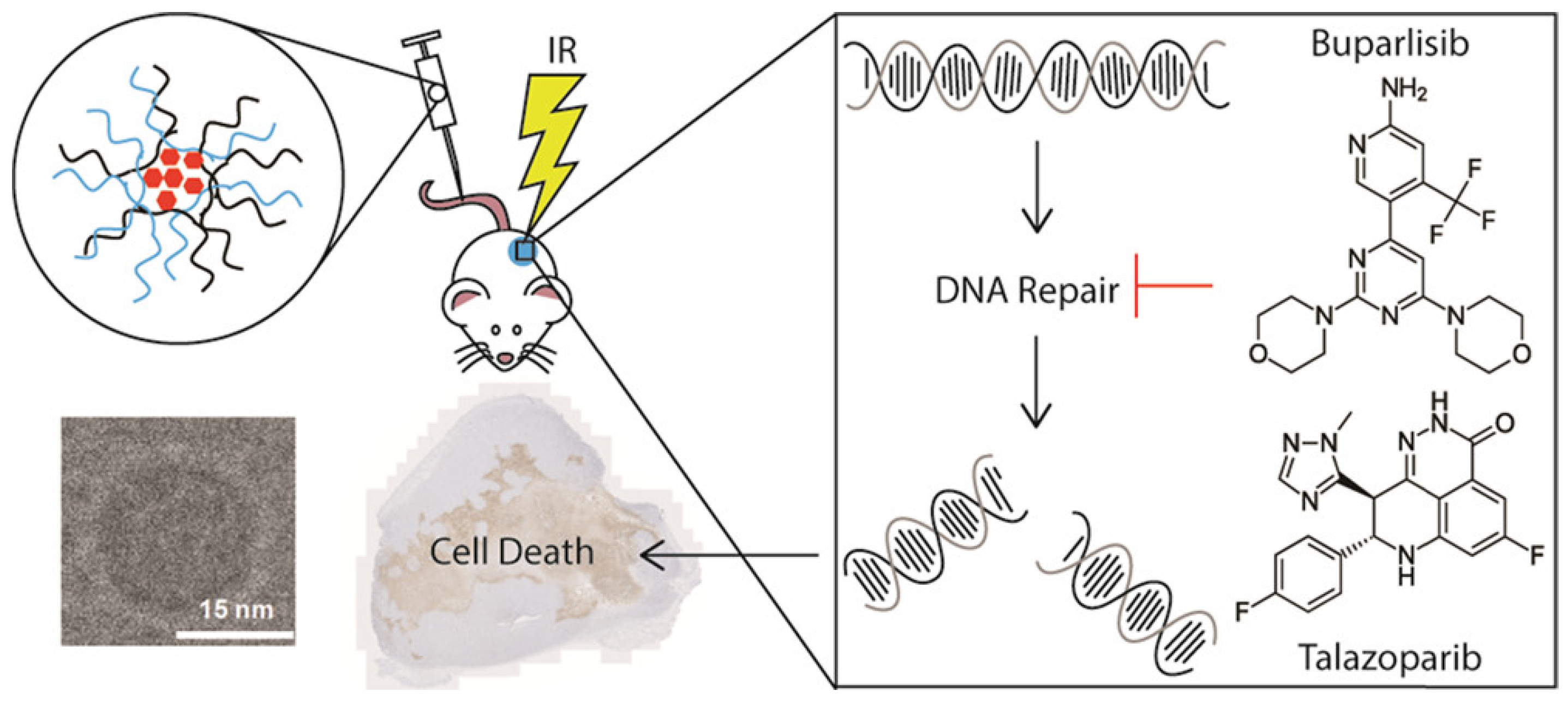
- DuRoss, A.N.; Neufeld, M.J.; Landry, M.R.; Rosch, J.G.; Eaton, C.T.; Sahay, G.; Thomas, C.R., Jr.; Sun, C. Micellar Formulation of Talazoparib and Buparlisib for Enhanced DNA Damage in Breast Cancer Chemoradiotherapy. ACS Appl. Mater. Interfaces 2019, 11, 12342–12356. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, M.J.; Duross, A.N.; Landry, M.R.; Winter, H.; Sun, C. Co-delivery of PARP and PI3K inhibitors by nanoscale metal–organic frameworks for enhanced tumor chemoradiation. Nano Res. 2019, 12, 3003–3017. [Google Scholar] [CrossRef]
- Lin, L.; Song, X.; Dong, X.; Li, B. Nano-photosensitizers for enhanced photodynamic therapy. Photodiagnosis Photodyn. Ther. 2021, 36, 102597. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, S.; Knap, B.; Przystupski, D.; Saczko, J.; Kędzierska, E.; Knap-Czop, K.; Kotlińska, J.; Michel, O.; Kotowski, K.; Kulbacka, J. Photodynamic therapy-mechanisms, photosensitizers and combinations. Biomed. Pharmacother. 2018, 106, 1098–1107. [Google Scholar] [CrossRef]
- Magalhães, J.A.; Arruda, D.C.; Baptista, M.S.; Tada, D.B. Co-Encapsulation of Methylene Blue and PARP-Inhibitor into Poly(Lactic-Co-Glycolic Acid) Nanoparticles for Enhanced PDT of Cancer. Nanomaterials 2021, 11, 1514. [Google Scholar] [CrossRef]
- Cheng, H.W.; Chiang, C.S.; Ho, H.Y.; Chou, S.H.; Lai, Y.H.; Shyu, W.C.; Chen, S.Y. Dextran-modified Quercetin-Cu(II)/hyaluronic acid nanomedicine with natural poly(ADP-ribose)polymerase inhibitor and dual targeting for programmed synthetic lethal therapy in triple-negative breast cancer. J. Control. Release 2021, 329, 136–147. [Google Scholar] [CrossRef]
- Gonzales, J.; Kossatz, S.; Roberts, S.; Pirovano, G.; Brand, C.; Pérez-Medina, C.; Donabedian, P.; de la Cruz, M.J.; Mulder, W.J.M.; Reiner, T. Nanoemulsion-Based Delivery of Fluorescent PARP Inhibitors in Mouse Models of Small Cell Lung Cancer. Bioconjug. Chem. 2018, 29, 3776–3782. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, L.; Xu, X.; Chen, W. The Current State of the Art in PARP Inhibitor-Based Delivery Nanosystems. Pharmaceutics 2022, 14, 1647. https://doi.org/10.3390/pharmaceutics14081647
Cai L, Xu X, Chen W. The Current State of the Art in PARP Inhibitor-Based Delivery Nanosystems. Pharmaceutics. 2022; 14(8):1647. https://doi.org/10.3390/pharmaceutics14081647
Chicago/Turabian StyleCai, Lisha, Xiaoling Xu, and Wei Chen. 2022. "The Current State of the Art in PARP Inhibitor-Based Delivery Nanosystems" Pharmaceutics 14, no. 8: 1647. https://doi.org/10.3390/pharmaceutics14081647
APA StyleCai, L., Xu, X., & Chen, W. (2022). The Current State of the Art in PARP Inhibitor-Based Delivery Nanosystems. Pharmaceutics, 14(8), 1647. https://doi.org/10.3390/pharmaceutics14081647