

Nanoparticle Delivery of Novel PDE4B Inhibitor for the Treatment of Alcoholic Liver Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Cytotoxicity Assay
2.4. Measurement of KVA-D88 Inhibitory Activity In Vitro
2.5. Measurement of cAMP Level
2.6. Quantitative Real Time RT-PCR and Western Blot
2.7. Measurement of TNF-α Level
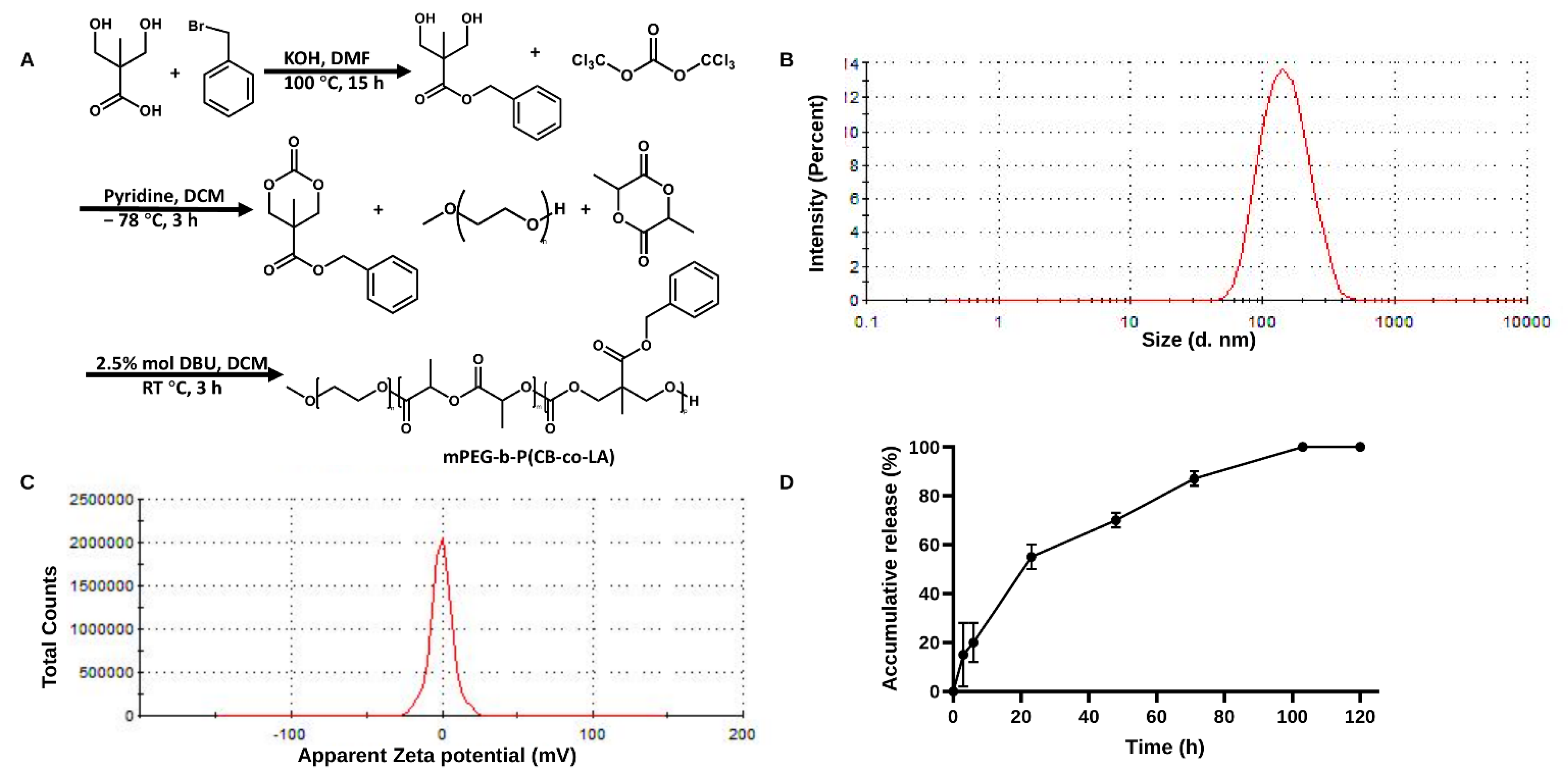
2.8. Preparation and Characterization of Nanoparticles
2.9. Measurement of Drug Loading and Drug Release
2.10. In Vivo Study
2.11. Measurement of Plasma Levels of Enzymes
2.12. Measurement of Hepatic Triglyceride Level
2.13. Histological and Immunohistochemistry Analysis
3. Results
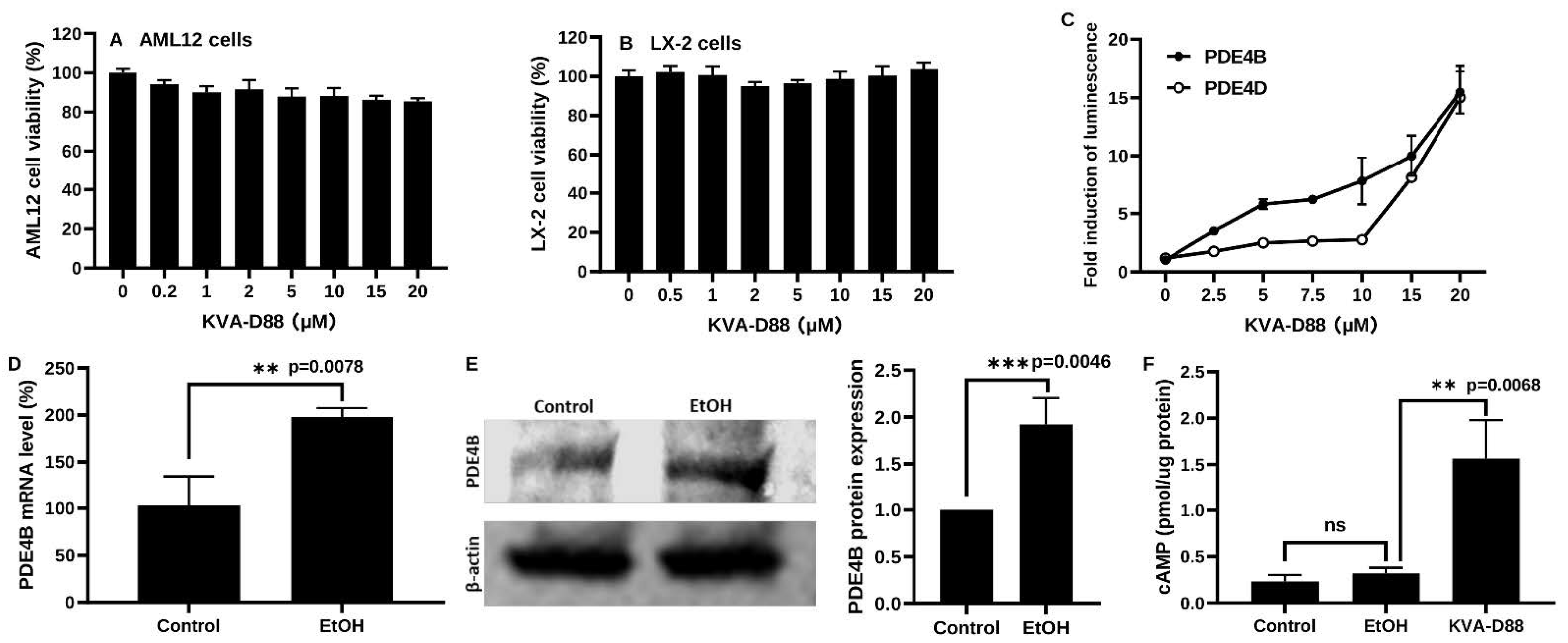
3.1. KVA-D88 Selectively Inhibits PDE4B Activity
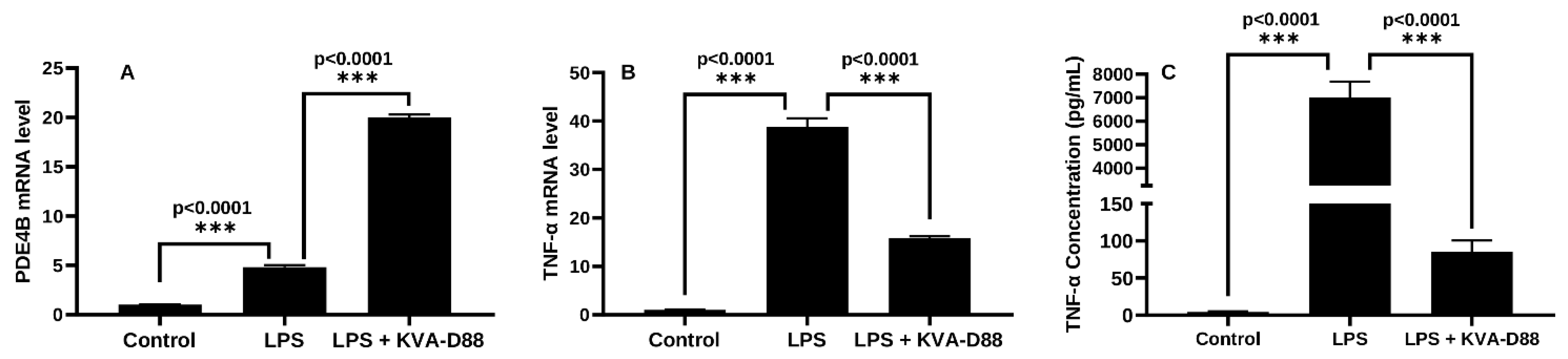
3.2. KVA-D88 Represses Inflammatory Responses in Macrophages
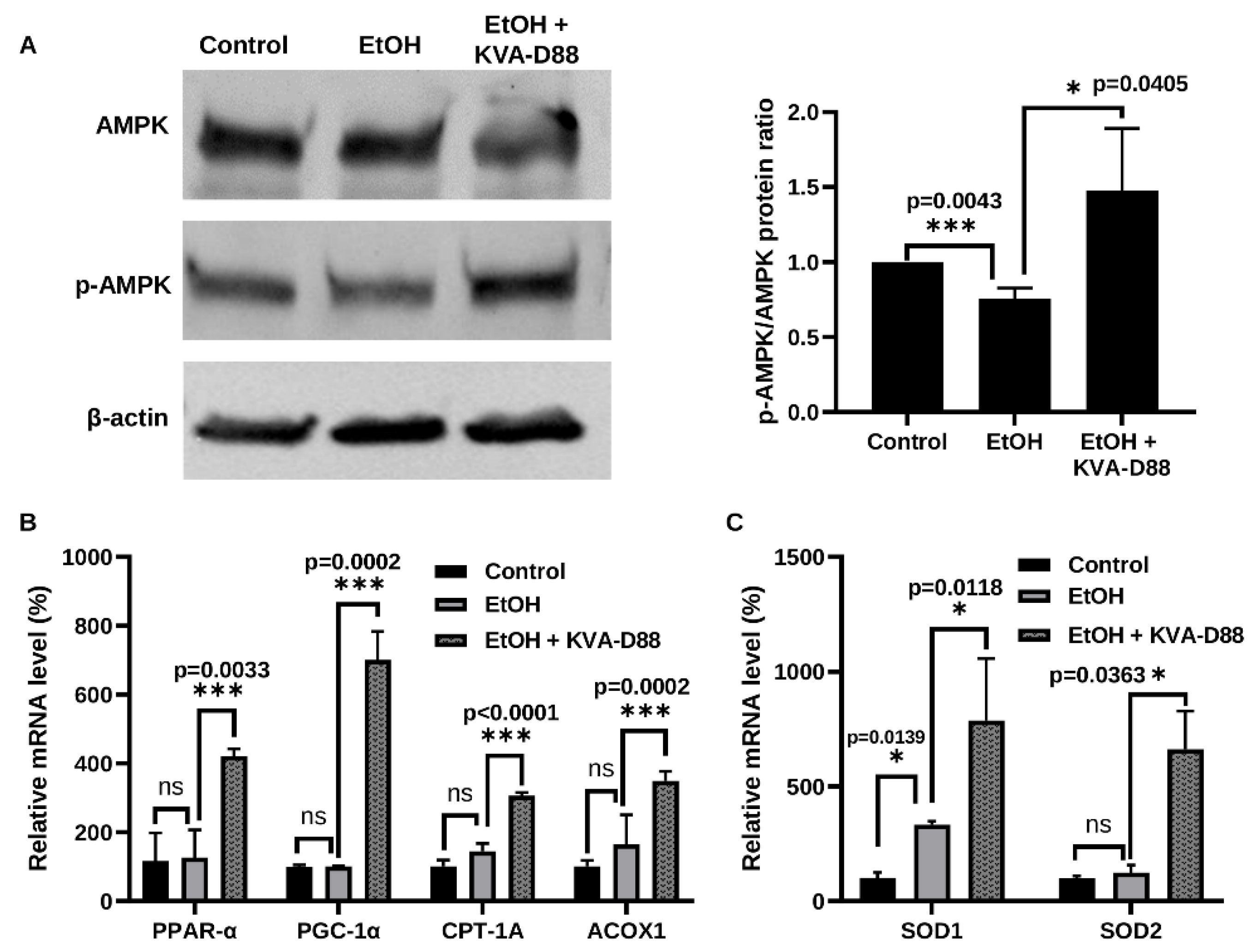
3.3. PDE4B Inhibition by KVA-D88 Promotes the Expression of Genes and Proteins Related to β-Oxidation and Antioxidants In Vitro
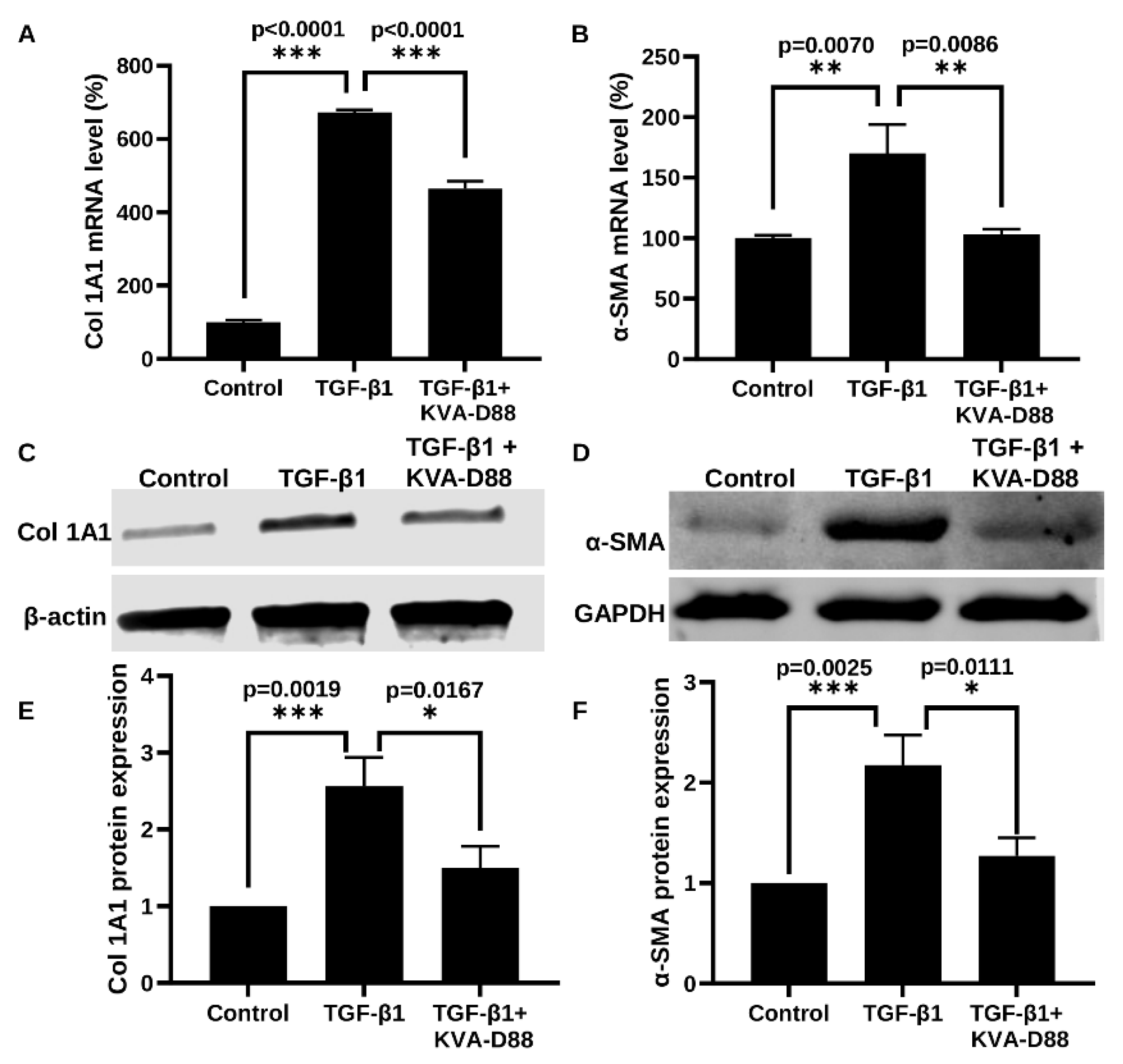
3.4. KVA-D88 Inhibits the Expression of Profibrotic Genes and Relative Protein
3.5. Preparation and Characterization of KVA-D88 Loaded Nanoparticles
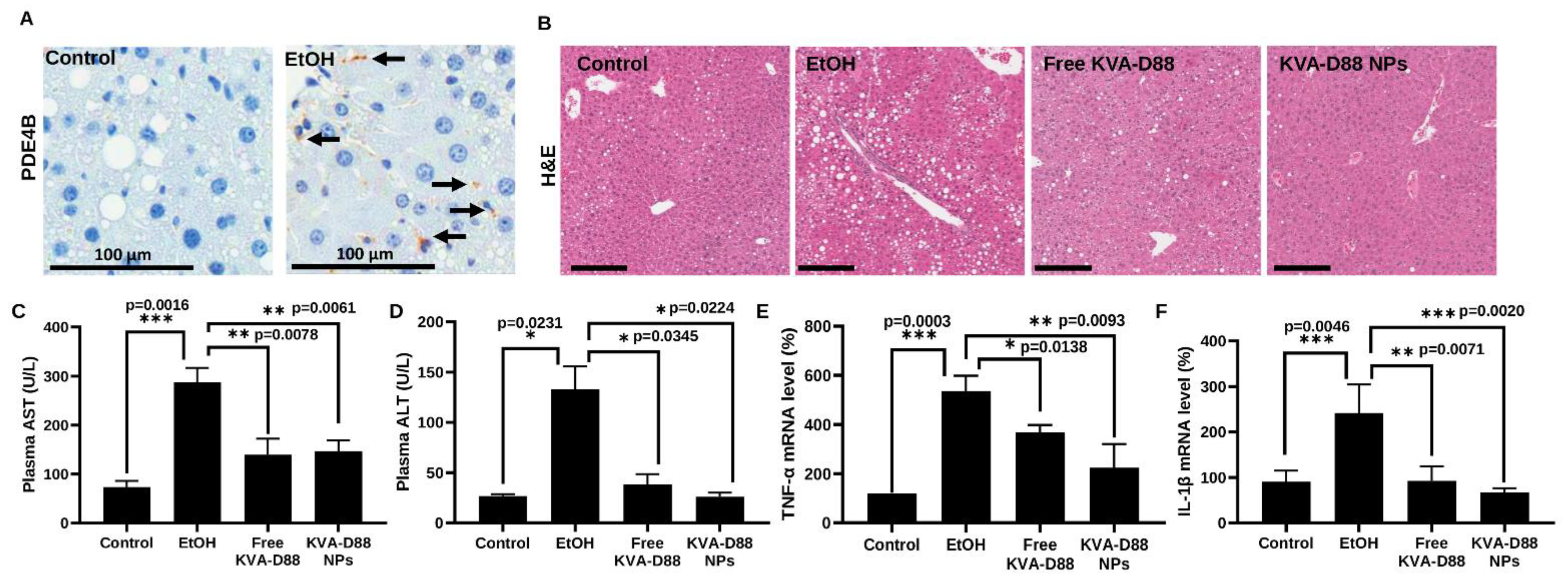
3.6. KVA-D88 Loaded Nanoparticles Alleviate Alcohol-Induced Liver Injury and Inflammation In Vivo
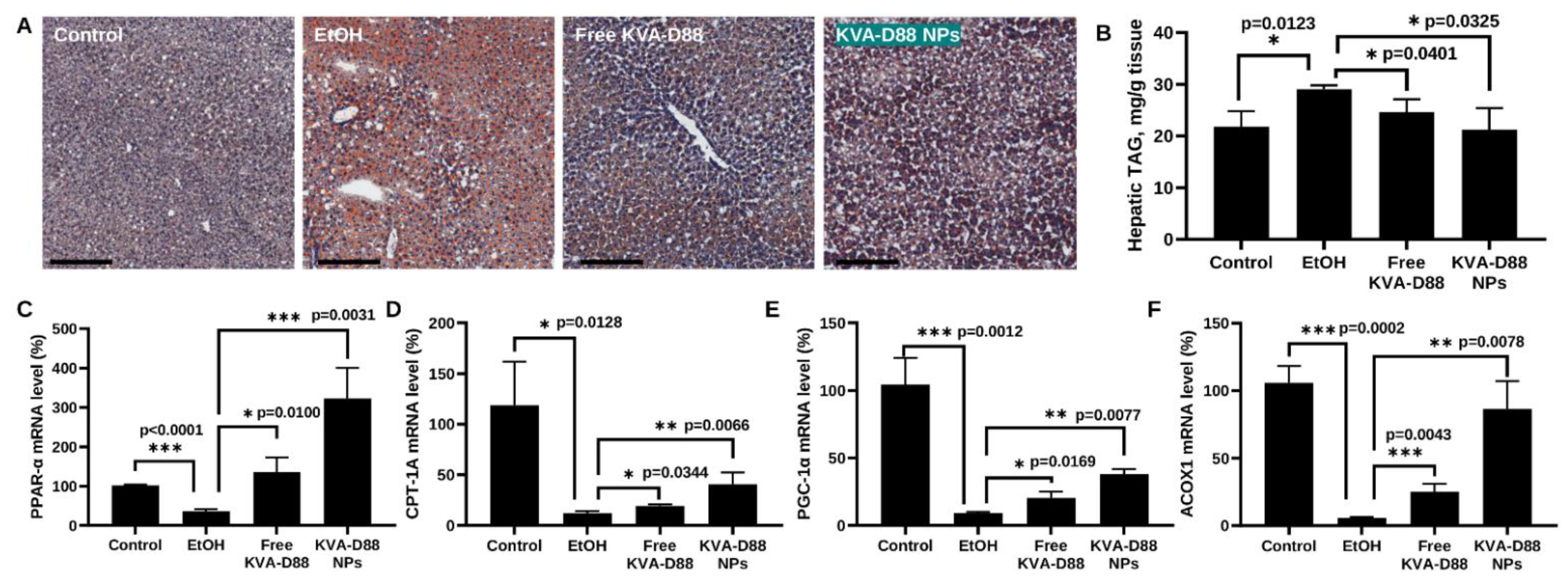
3.7. KVA-D88 Loaded Nanoparticles Alleviate Alcohol-Induced Steatosis and Promote the Expression of Genes Involved in β-Oxidation In Vivo
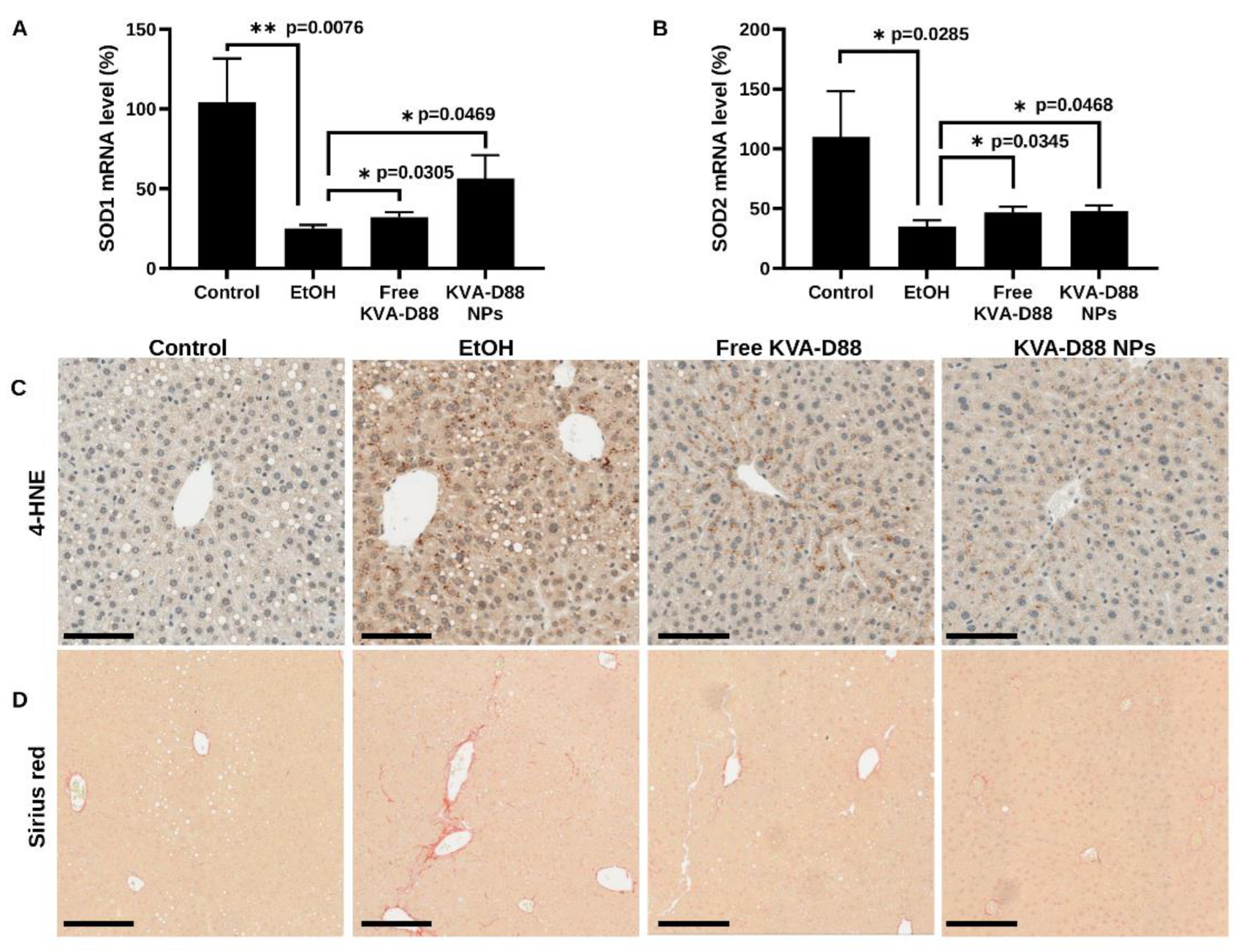
3.8. KVA-D88 Loaded Nanoparticles Alleviate Alcohol-Induced Oxidative Stress and Fibrosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- About Underlying Cause of Death, 1999–2020. Available online: https://wonder.cdc.gov/ucd-icd10.html (accessed on 19 May 2022).
- Crabb, D.W.; Im, G.Y.; Szabo, G.; Mellinger, J.L.; Lucey, M.R. Diagnosis and treatment of alcohol-associated liver diseases: 2019 practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2020, 71, 306–333. [Google Scholar] [CrossRef]
- Daniel, P.B.; Walker, W.H.; Habener, J.F. Cyclic AMP signaling and gene regulation. Annu. Rev. Nutr. 1998, 18, 353–383. [Google Scholar] [CrossRef] [PubMed]
- Wahlang, B.; McClain, C.; Barve, S.; Gobejishvili, L. Role of cAMP and phosphodiesterase signaling in liver health and disease. Cell. Signal. 2018, 49, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Fertig, B.A.; Baillie, G.S. PDE4-mediated cAMP signalling. J. Cardiovasc. Dev. Dis. 2018, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, W.E.; Wahlang, B.; Wang, Y.; Zhang, J.; Vadhanam, M.V.; Joshi-Barve, S.; Bauer, P.; Cannon, R.; Ahmadi, A.R.; Sun, Z. Phosphodiesterase 4 Inhibition as a Therapeutic Target for Alcoholic Liver Disease: From Bedside to Bench. Hepatology 2019, 70, 1958–1971. [Google Scholar] [CrossRef]
- Calverley, P.M.; Rabe, K.F.; Goehring, U.-M.; Kristiansen, S.; Fabbri, L.M.; Martinez, F.J. Roflumilast in symptomatic chronic obstructive pulmonary disease: Two randomised clinical trials. Lancet 2009, 374, 685–694. [Google Scholar] [CrossRef]
- Zhang, C.; Xu, Y.; Zhang, H.-T.; Gurney, M.E.; O’Donnell, J.M. Comparison of the pharmacological profiles of selective PDE4B and PDE4D inhibitors in the central nervous system. Sci. Rep. 2017, 7, 40115. [Google Scholar] [CrossRef]
- Vadukoot, A.K.; Sharma, S.; Aretz, C.D.; Kumar, S.; Gautam, N.; Alnouti, Y.; Aldrich, A.L.; Heim, C.E.; Kielian, T.; Hopkins, C.R. Synthesis and SAR Studies of 1 H-Pyrrolo [2,3-b] pyridine-2-carboxamides as Phosphodiesterase 4B (PDE4B) Inhibitors. ACS Med. Chem. Lett. 2020, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Burkovetskaya, M.E.; Liu, Q.; Vadukoot, A.K.; Gautam, N.; Alnouti, Y.; Kumar, S.; Miczek, K.; Buch, S.; Hopkins, C.R.; Guo, M. KVA-D-88, a novel preferable phosphodiesterase 4B inhibitor, decreases cocaine-mediated reward properties in vivo. ACS Chem. Neurosci. 2020, 11, 2231–2242. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Lillard, J.W., Jr. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-N.; Poon, W.; Tavares, A.J.; McGilvray, I.D.; Chan, W.C. Nanoparticle–liver interactions: Cellular uptake and hepatobiliary elimination. J. Control. Release 2016, 240, 332–348. [Google Scholar] [CrossRef] [PubMed]
- Wani, T.U.; Raza, S.N.; Khan, N.A. Nanoparticle opsonization: Forces involved and protection by long chain polymers. Polym. Bull. 2020, 77, 3865–3889. [Google Scholar] [CrossRef]
- Bariwal, J.; Kumar, V.; Chen, H.; Bhattarai, R.S.; Peng, Y.; Li, W.; Mahato, R.I. Nanoparticulate delivery of potent microtubule inhibitor for metastatic melanoma treatment. J. Control. Release 2019, 309, 231–243. [Google Scholar] [CrossRef]
- Bertola, A.; Mathews, S.; Ki, S.H.; Wang, H.; Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 2013, 8, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Kumar, V.; Lin, F.; Sethi, B.; Coulter, D.W.; McGuire, T.R.; Mahato, R.I. ApoE mimetic peptide targeted nanoparticles carrying a BRD4 inhibitor for treating Medulloblastoma in mice. J. Control. Release 2020, 323, 463–474. [Google Scholar] [CrossRef]
- Peng, Y.; Wen, D.; Lin, F.; Mahato, R.I. Co-delivery of siAlox15 and sunitinib for reversing the new-onset of type 1 diabetes in non-obese diabetic mice. J. Control. Release 2018, 292, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Gao, B.; Zakhari, S.; Nagy, L.E. Inflammation in alcoholic liver disease. Annu. Rev. Nutr. 2012, 32, 343–368. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tan, H.-Y.; Wang, N.; Feng, Y.; Wang, X.; Feng, Y. Recent insights into the role of immune cells in alcoholic liver disease. Front. Immunol. 2019, 10, 1328. [Google Scholar] [CrossRef]
- Gao, B.; Seki, E.; Brenner, D.A.; Friedman, S.; Cohen, J.I.; Nagy, L.; Szabo, G.; Zakhari, S. Innate immunity in alcoholic liver disease. Am. J. Physiol.-Gastrointest. Liver Physiol. 2011, 300, G516–G525. [Google Scholar] [CrossRef] [PubMed]
- Dastidar, S.G.; Rajagopal, D.; Ray, A. Therapeutic benefit of PDE4 inhibitors in inflammatory diseases. Curr. Opin. Investig. Drugs 2007, 8, 364. [Google Scholar]
- Ceni, E.; Mello, T.; Galli, A. Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J. Gastroenterol. WJG 2014, 20, 17756. [Google Scholar] [CrossRef]
- Zhu, R.; Wang, Y.; Zhang, L.; Guo, Q. Oxidative stress and liver disease. Hepatol. Res. 2012, 42, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Ishii, H.; Kurose, I.; Kato, S. Pathogenesis of alcoholic liver disease with particular emphasis on oxidative stress. J. Gastroenterol. Hepatol. 1997, 12, S272–S282. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Arteel, G.E. Effect of ethanol on lipid metabolism. J. Hepatol. 2019, 70, 237–248. [Google Scholar] [CrossRef]
- Crabb, D.W.; Liangpunsakul, S. Alcohol and lipid metabolism. J. Gastroenterol. Hepatol. 2006, 21, S56–S60. [Google Scholar] [CrossRef]
- You, M.; Matsumoto, M.; Pacold, C.M.; Cho, W.K.; Crabb, D.W. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology 2004, 127, 1798–1808. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.; Chinetti, G.; Fruchart, J.-C.; Staels, B. Sorting out the roles of PPARα in energy metabolism and vascular homeostasis. J. Clin. Investig. 2006, 116, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.-G.; Jeong, W.-I. Hepatic stellate cells and innate immunity in alcoholic liver disease. World J. Gastroenterol. WJG 2011, 17, 2543. [Google Scholar] [CrossRef]
- Spina, D. PDE4 inhibitors: Current status. Br. J. Pharmacol. 2008, 155, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Bischoff, E. Potency, selectivity, and consequences of nonselectivity of PDE inhibition. Int. J. Impot. Res. 2004, 16, S11–S14. [Google Scholar] [CrossRef]
- Zhang, D.; Tong, X.; Nelson, B.B.; Jin, E.; Sit, J.; Charney, N.; Yang, M.; Omary, M.B.; Yin, L. The hepatic BMAL1/AKT/lipogenesis axis protects against alcoholic liver disease in mice via promoting PPARα pathway. Hepatology 2018, 68, 883–896. [Google Scholar] [CrossRef]
- Bukong, T.N.; Iracheta-Vellve, A.; Saha, B.; Ambade, A.; Satishchandran, A.; Gyongyosi, B.; Lowe, P.; Catalano, D.; Kodys, K.; Szabo, G. Inhibition of spleen tyrosine kinase activation ameliorates inflammation, cell death, and steatosis in alcoholic liver disease. Hepatology 2016, 64, 1057–1071. [Google Scholar] [CrossRef]
- Kumar, V.; Dong, Y.; Kumar, V.; Almawash, S.; Mahato, R.I. The use of micelles to deliver potential hedgehog pathway inhibitor for the treatment of liver fibrosis. Theranostics 2019, 9, 7537. [Google Scholar] [CrossRef]
- Kumar, V.; Mundra, V.; Mahato, R.I. Nanomedicines of Hedgehog Inhibitor and PPAR-γ Agonist for Treating Liver Fibrosis. Pharm. Res. 2014, 31, 1158–1169. [Google Scholar] [CrossRef]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride metabolism in the liver. Compr. Physiol. 2011, 8, 1–8. [Google Scholar]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef]
- Chen, H.; Shen, F.; Sherban, A.; Nocon, A.; Li, Y.; Wang, H.; Xu, M.J.; Rui, X.; Han, J.; Jiang, B. DEP domain–containing mTOR–interacting protein suppresses lipogenesis and ameliorates hepatic steatosis and acute-on-chronic liver injury in alcoholic liver disease. Hepatology 2018, 68, 496–514. [Google Scholar] [CrossRef]
- Ambade, A.; Lowe, P.; Kodys, K.; Catalano, D.; Gyongyosi, B.; Cho, Y.; Iracheta-Vellve, A.; Adejumo, A.; Saha, B.; Calenda, C. Pharmacological inhibition of CCR2/5 signaling prevents and reverses alcohol-induced liver damage, steatosis, and inflammation in mice. Hepatology 2019, 69, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Yin, H.; Mitra, M.S.; Liang, X.; Ajmo, J.M.; Nadra, K.; Chrast, R.; Finck, B.N.; You, M. Hepatic-specific lipin-1 deficiency exacerbates experimental alcohol-induced steatohepatitis in mice. Hepatology 2013, 58, 1953–1963. [Google Scholar] [CrossRef]
- Ambade, A.; Mandrekar, P. Oxidative stress and inflammation: Essential partners in alcoholic liver disease. Int. J. Hepatol. 2012, 2012, 853175. [Google Scholar] [CrossRef]
- Smathers, R.L.; Galligan, J.J.; Stewart, B.J.; Petersen, D.R. Overview of lipid peroxidation products and hepatic protein modification in alcoholic liver disease. Chem. -Biol. Interact. 2011, 192, 107–112. [Google Scholar] [CrossRef]
- Van Kuijk, F.J.; Holte, L.L.; Dratz, E.A. 4-Hydroxyhexenal: A lipid peroxidation product derived from oxidized docosahexaenoic acid. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab. 1990, 1043, 116–118. [Google Scholar] [CrossRef]
- Tomita, K.; Azuma, T.; Kitamura, N.; Nishida, J.; Tamiya, G.; Oka, A.; Inokuchi, S.; Nishimura, T.; Suematsu, M.; Ishii, H. Pioglitazone prevents alcohol-induced fatty liver in rats through up-regulation of c-Met. Gastroenterology 2004, 126, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Alcoholic liver disease: Pathogenesis and current management. Alcohol Res. Curr. Rev. 2017, 38, 147. [Google Scholar]
- Liu, D.; Ahmet, A.; Ward, L.; Krishnamoorthy, P.; Mandelcorn, E.D.; Leigh, R.; Brown, J.P.; Cohen, A.; Kim, H. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma Clin. Immunol. 2013, 9, 30. [Google Scholar] [CrossRef]
- Chan, A.C.; Fan, S.T.; Lo, C.M.; Liu, C.L.; Chan, S.C.; Ng, K.K.; Yong, B.H.; Chiu, A.; Lam, B.K. Liver transplantation for acute-on-chronic liver failure. Hepatol. Int. 2009, 3, 571–581. [Google Scholar] [CrossRef]
- Kumar, V.; Xin, X.; Ma, J.; Tan, C.; Osna, N.; Mahato, R.I. Therapeutic Targets, Novel Drugs, and Delivery Systems for Diabetes associated NAFLD and Liver Fibrosis. Adv. Drug Deliv. Rev. 2021, 176, 113888. [Google Scholar] [CrossRef]
- Heymann, F.; Trautwein, C.; Tacke, F. Monocytes and macrophages as cellular targets in liver fibrosis. Inflamm. Allergy-Drug Targets 2009, 8, 307–318. [Google Scholar] [CrossRef]
- Bukara, M.; Bautista, A.P. Acute alcohol intoxication and gadolinium chloride attenuate endotoxin-induced release of CC chemokines in the rat. Alcohol 2000, 20, 193–203. [Google Scholar] [CrossRef]
- Lowe, P.P.; Gyongyosi, B.; Satishchandran, A.; Iracheta-Vellve, A.; Cho, Y.; Ambade, A.; Szabo, G. Reduced gut microbiome protects from alcohol-induced neuroinflammation and alters intestinal and brain inflammasome expression. J. Neuroinflamm. 2018, 15, 298. [Google Scholar] [CrossRef]
- Brownsey, R.; Boone, A.; Elliott, J.; Kulpa, J.; Lee, W. Regulation of acetyl-CoA carboxylase. Biochem. Soc. Trans. 2006, 34, 223–227. [Google Scholar] [CrossRef]
- Aroor, A.R.; Jackson, D.E.; Shukla, S.D. Dysregulated phosphorylation and nuclear translocation of cyclic AMP response element binding protein (CREB) in rat liver after chronic ethanol binge. Eur. J. Pharmacol. 2012, 679, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Elnagdy, M.; Barve, S.; McClain, C.; Gobejishvili, L. cAMP Signaling in Pathobiology of Alcohol Associated Liver Disease. Biomolecules 2020, 10, 1433. [Google Scholar] [CrossRef]
- Patel, S.; Behara, R.; Swanson, G.R.; Forsyth, C.B.; Voigt, R.M.; Keshavarzian, A. Alcohol and the Intestine. Biomolecules 2015, 5, 2573–2588. [Google Scholar] [CrossRef] [PubMed]
- Shames, B.D.; McIntyre, R.C., Jr.; Bensard, D.D.; Pulido, E.J.; Selzman, C.H.; Reznikov, L.L.; Harken, A.H.; Meng, X. Suppression of tumor necrosis factor α production by cAMP in human monocytes: Dissociation with mRNA level and independent of interleukin-10. J. Surg. Res. 2001, 99, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Iimuro, Y.; Gallucci, R.M.; Luster, M.I.; Kono, H.; Thurman, R.G. Antibodies to tumor necrosis factor alfa attenuate hepatic necrosis and inflammation caused by chronic exposure to ethanol in the rat. Hepatology 1997, 26, 1530–1537. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, M.; Nelson, E.J.; Hoffman, P.L.; Tabakoff, B. Role of protein kinase C in ethanol-induced activation of adenylyl cyclase. Alcohol. Clin. Exp. Res. 1999, 23, 77–86. [Google Scholar] [CrossRef]
- Takahashi, M.; Terwilliger, R.; Lane, C.; Mezes, P.S.; Conti, M.; Duman, R.S. Chronic antidepressant administration increases the expression of cAMP-specific phosphodiesterase 4A and 4B isoforms. J. Neurosci. 1999, 19, 610–618. [Google Scholar] [CrossRef]
- Li, H.; Zuo, J.; Tang, W. Phosphodiesterase-4 inhibitors for the treatment of inflammatory diseases. Front. Pharmacol. 2018, 9, 1048. [Google Scholar] [CrossRef] [PubMed]
- Logrip, M.L. Phosphodiesterase regulation of alcohol drinking in rodents. Alcohol 2015, 49, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, L.M.; Calverley, P.M.; Izquierdo-Alonso, J.L.; Bundschuh, D.S.; Brose, M.; Martinez, F.J.; Rabe, K.F. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: Two randomised clinical trials. Lancet 2009, 374, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.; Leray, V.; Diez, M.; Serisier, S.; Bloc’h, J.L.; Siliart, B.; Dumon, H. Liver lipid metabolism. J. Anim. Physiol. Anim. Nutr. 2008, 92, 272–283. [Google Scholar] [CrossRef]
- Bradbury, M.W. Lipid metabolism and liver inflammation. I. Hepatic fatty acid uptake: Possible role in steatosis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2006, 290, G194–G198. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.K.; Sambasiva Rao, M. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am. J. Physiol.-Gastrointest. Liver Physiol. 2006, 290, G852–G858. [Google Scholar] [CrossRef]
- Jeon, S.; Carr, R. Alcohol effects on hepatic lipid metabolism. J. Lipid Res. 2020, 61, 470–479. [Google Scholar] [CrossRef]
- Kumar, V.; Kumar, V.; Luo, J.; Mahato, R.I. Therapeutic potential of OMe-PS-miR-29b1 for treating liver fibrosis. Mol. Ther. 2018, 26, 2798–2811. [Google Scholar] [CrossRef]
- Houglum, K.; Lee, K.S.; Chojkier, M. Proliferation of hepatic stellate cells is inhibited by phosphorylation of CREB on serine 133. J. Clin. Investig. 1997, 99, 1322–1328. [Google Scholar] [CrossRef]
- Yu, S.; Pearson, A.D.; Lim, R.K.; Rodgers, D.T.; Li, S.; Parker, H.B.; Weglarz, M.; Hampton, E.N.; Bollong, M.J.; Shen, J. Targeted delivery of an anti-inflammatory PDE4 inhibitor to immune cells via an antibody–drug conjugate. Mol. Ther. 2016, 24, 2078–2089. [Google Scholar] [CrossRef]
- Chapman, R.W.; House, A.; Richard, J.; Prelusky, D.; Lamca, J.; Wang, P.; Lundell, D.; Wu, P.; Ting, P.C.; Lee, J.F. Pharmacology of a potent and selective inhibitor of PDE4 for inhaled administration. Eur. J. Pharmacol. 2010, 643, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Gref, R.; Lück, M.; Quellec, P.; Marchand, M.; Dellacherie, E.; Harnisch, S.; Blunk, T.; Müller, R. ‘Stealth’corona-core nanoparticles surface modified by polyethylene glycol (PEG): Influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf. B Biointerfaces 2000, 18, 301–313. [Google Scholar] [CrossRef]
- Dutta, R.; Kumar, V.; Peng, Y.; Evande, R.E.; Grem, J.L.; Mahato, R.I. Pharmacokinetics and biodistribution of GDC-0449 loaded micelles in normal and liver fibrotic mice. Pharm. Res. 2017, 34, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Mondal, G.; Dutta, R.; Mahato, R.I. Co-delivery of small molecule hedgehog inhibitor and miRNA for treating liver fibrosis. Biomaterials 2016, 76, 144–156. [Google Scholar] [CrossRef]
- Albano, E. Alcohol, oxidative stress and free radical damage. Proc. Nutr. Soc. 2006, 65, 278–290. [Google Scholar] [CrossRef]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Wu, J.; Zern, M.A. Hepatic stellate cells: A target for the treatment of liver fibrosis. J. Gastroenterol. 2000, 35, 665–672. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, J.; Kumar, V.; Mahato, R.I. Nanoparticle Delivery of Novel PDE4B Inhibitor for the Treatment of Alcoholic Liver Disease. Pharmaceutics 2022, 14, 1894. https://doi.org/10.3390/pharmaceutics14091894
Ma J, Kumar V, Mahato RI. Nanoparticle Delivery of Novel PDE4B Inhibitor for the Treatment of Alcoholic Liver Disease. Pharmaceutics. 2022; 14(9):1894. https://doi.org/10.3390/pharmaceutics14091894
Chicago/Turabian StyleMa, Jingyi, Virender Kumar, and Ram I. Mahato. 2022. "Nanoparticle Delivery of Novel PDE4B Inhibitor for the Treatment of Alcoholic Liver Disease" Pharmaceutics 14, no. 9: 1894. https://doi.org/10.3390/pharmaceutics14091894
APA StyleMa, J., Kumar, V., & Mahato, R. I. (2022). Nanoparticle Delivery of Novel PDE4B Inhibitor for the Treatment of Alcoholic Liver Disease. Pharmaceutics, 14(9), 1894. https://doi.org/10.3390/pharmaceutics14091894