
Evaluation of a Cardiovascular Systems Model for Design and Analysis of Hemodynamic Safety Studies

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
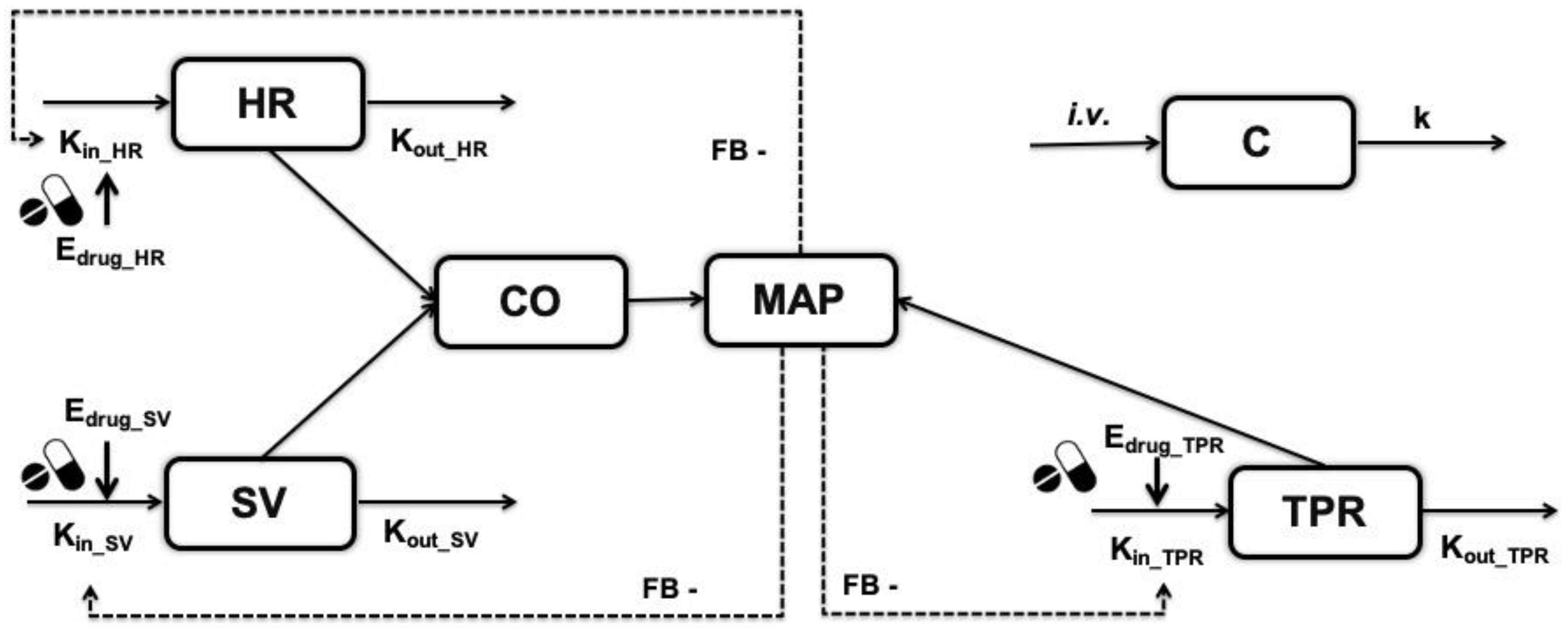
2.1. Model Structure
2.2. Simulation Scenarios
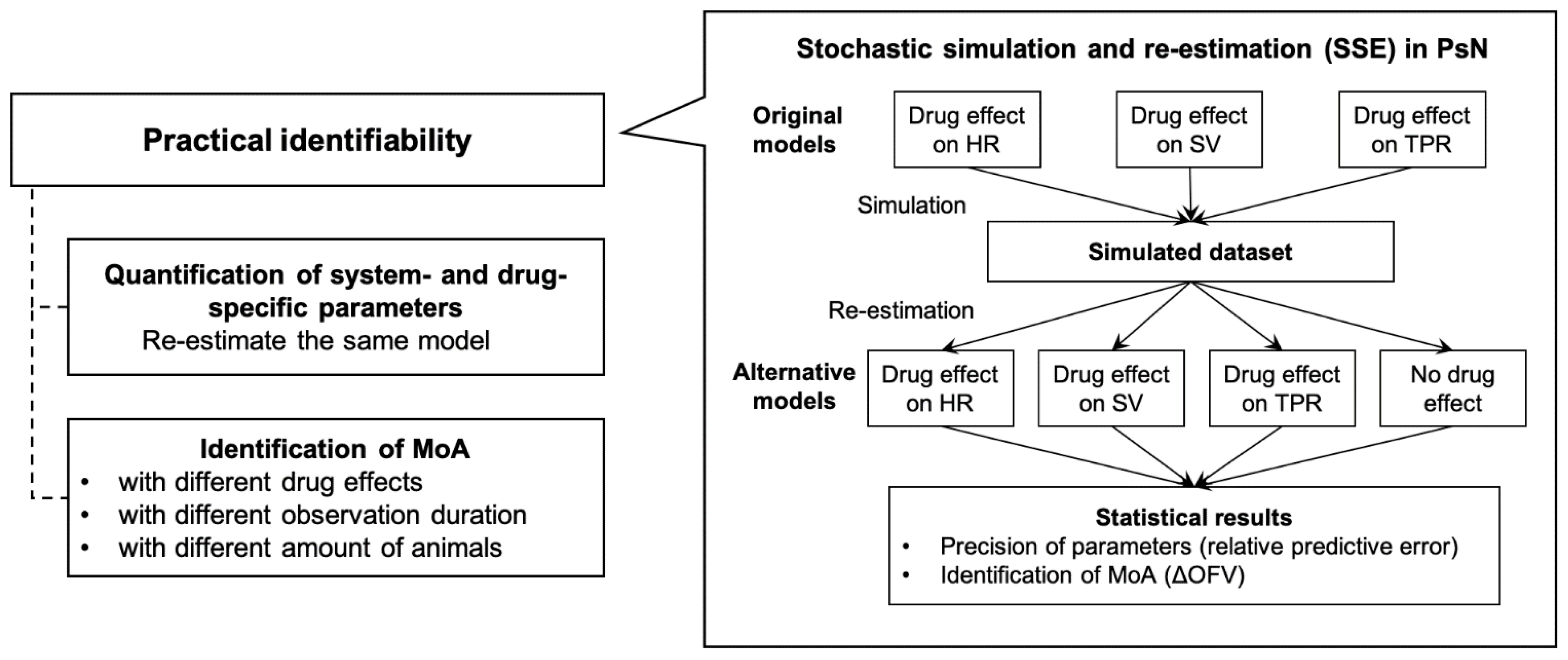
2.3. Practical Identifiability Analysis
2.3.1. Stochastic Stimulation and Re-Estimation
2.3.2. Identifiability of Drug- and System-Specific Parameters
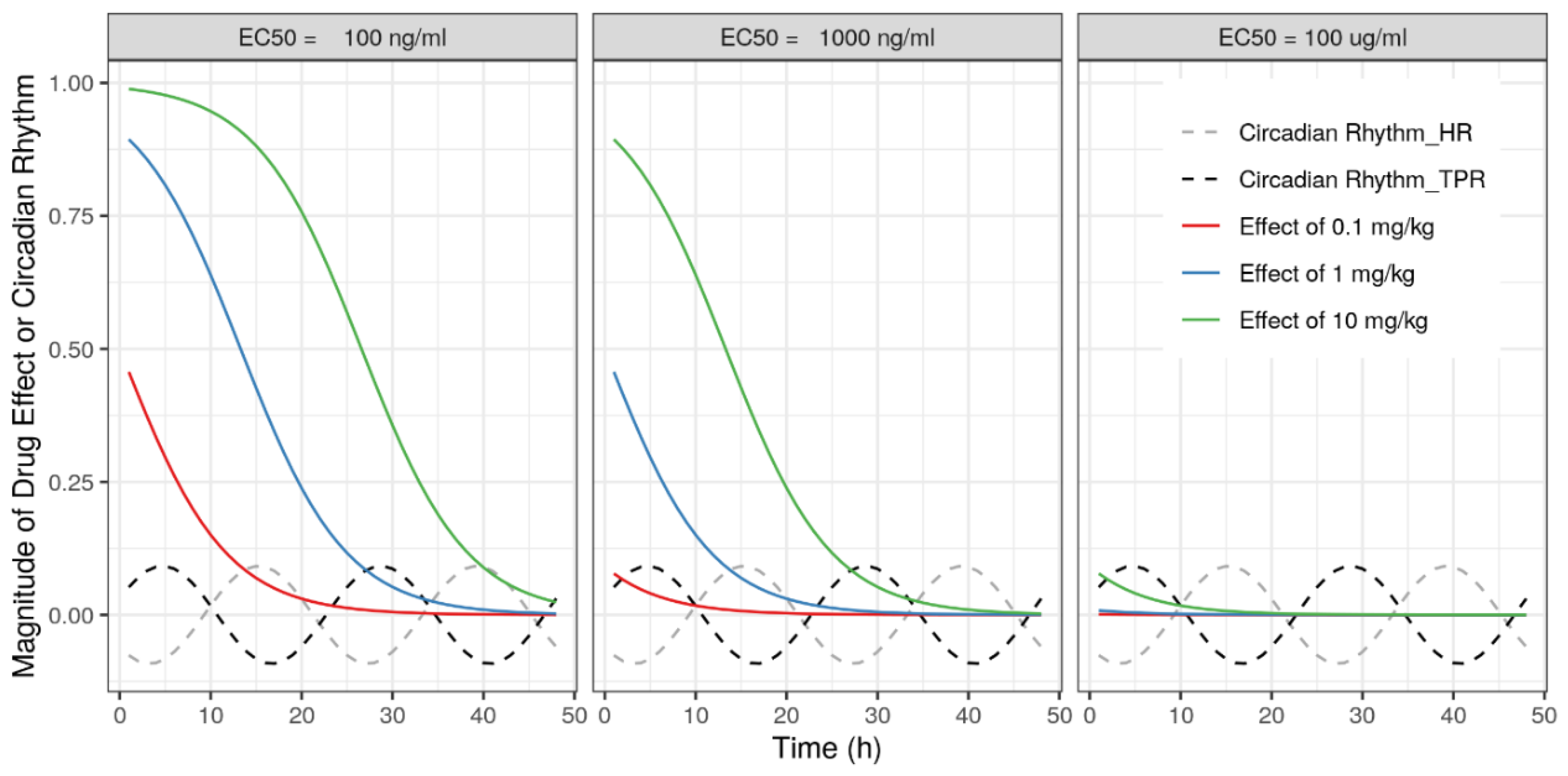
2.3.3. Drug Effect Magnitudes
2.3.4. Observation Duration
2.3.5. Number of Animals
3. Results
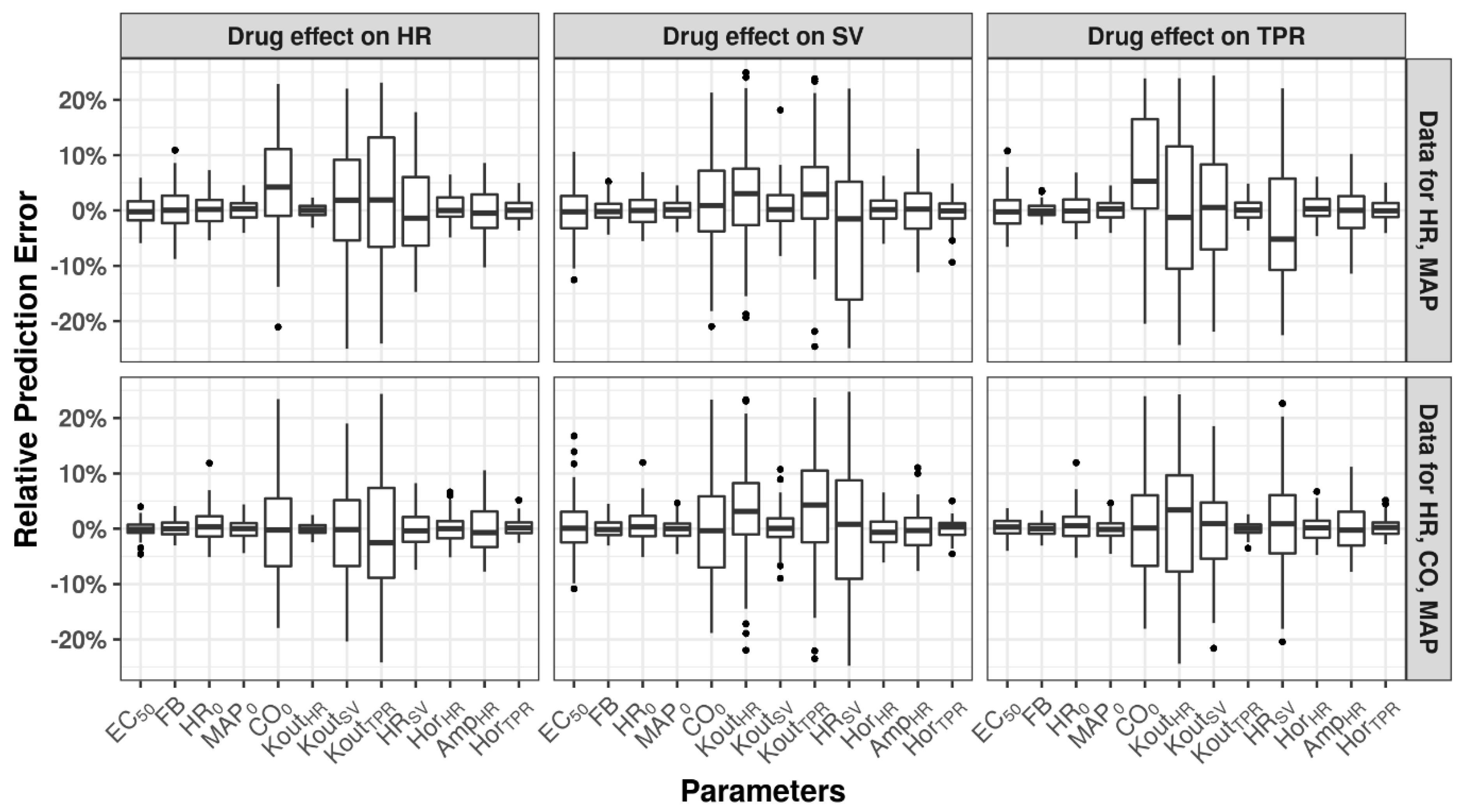
3.1. Identifiability of Drug- and System-Specific Parameters
3.2. Drug Effect Magnitudes
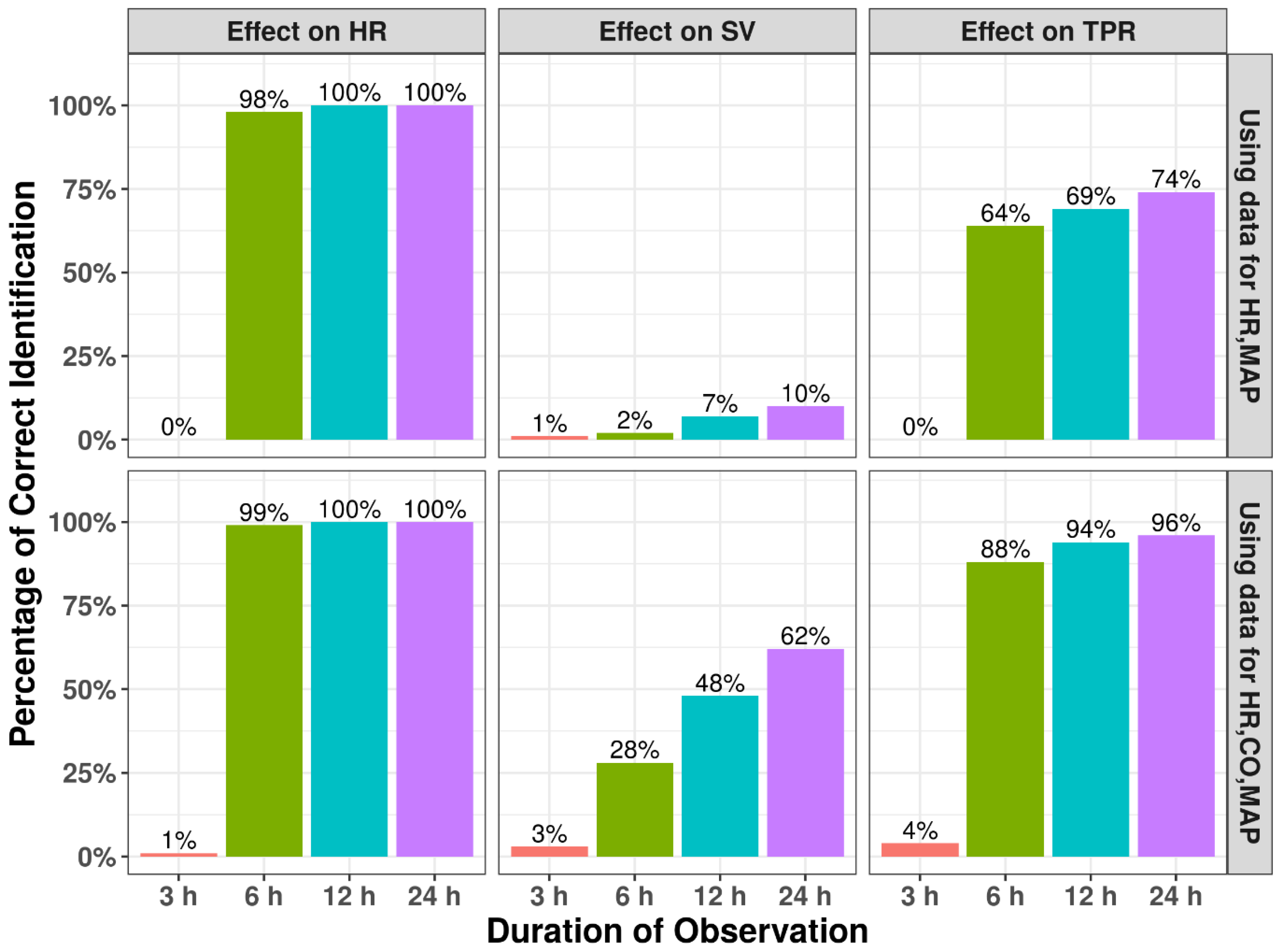
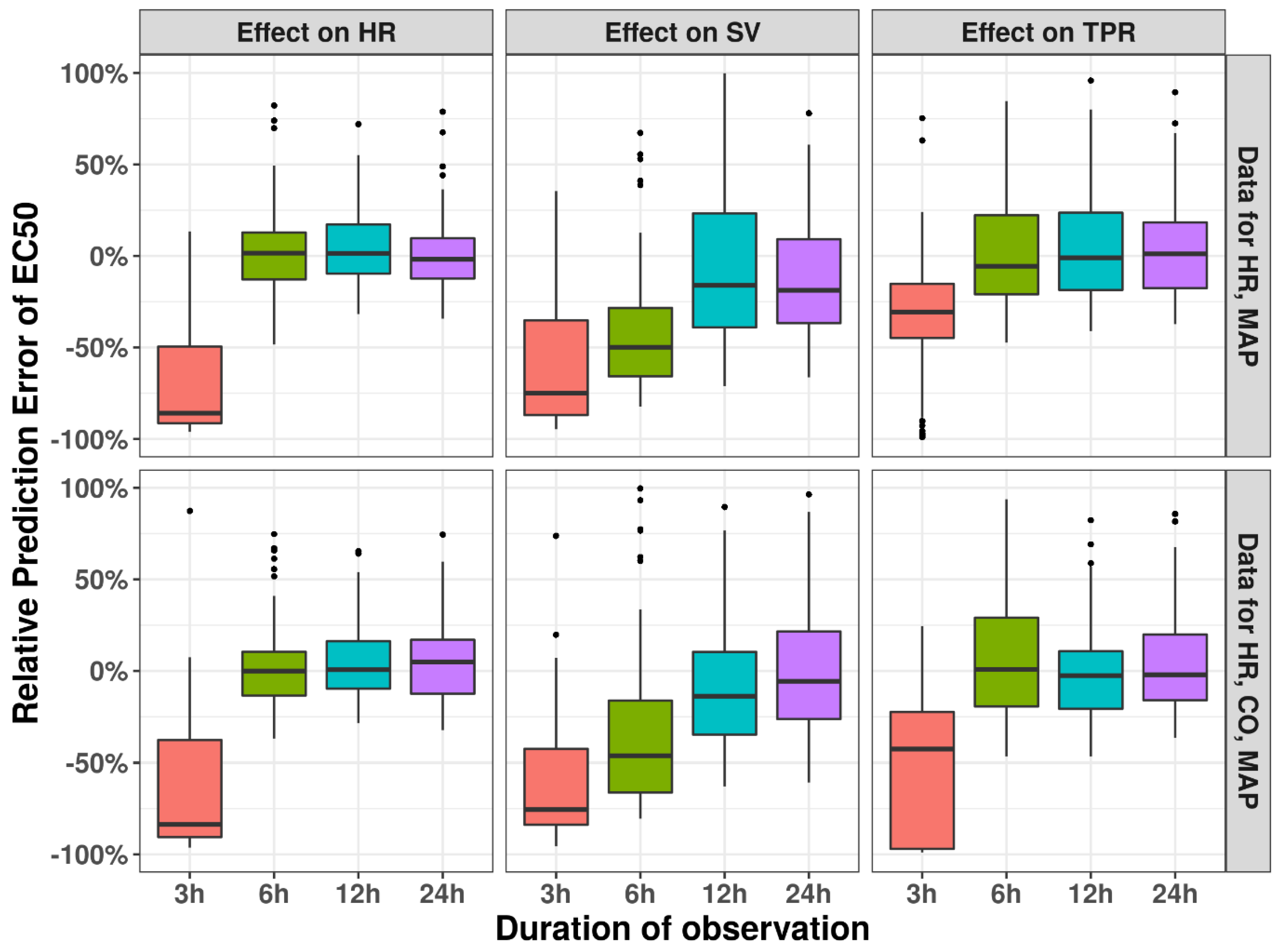
3.3. Observation Duration
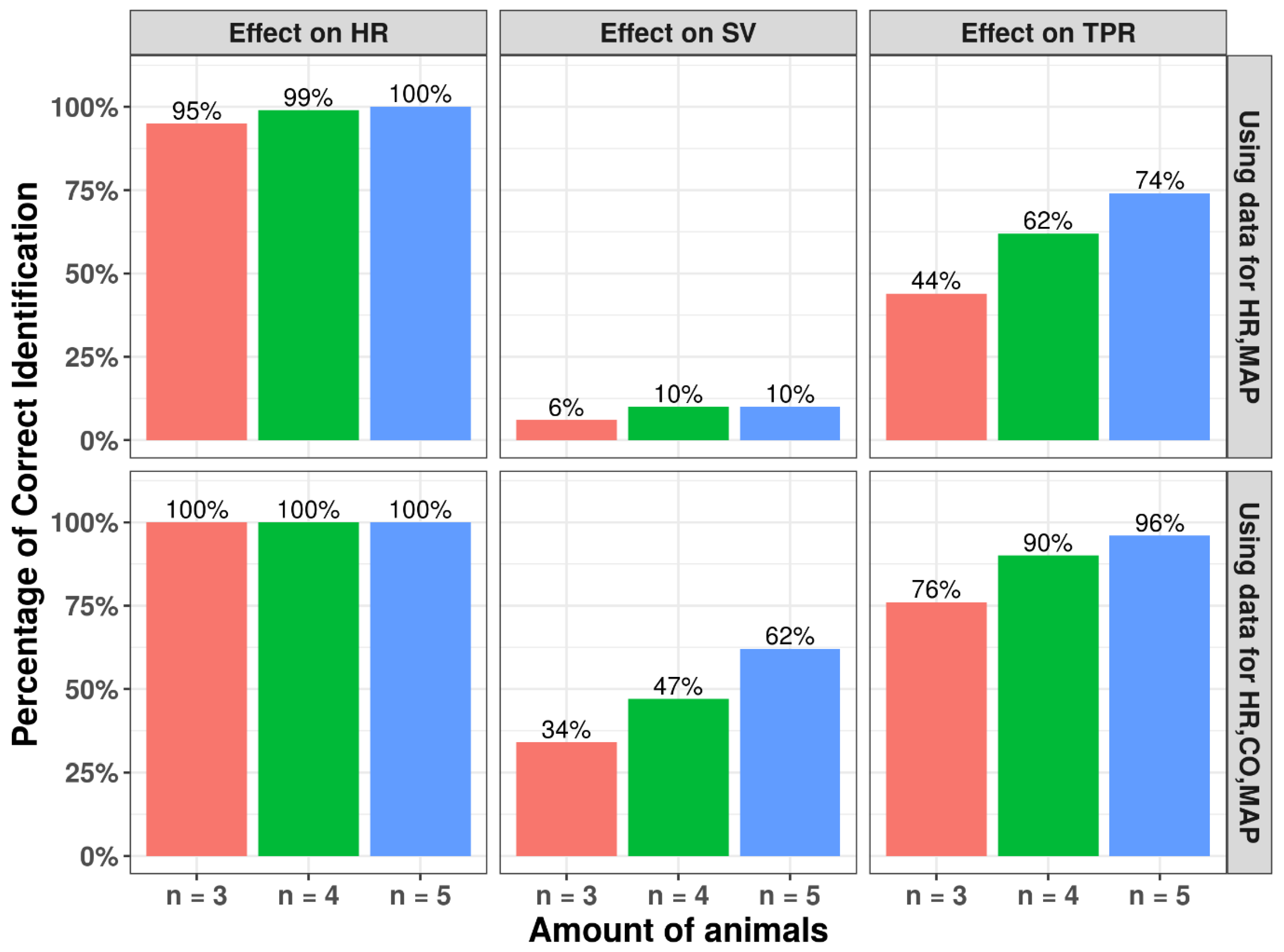
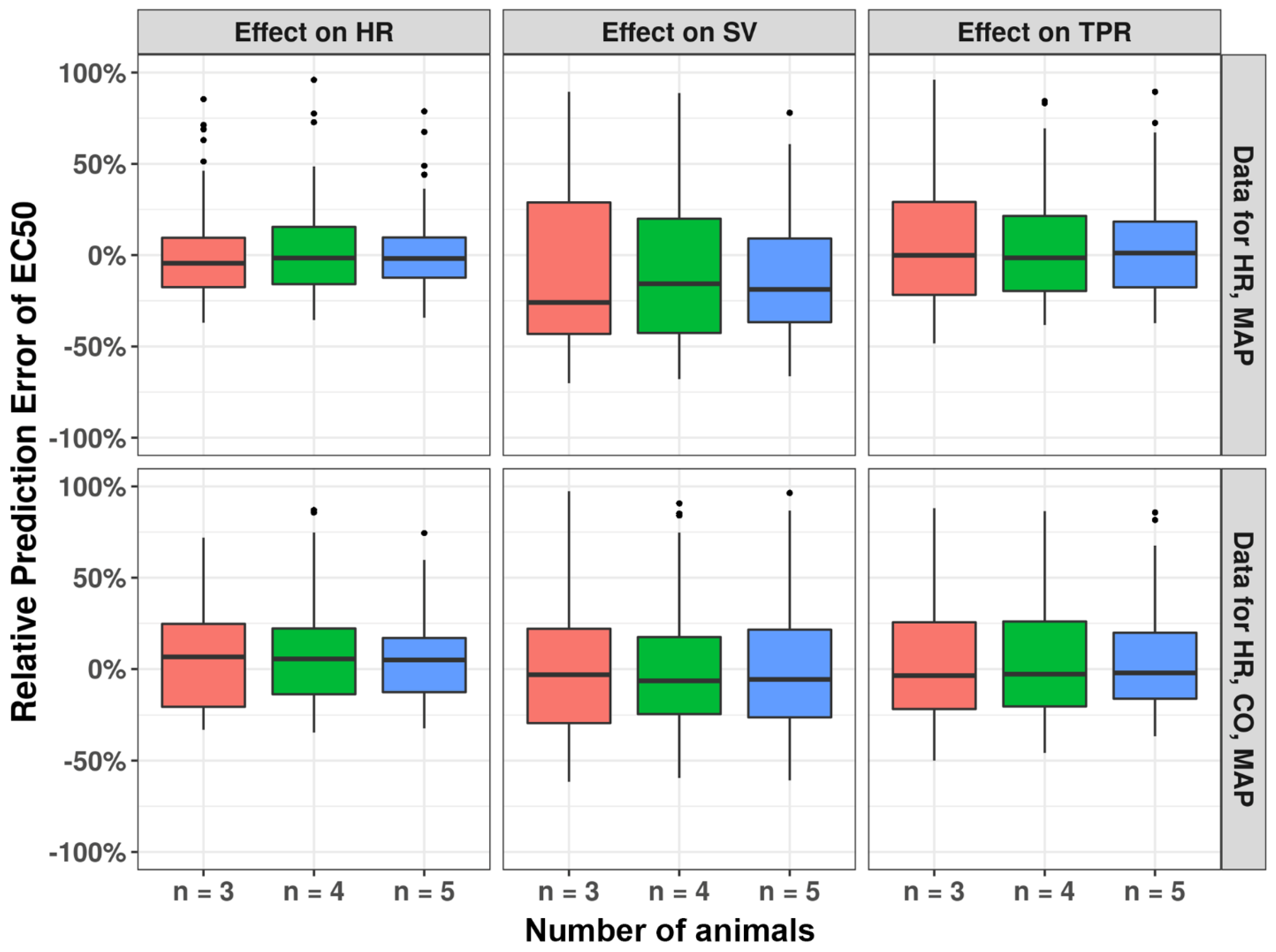
3.4. Number of Animals
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Laverty, H.G.; Benson, C.; Cartwright, E.; Cross, M.; Garland, C.; Hammond, T.; Holloway, C.; McMahon, N.; Milligan, J.; Park, B.; et al. How can we improve our understanding of cardiovascular safety liabilities to develop safer medicines? Br. J. Pharmacol. 2011, 163, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Munos, B. Lessons from 60 years of pharmaceutical innovation. Nat. Rev. Drug Discov. 2009, 8, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.; Brown, D.; Alexander, R.; March, R.; Morgan, P.; Satterthwaite, G.; Pangalos, M.N. Lessons learned from the fate of AstraZeneca’s drug pipeline: A five-dimensional framework. Nat. Rev. Drug Discov. 2014, 13, 419–431. [Google Scholar] [CrossRef] [PubMed]
- FDA Assessment of Pressor Effects of Drugs Guidance for Industry DRAFT GUIDANCE. 2022. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/assessment-pressor-effects-drugs-guidance-industry (accessed on 1 January 2023).
- Oparil, S.; Zaman, M.A.; Calhoun, D.A. Pathogenesis of Hypertension. Ann. Intern. Med. 2003, 139, 761. [Google Scholar] [CrossRef] [PubMed]
- Snelder, N.; Ploeger, A.B.; Luttringer, O.; Rigel, D.F.; Webb, R.L.; Feldman, D.; Fu, F.; Beil, M.; Jin, L.; Stanski, D.R.; et al. PKPD modelling of the interrelationship between mean arterial BP, cardiac output and total peripheral resistance in conscious rats. Br. J. Pharmacol. 2013, 169, 1510–1524. [Google Scholar] [CrossRef] [PubMed]
- Snelder, N.; Ploeger, A.B.; Luttringer, O.; Rigel, D.F.; Fu, F.; Beil, M.; Stanski, D.R.; Danhof, M. Drug effects on the CVS in conscious rats: Separating cardiac output into heart rate and stroke volume using PKPD modelling. Br. J. Pharmacol. 2014, 171, 5076–5092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatasubramanian, R.; Collins, T.A.; Lesko, L.J.; Mettetal, J.T.; Trame, M.N. Semi-mechanistic modelling platform to assess cardiac contractility and haemodynamics in preclinical cardiovascular safety profiling of new molecular entities. Br. J. Pharmacol. 2020, 177, 3568–3590. [Google Scholar] [CrossRef] [PubMed]
- Bahnasawy, S.; Al-Sallami, H.; Duffull, S. A minimal model to describe short-term haemodynamic changes of the cardiovascular system. Br. J. Clin. Pharmacol. 2020, 87, 1411–1421. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; du Sert, N.P.; Vollert, J.; Rice, A.S.C. General Principles of Preclinical Study Design. Good Res. Pract. Non Clin. Pharmacol. Biomed. 2019, 257, 55–69. [Google Scholar]
- Landis, S.C.; Amara, S.G.; Asadullah, K.; Austin, C.P.; Blumenstein, R.; Bradley, E.W.; Crystal, R.G.; Darnell, R.B.; Ferrante, R.J.; Fillit, H.; et al. A call for transparent reporting to optimize the predictive value of preclinical research. Nature 2012, 490, 187–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, M.J.; Hancox, J.C.; Farkas, A.; Wainwright, C.L.; Stables, C.L.; Saint, D.A.; Clements-Jewery, H.; Lambiase, P.D.; Billman, G.E.; Janse, M.J.; et al. The Lambeth Conventions (II): Guidelines for the study of animal and human ventricular and supraventricular arrhythmias. Pharmacol. Ther. 2013, 139, 213–248. [Google Scholar] [CrossRef] [PubMed]
- Ventura, H.O.; Taler, S.J.; Strobeck, J.E. Hypertension as a hemodynamic disease: The role of impedance cardiography in diagnostic, prognostic, and therapeutic decision making. Am. J. Hypertens. 2005, 18, 26–43. [Google Scholar] [CrossRef] [PubMed]
- Koobi, T.; Kaukinen, N.A.; Turjanmaa, V.M.H.; Uusitalo, A.J. Whole-body impedance cardiography in the measurement of cardiac output. Crit. Care Med. 1997, 25, 779–785. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, Y.; Snelder, N.; Guo, T.; van der Graaf, P.H.; van Hasselt, J.G.C. Evaluation of a Cardiovascular Systems Model for Design and Analysis of Hemodynamic Safety Studies. Pharmaceutics 2023, 15, 1175. https://doi.org/10.3390/pharmaceutics15041175
Fu Y, Snelder N, Guo T, van der Graaf PH, van Hasselt JGC. Evaluation of a Cardiovascular Systems Model for Design and Analysis of Hemodynamic Safety Studies. Pharmaceutics. 2023; 15(4):1175. https://doi.org/10.3390/pharmaceutics15041175
Chicago/Turabian StyleFu, Yu, Nelleke Snelder, Tingjie Guo, Piet H. van der Graaf, and Johan. G. C. van Hasselt. 2023. "Evaluation of a Cardiovascular Systems Model for Design and Analysis of Hemodynamic Safety Studies" Pharmaceutics 15, no. 4: 1175. https://doi.org/10.3390/pharmaceutics15041175
APA StyleFu, Y., Snelder, N., Guo, T., van der Graaf, P. H., & van Hasselt, J. G. C. (2023). Evaluation of a Cardiovascular Systems Model for Design and Analysis of Hemodynamic Safety Studies. Pharmaceutics, 15(4), 1175. https://doi.org/10.3390/pharmaceutics15041175