Furfural Analogs as Sustainable Corrosion Inhibitors—Predictive Efficiency Using DFT and Monte Carlo Simulations on the Cu(111), Fe(110), Al(111) and Sn(111) Surfaces in Acid Media

 , ,
, ,  ,
, 
Abstract
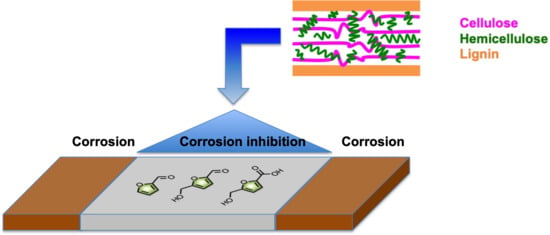
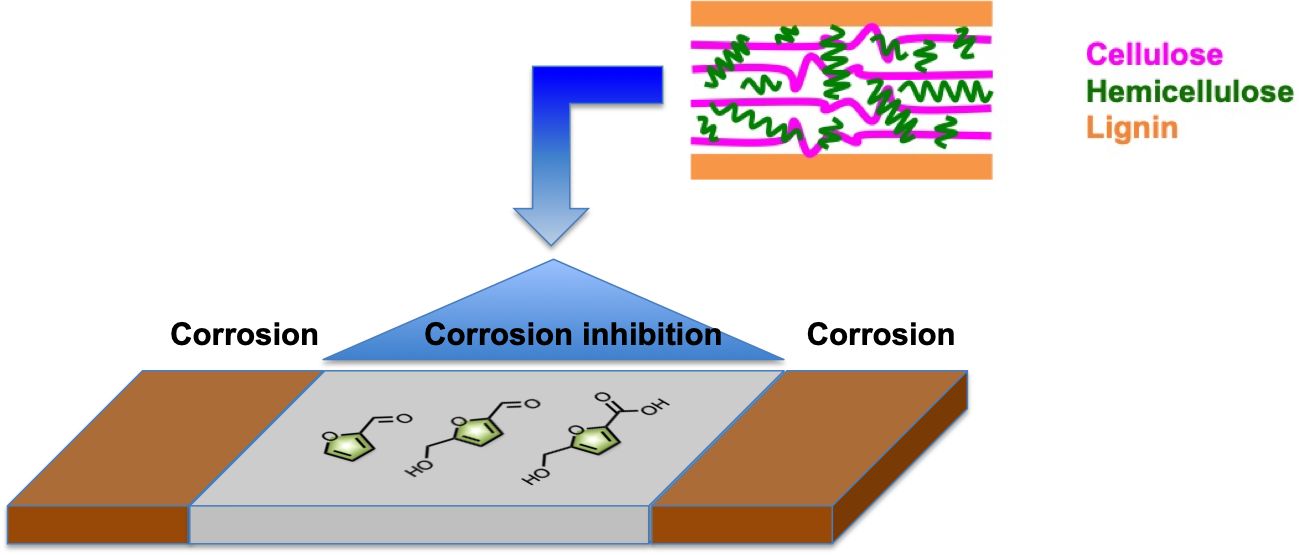
:
1. Introduction
2. Computational Details
2.1. DFT Calculations
2.2. Monte Carlo Simulations
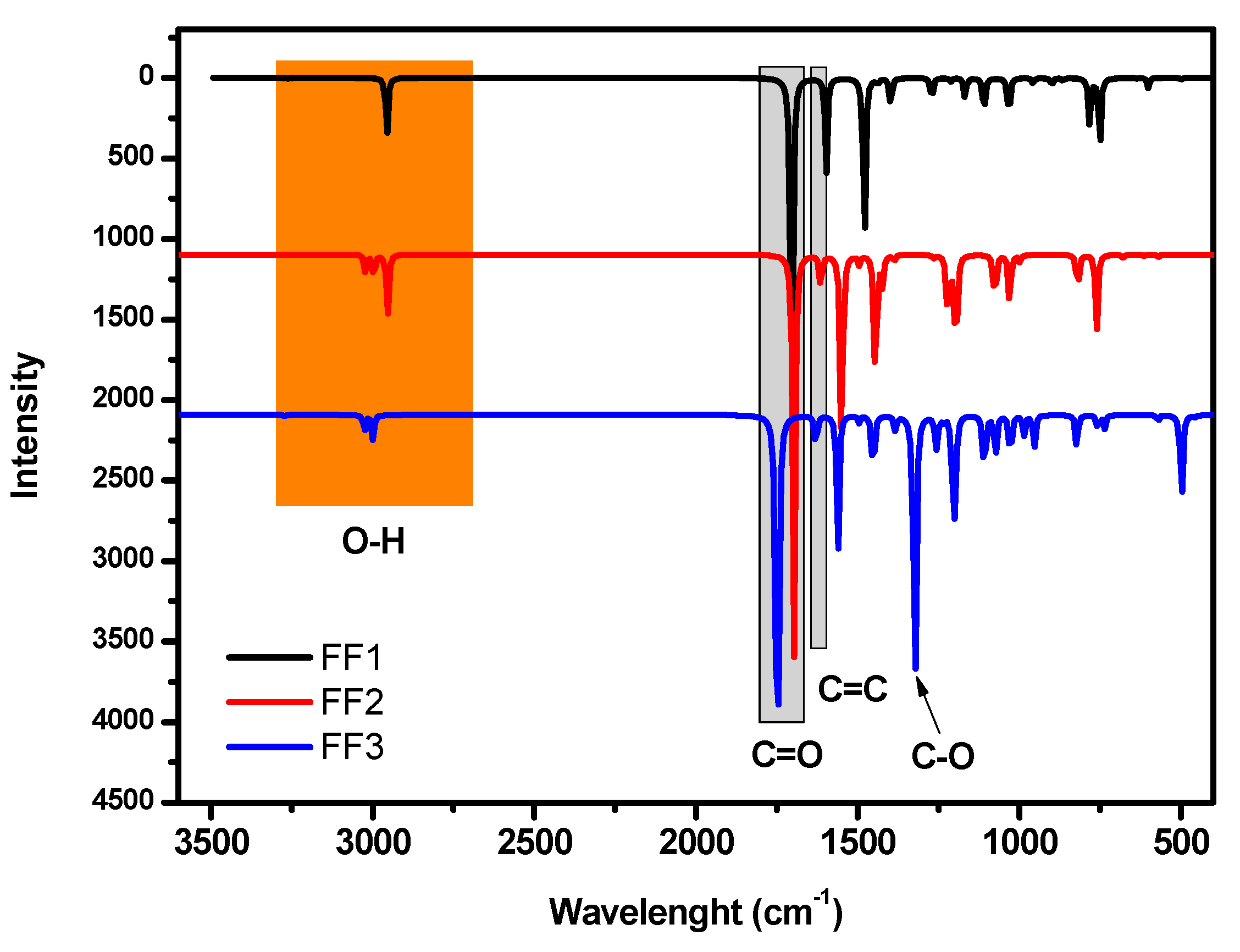
3. DFT Performances
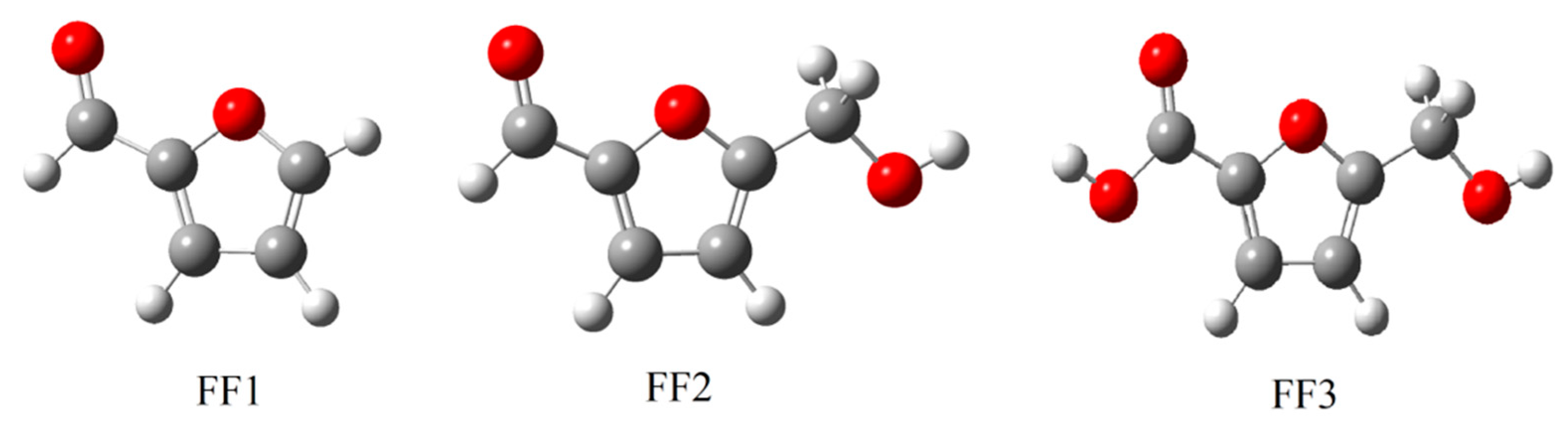
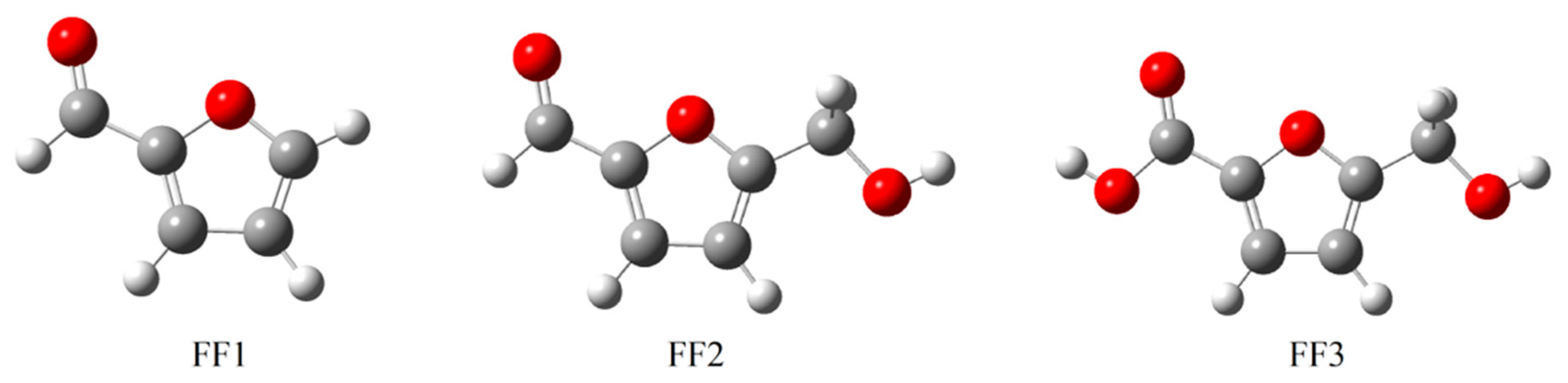
3.1. Optimized Structures
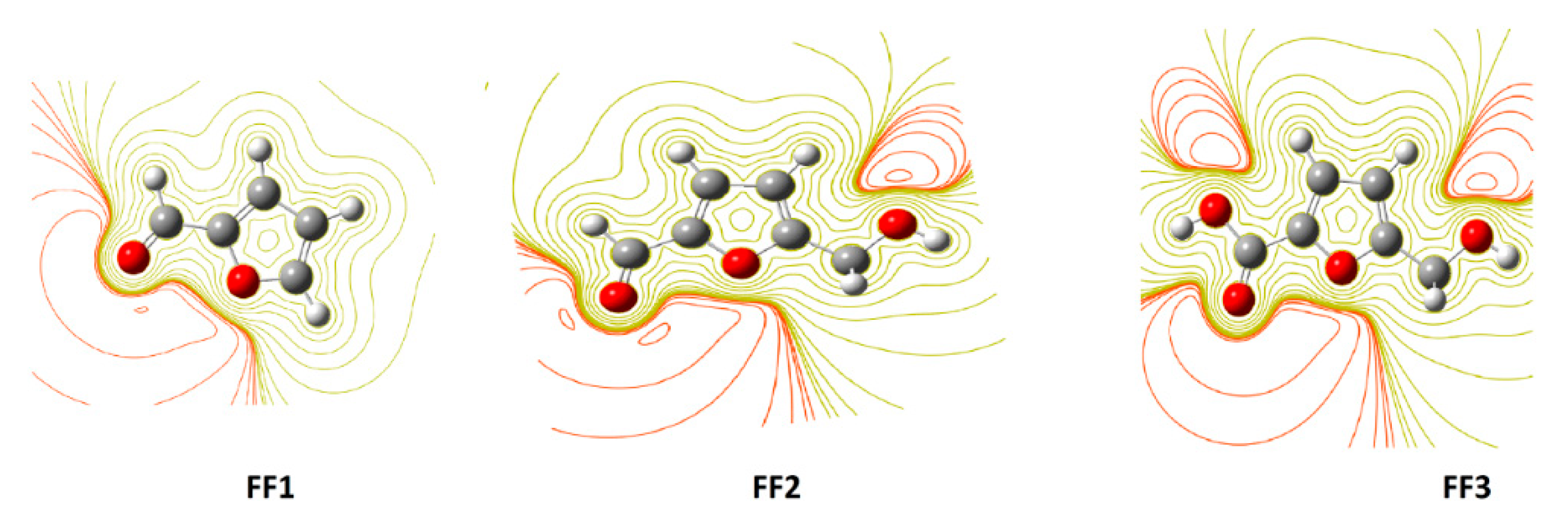
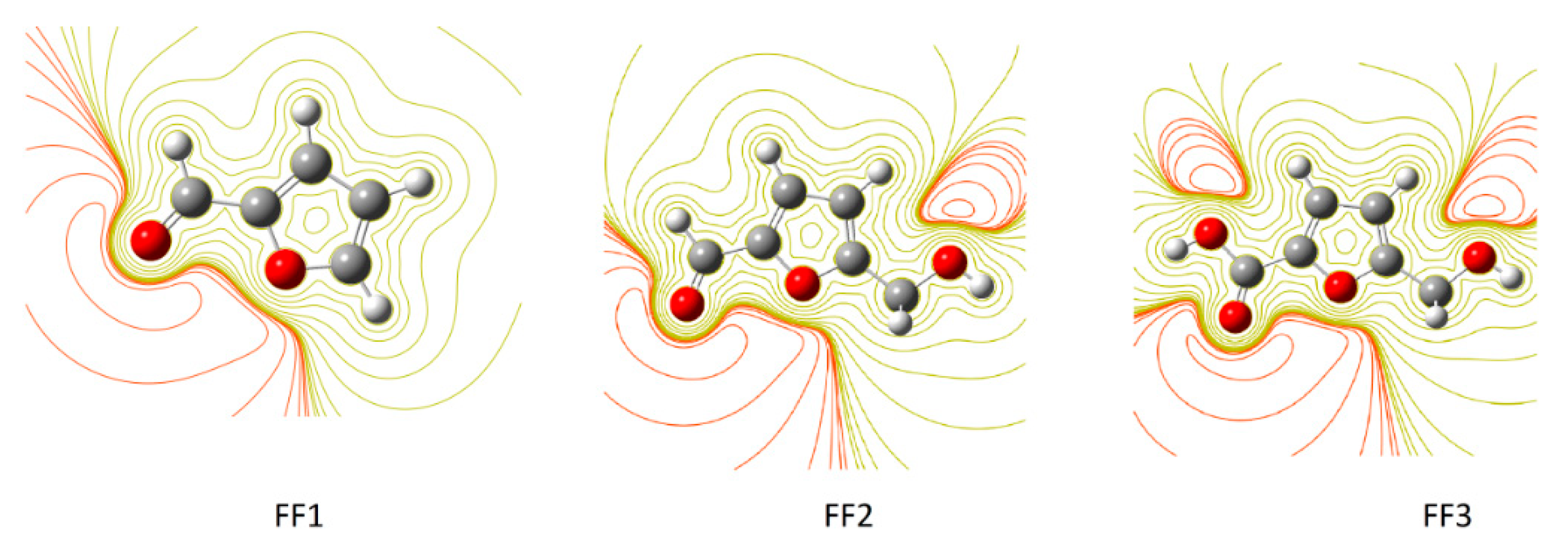
3.2. Electrostatic Potential (ESP) 2D Maps
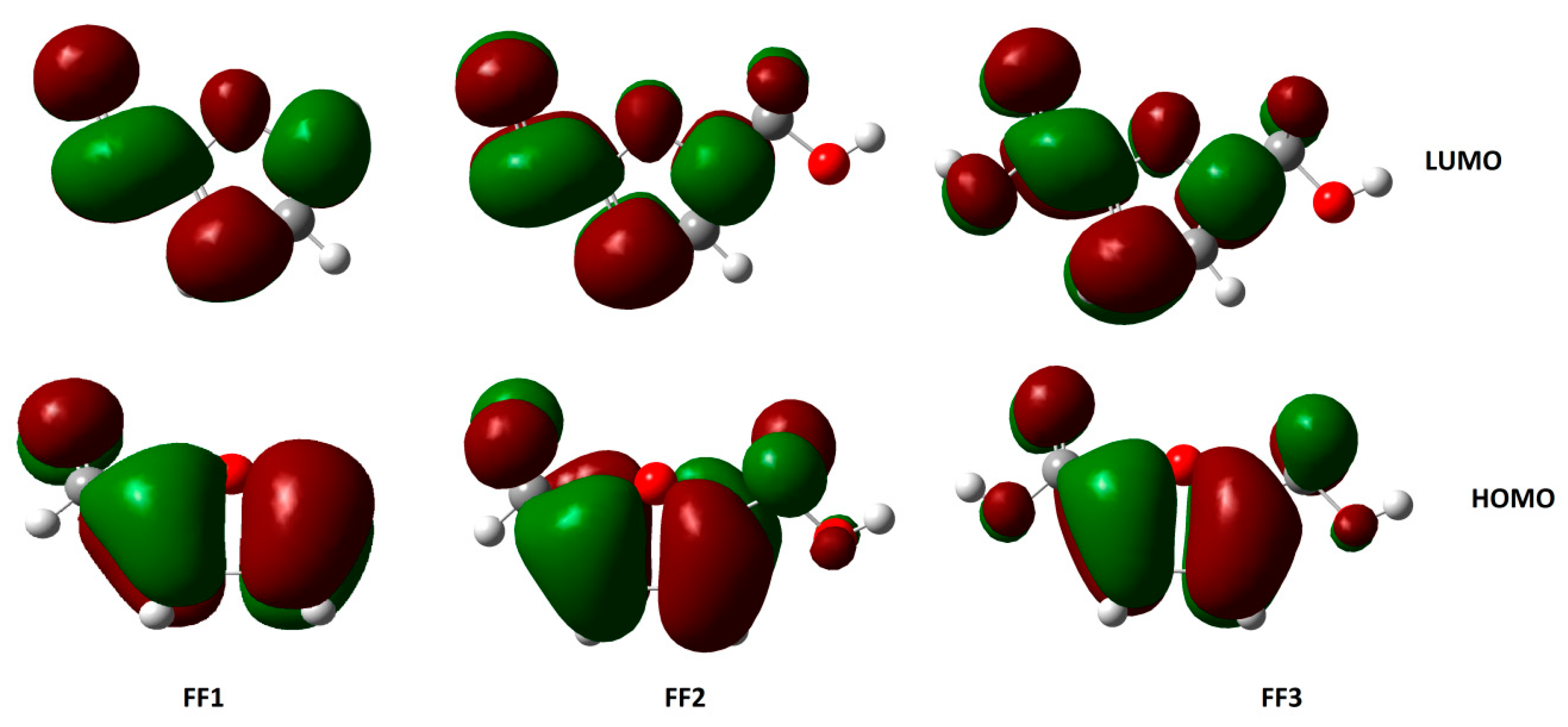
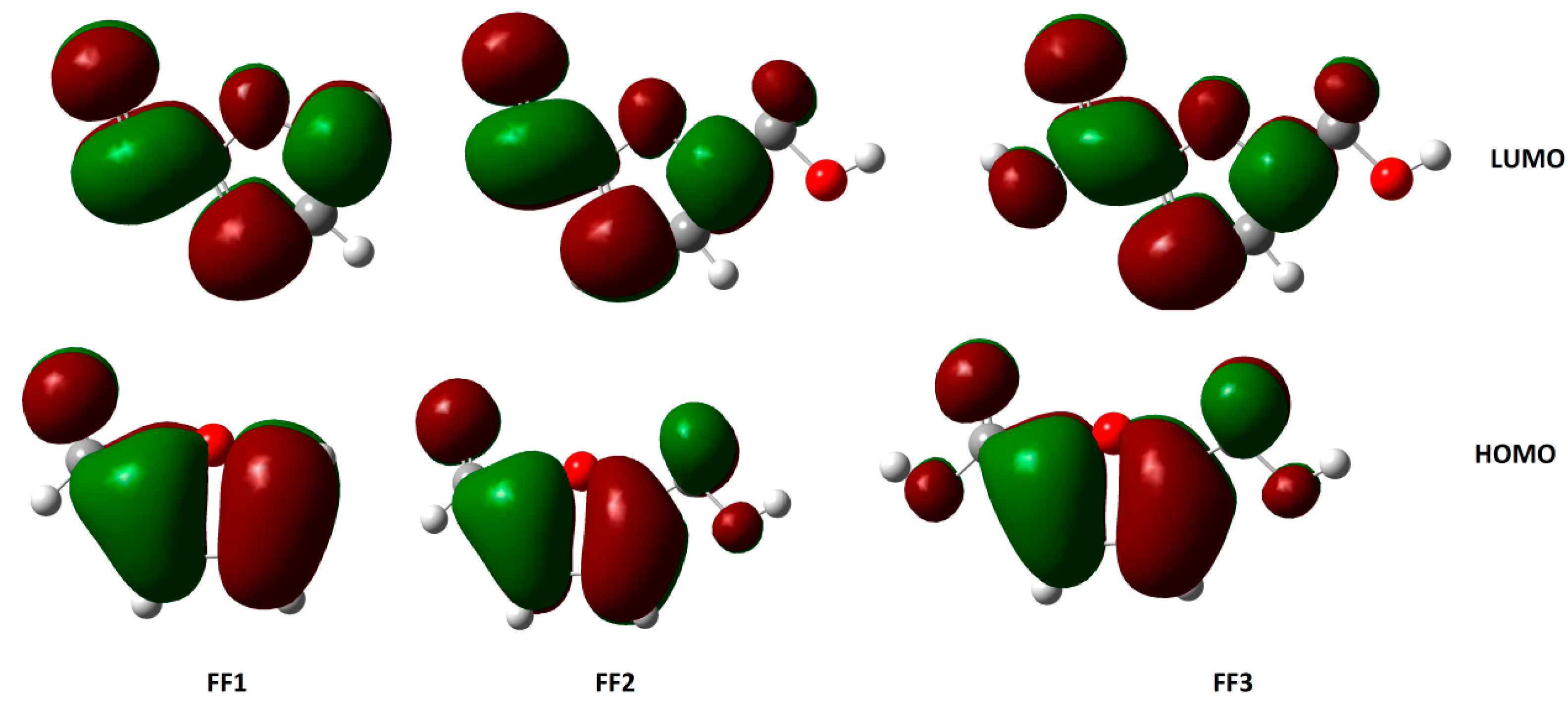
3.3. HOMO and LUMO Energies and Derived Parameters
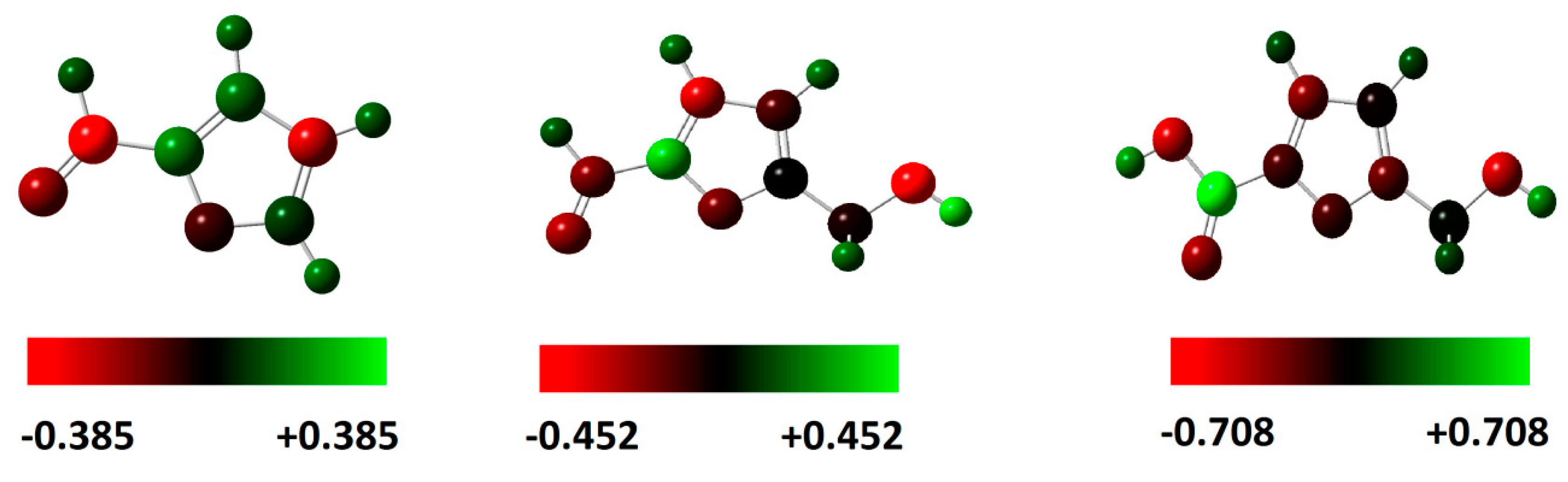
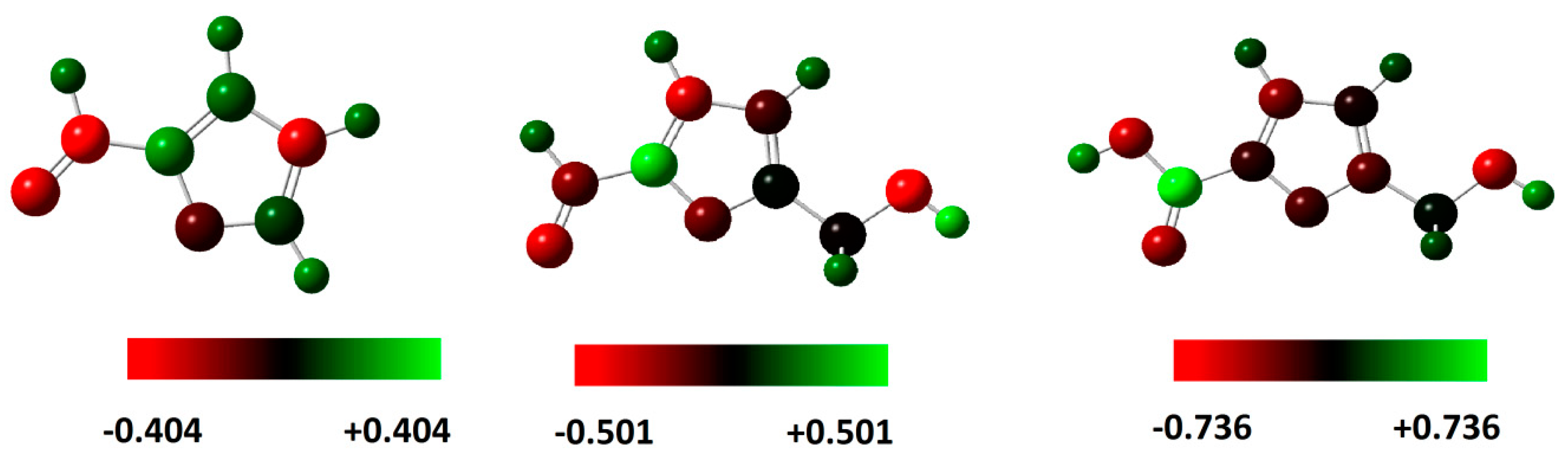
4. Charge Distribution
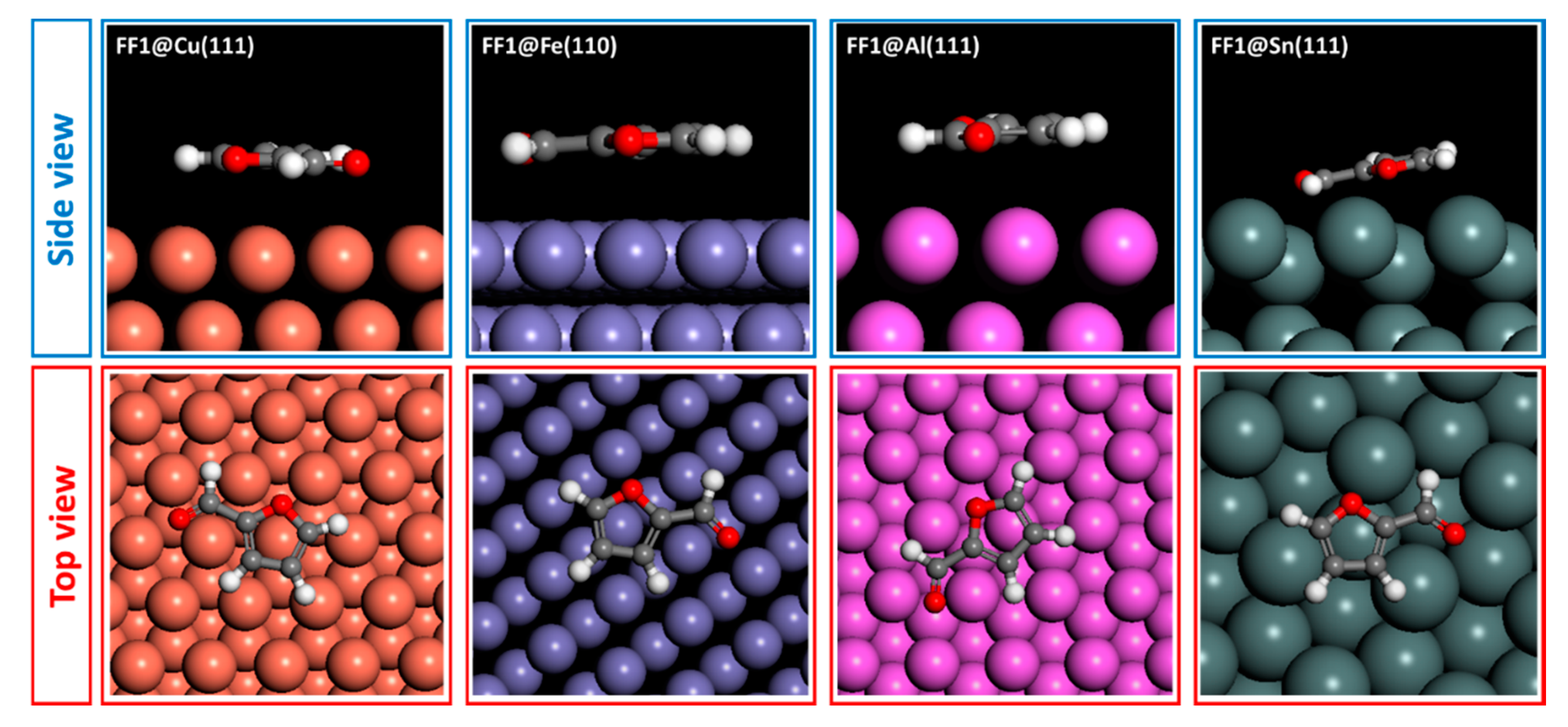
5. Morphology of Studied Metal Surfaces
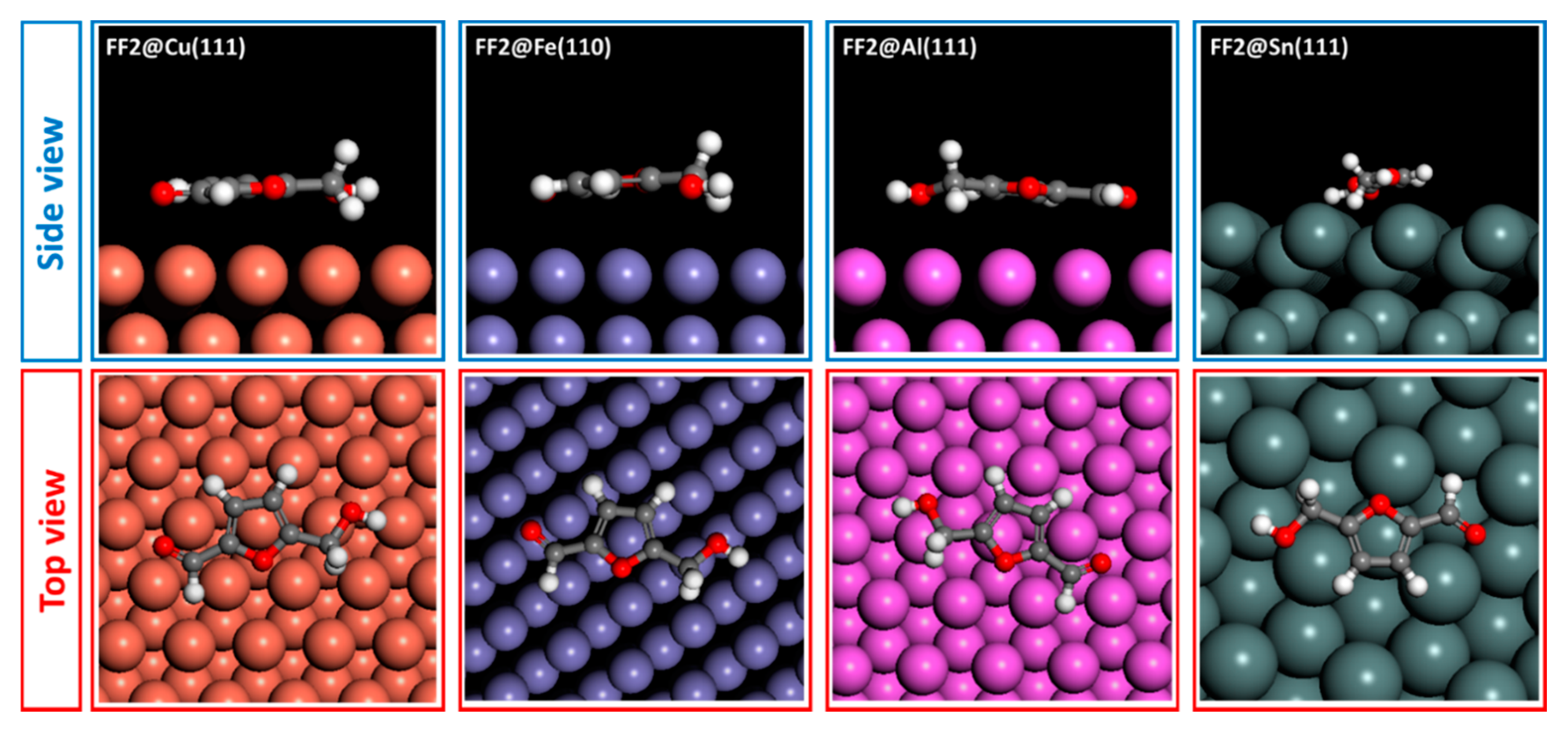
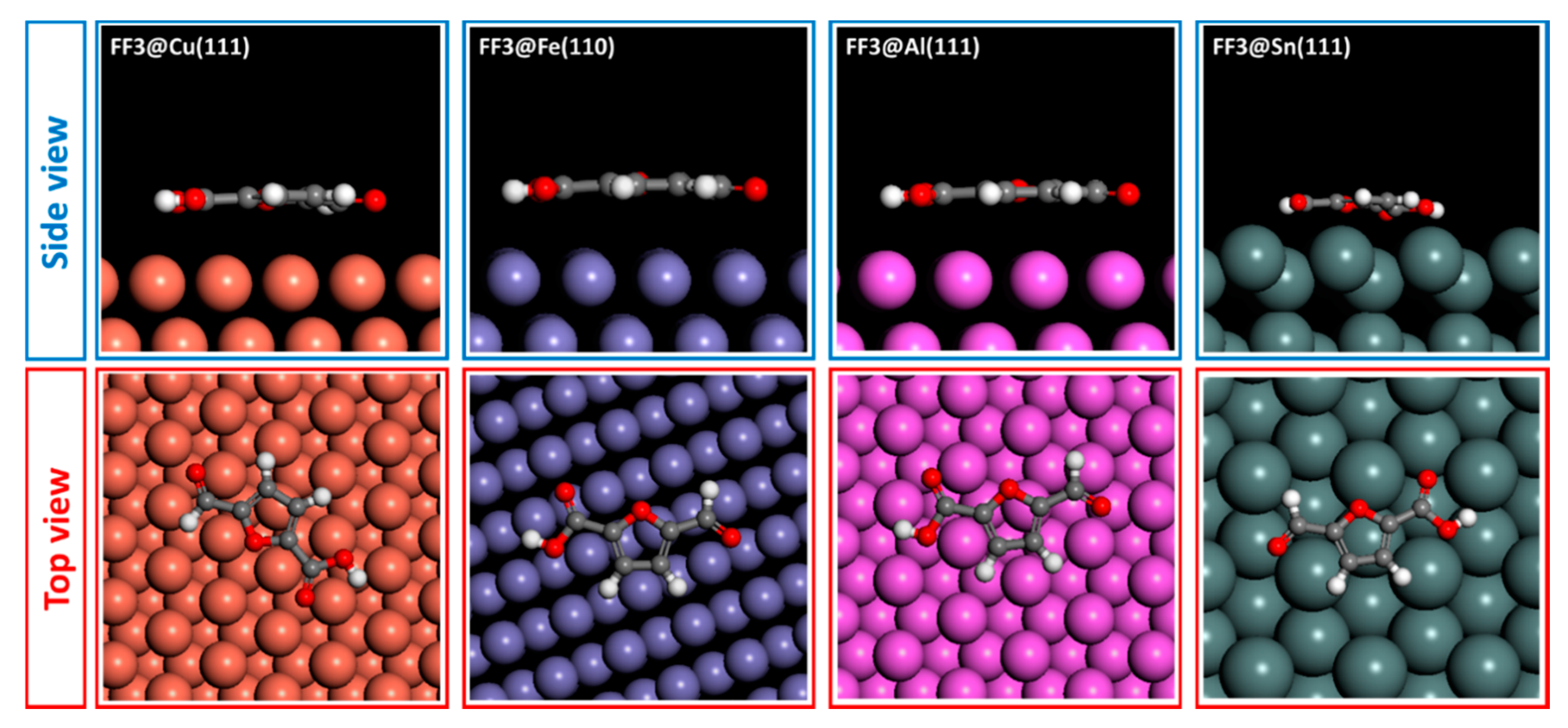
6. Monte Carlo Simulations
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- King, D.; Inderwildi, O.R.; Williams, A.; Hagan, A. The future of industrial biorefineries. In Proceedings of the World Economic Forum White Paper; World Economic Forum: Cologn/Geneva, Switzerland, 2010. [Google Scholar]
- Koch, G.H.; Brongers, M.P.H.; Thompson, N.G.; Virmani, Y.P.; Payer, J.H. Corrosion Cost and Preventive Strategies in the United States; Federal Highway Administration: McLean, VA, USA, 2002.
- Shaw, B.A.; Kelly, R.G. What is corrosion? Electrochem. Soc. Interface 2006, 15, 24–27. [Google Scholar]
- Connatser, R.M.; Frith, M.G.; Jun, J.; Lewis Sr, S.A.; Brady, M.P.; Keiser, J.R. Approaches to investigate the role of chelation in the corrosivity of biomass-derived oils. Biomass Bioenergy 2020, 133, 105446. [Google Scholar] [CrossRef]
- Hsissou, R.; Dagdag, O.; Abbout, S.; Benhiba, F.; Berradi, M.; El Bouchti, M.; Berisha, A.; Hajjaji, N.; Elharfi, A. Novel derivative epoxy resin TGETET as a corrosion inhibition of E24 carbon steel in 1.0 M HCl solution. Experimental and computational (DFT and MD simulations) methods. J. Mol. Liq. 2019, 284, 182–192. [Google Scholar] [CrossRef]
- Dohare, P.; Quraishi, M.A.; Lgaz, H.; Salghi, R. Electrochemical DFT and MD simulation study of substituted imidazoles as novel corrosion inhibitors for mild steel. Port. Electrochim. Acta 2019, 37, 217–239. [Google Scholar] [CrossRef]
- Bahlakeh, G.; Dehghani, A.; Ramezanzadeh, B.; Ramezanzadeh, M. Highly effective mild steel corrosion inhibition in 1 M HCl solution by novel green aqueous mustard seed extract: Experimental, electronic-scale DFT and atomic-scale MC/MD explorations. J. Mol. Liq. 2019, 293, 111559. [Google Scholar] [CrossRef]
- El Ibrahimi, B.; El Mouaden, K.; Jmiai, A.; Baddouh, A.; El Issami, S.; Bazzi, L.; Hilali, M. Understanding the influence of solution’s pH on the corrosion of tin in saline solution containing functional amino acids using electrochemical techniques and molecular modeling. Surf. Interfaces 2019, 17, 100343. [Google Scholar] [CrossRef]
- Qiang, Y.; Zhang, S.; Tan, B.; Chen, S. Evaluation of Ginkgo leaf extract as an eco-friendly corrosion inhibitor of X70 steel in HCl solution. Corros. Sci. 2018, 133, 6–16. [Google Scholar] [CrossRef]
- Motamedi, M.; Ramezanzadeh, B.; Mahdavian, M. Corrosion inhibition properties of a green hybrid pigment based on Pr-Urtica Dioica plant extract. J. Ind. Eng. Chem. 2018, 66, 116–125. [Google Scholar] [CrossRef]
- Saxena, A.; Prasad, D.; Haldhar, R.; Singh, G.; Kumar, A. Use of Sida cordifolia extract as green corrosion inhibitor for mild steel in 0.5 M H2SO4. J. Environ. Chem. Eng. 2018, 6, 694–700. [Google Scholar] [CrossRef]
- Ebenso, E.E.; Kabanda, M.M.; Arslan, T.; Saracoglu, M.; Kandemirli, F.; Murulana, L.C.; Singh, A.K.; Shukla, S.K.; Hammouti, B.; Khaled, K.F.; et al. Quantum chemical investigations on quinoline derivatives as effective corrosion inhibitors for mild steel in acidic medium. Int. J. Electrochem. Sci. 2012, 7, 5643–5676. [Google Scholar]
- Iruthayaraj, A.; Chinnasamy, K.; Jha, K.K.; Munshi, P.; Pavan, M.S.; Kumaradhas, P. Topology of electron density and electrostatic potential of HIV reverse transcriptase inhibitor zidovudine from high resolution X-ray diffraction and charge density analysis. J. Mol. Struct. 2019, 1180, 683–697. [Google Scholar] [CrossRef]
- Elusta, M.I.; Başaran, M.A.; Kandemirli, F. Theoretical studies on mild steel corrosion inhibition by 5-substituted 1H-tetrazoles in acidic media. Int. J. Electrochem. Sci. 2019, 14, 2743–2756. [Google Scholar] [CrossRef]
- Oukhrib, R.; El Ibrahimi, B.; Bourzi, H.; El Mouaden, K.; Jmiai, A.; El Issami, S.; Bammou, L.; Bazzi, L. Quantum chemical calculations and corrosion inhibition efficiency of biopolymer “chitosan” on copper surface in 3%NaCl. J. Mater. Environ. Sci. 2017, 8, 195–208. [Google Scholar]
- Gao, G.; Liang, C. Electrochemical and DFT studies of β-amino-alcohols as corrosion inhibitors for brass. Electrochim. Acta 2007, 52, 4554–4559. [Google Scholar] [CrossRef]
- Ahmed, S.K.; Ali, W.B.; Khadom, A.A. Synthesis and investigations of heterocyclic compounds as corrosion inhibitors for mild steel in hydrochloric acid. Int. J. Ind. Chem. 2019, 10, 159–173. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Ansari, K.R.; Quraishi, M.A.; Kaya, S.; Banerjee, P. The effect of an N-heterocyclic compound on corrosion inhibition of J55 steel in sweet corrosive medium. New J. Chem. 2019, 43, 6303–6313. [Google Scholar] [CrossRef]
- Al Zoubi, W.; Ko, Y.G. Self-assembly of hierarchical N-heterocycles-inorganic materials into three-dimensional structure for superior corrosion protection. Chem. Eng. J. 2019, 356, 850–856. [Google Scholar] [CrossRef]
- Benmahammed, I.; Douadi, T.; Issaadi, S.; Al-Noaimi, M.; Chafaa, S. Heterocyclic Schiff bases as corrosion inhibitors for carbon steel in 1 M HCl solution: Hydrodynamic and synergetic effect. J. Dispersion Sci. Technol. 2019, 1–20. Available online: https://doi.org/10.1080/01932691.2019.1614038 (accessed on 13 April 2020). [CrossRef]
- Al-abdali, F.H.; Abdallah, M.; El-Sayed, R. Corrosion inhibition of aluminum using nonionic surfactant compounds with a six membered heterocyclic ring in 1.0 M HCl solution. Int. J. Electrochem. Sci. 2019, 14, 3509–3523. [Google Scholar] [CrossRef]
- Verma, C.; Ebenso, E.E.; Quraishi, M.A. Alkaloids as green and environmental benign corrosion inhibitors: An overview. Int. J. Corros. Scale Inhib. 2019, 8, 512–528. [Google Scholar]
- Cai, C.M.; Zhang, T.; Kumar, R.; Wyman, C.E. Integrated furfural production as a renewable fuel and chemical platform from lignocellulosic biomass. J. Chem. Technol. Biotechnol. 2014, 89, 2–10. [Google Scholar] [CrossRef]
- Montané, D.; Salvadó, J.; Torras, C.; Farriol, X. High-temperature dilute-acid hydrolysis of olive stones for furfural production. Biomass Bioenergy 2002, 22, 295–304. [Google Scholar] [CrossRef]
- Mansilla, H.D.; Baeza, J.; Urzua, S.; Maturana, G.; Villaseñor, J.; Durán, N. Acid-catalysed hydrolysis of rice hull: Evaluation of furfural production. Bioresour. Technol. 1998, 66, 189–193. [Google Scholar] [CrossRef]
- Garcia-Olmo, A.J.; Yepez, A.; Balu, A.M.; Prinsen, P.; Garcia, A.; Maziere, A.; Len, C.; Luque, R. Activity of continuous flow synthesized Pd-based nanocatalysts in the flow hydroconversion of furfural. Tetrahedron 2017, 73, 5599–5604. [Google Scholar] [CrossRef]
- Delbecq, F.; Wang, Y.; Muralidhara, A.; El Ouardi, K.; Marlair, G.; Len, C. Hydrolysis of hemicellulose and derivatives—A review of recent advances in the production of furfural. Front. Chem. 2018, 6, 146. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Delbecq, F.; Kwapinski, W.; Len, C. Application of sulfonated carbon-based catalyst for the furfural production from d-xylose and xylan in a microwave-assisted biphasic reaction. Mol. Catal. 2017, 438, 167–172. [Google Scholar] [CrossRef]
- Le Guenic, S.; Delbecq, F.; Ceballos, C.; Len, C. Microwave-assisted dehydration of D-xylose into furfural by diluted inexpensive inorganic salts solution in a biphasic system. J. Mol. Catal. A Chem. 2015, 410, 1–7. [Google Scholar] [CrossRef]
- Delbecq, F.; Takahashi, Y.; Kondo, T.; Corbas, C.C.; Ramos, E.R.; Len, C. Microwave assisted efficient furfural production using nano-sized surface-sulfonated diamond powder. Catal. Commun. 2018, 110, 74–78. [Google Scholar] [CrossRef]
- Delbecq, F.; Wang, Y.; Len, C. Conversion of xylose, xylan and rice husk into furfural via betaine and formic acid mixture as novel homogeneous catalyst in biphasic system by microwave-assisted dehydration. J. Mol. Catal. A Chem. 2016, 423, 520–525. [Google Scholar] [CrossRef]
- Le Guenic, S.; Gergela, D.; Ceballos, C.; Delbecq, F.; Len, C. Furfural production from D-xylose and xylan by using stable Nafion NR50 and NaCl in a microwave-assisted biphasic reaction. Molecules 2016, 21, 1102. [Google Scholar] [CrossRef]
- Obot, I.B.; Obi-Egbedi, N.O. Theoretical study of benzimidazole and its derivatives and their potential activity as corrosion inhibitors. Corros. Sci. 2010, 52, 657–660. [Google Scholar] [CrossRef]
- Obot, I.B.; Obi-Egbedi, N.O.; Umoren, S.A. Antifungal drugs as corrosion inhibitors for aluminium in 0.1 M HCl. Corros. Sci. 2009, 51, 1868–1875. [Google Scholar] [CrossRef]
- Kaya, S.; Kaya, C.; Guo, L.; Kandemirli, F.; Tüzün, B.; Uğurlu, İ.; Madkour, L.H.; Saraçoğlu, M. Quantum chemical and molecular dynamics simulation studies on inhibition performances of some thiazole and thiadiazole derivatives against corrosion of iron. J. Mol. Liq. 2016, 219, 497–504. [Google Scholar] [CrossRef]
- Erdoğan, Ş.; Safi, Z.S.; Kaya, S.; Işın, D.Ö.; Guo, L.; Kaya, C. A computational study on corrosion inhibition performances of novel quinoline derivatives against the corrosion of iron. J. Mol. Struct. 2017, 1134, 751–761. [Google Scholar] [CrossRef]
- El Belghiti, M.; Karzazi, Y.; Dafali, A.; Hammouti, B.; Bentiss, F.; Obot, I.B.; Bahadur, I.; Ebenso, E.E. Experimental, quantum chemical and Monte Carlo simulation studies of 3, 5-disubstituted-4-amino-1, 2, 4-triazoles as corrosion inhibitors on mild steel in acidic medium. J. Mol. Liq. 2016, 218, 281–293. [Google Scholar] [CrossRef]
- Obot, I.B.; Kaya, S.; Kaya, C.; Tüzün, B. Density Functional Theory (DFT) modeling and Monte Carlo simulation assessment of inhibition performance of some carbohydrazide Schiff bases for steel corrosion. Phys. E Low Dimens. Syst. Nanostruct. 2016, 80, 82–90. [Google Scholar] [CrossRef]
- Ledieu, A.; Devreux, F.; Barboux, P. Monte Carlo simulations of borosilicate glass corrosion: Predictions for morphology and kinetics. J. Non Cryst. Solids 2004, 345, 715–719. [Google Scholar] [CrossRef]
- Verma, C.; Obot, I.B.; Bahadur, I.; Sherif, E.S.M.; Ebenso, E.E. Choline based ionic liquids as sustainable corrosion inhibitors on mild steel surface in acidic medium: Gravimetric, electrochemical, surface morphology, DFT and Monte Carlo simulation studies. Appl. Surf. Sci. 2018, 457, 134–149. [Google Scholar] [CrossRef]
- Verma, C.; Lgaz, H.; Verma, D.K.; Ebenso, E.E.; Bahadur, I.; Quraishi, M.A. Molecular dynamics and Monte Carlo simulations as powerful tools for study of interfacial adsorption behavior of corrosion inhibitors in aqueous phase: A review. J. Mol. Liq. 2018, 260, 99–120. [Google Scholar] [CrossRef]
- El Faydy, M.; Touir, R.; Touhami, M.E.; Zarrouk, A.; Jama, C.; Lakhrissi, B.; Olasunkanmi, L.O.; Ebenso, E.E.; Bentiss, F. Corrosion inhibition performance of newly synthesized 5-alkoxymethyl-8-hydroxyquinoline derivatives for carbon steel in 1 M HCl solution: Experimental, DFT and Monte Carlo simulation studies. Phys. Chem. Chem. Phys. 2018, 20, 20167–20187. [Google Scholar] [CrossRef]
- El Basiony, N.M.; Elgendy, A.; Nady, H.; Migahed, M.A.; Zaki, E.G. Adsorption characteristics and inhibition effect of two Schiff base compounds on corrosion of mild steel in 0.5 M HCl solution: Experimental, DFT studies, and Monte Carlo simulation. RSC Adv. 2019, 9, 10473–10485. [Google Scholar] [CrossRef] [Green Version]
- Caleyo, F.; Velázquez, J.C.; Valor, A.; Hallen, J.M. Probability distribution of pitting corrosion depth and rate in underground pipelines: A Monte Carlo study. Corros. Sci. 2009, 51, 1925–1934. [Google Scholar] [CrossRef]
- Khaled, K.F. Monte Carlo simulations of corrosion inhibition of mild steel in 0.5 M sulphuric acid by some green corrosion inhibitors. J. Solid State Electrochem. 2009, 13, 1743–1756. [Google Scholar] [CrossRef]
- Dong, H.; Zheng, Y.; Hu, P. DFT study of furfural conversion on a Re/Pt bimetallic surface: Synergetic effect on the promotion of hydrodeoxygenation. Phys. Chem. Chem. Phys. 2019, 21, 8384–8393. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zheng, Y.; Hu, P. A DFT study of direct furfural conversion to 2-methylfuran on the Ru/Co3O4 surface. Phys. Chem. Chem. Phys. 2019, 21, 1597–1605. [Google Scholar] [CrossRef]
- Agrawal, N.; Gong, L.; Roman, A.; Mark, L.; Medlin, W.; Holewinski, A.; Janik, M.J. Mechanistic Investigations for Electrocatalytic Oxidation of Furfural Using Density Functional Theory. In Proceedings of the Meeting Abstracts; The Electrochemical Society: Pennington, NJ, USA, 2019; p. 1030. [Google Scholar]
- Lin, Z.; Wan, W.; Yao, S.; Chen, J.G. Cobalt-modified molybdenum carbide as a selective catalyst for hydrodeoxygenation of furfural. Appl. Catal. B 2018, 233, 160–166. [Google Scholar] [CrossRef]
- Wan, W.; Jiang, Z.; Chen, J.G. A Comparative study of hydrodeoxygenation of furfural Over Fe/Pt (111) and Fe/Mo 2 C surfaces. Top. Catal. 2018, 61, 439–445. [Google Scholar] [CrossRef]
- Obot, I.B.; Macdonald, D.D.; Gasem, Z.M. Density functional theory (DFT) as a powerful tool for designing new organic corrosion inhibitors. Part 1: An overview. Corros. Sci. 2015, 99, 1–30. [Google Scholar] [CrossRef]
- Kovačević, N.; Milošev, I.; Kokalj, A. The roles of mercapto, benzene, and methyl groups in the corrosion inhibition of imidazoles on copper: II. Inhibitor–copper bonding. Corros. Sci. 2015, 98, 457–470. [Google Scholar] [CrossRef]
- El Ibrahimi, B.; Jmiai, A.; El Mouaden, K.; Baddouh, A.; El Issami, S.; Bazzi, L.; Hilali, M. Effect of solution’s pH and molecular structure of three linear α-amino acids on the corrosion of tin in salt solution: A combined experimental and theoretical approach. J. Mol. Struct. 2019, 1196, 105–118. [Google Scholar] [CrossRef]
- Hölzl, J.; Schulte, F.K. Work function of metals. In Solid Surface Physics; Springer: Berlin, Germany, 1979; pp. 1–150. [Google Scholar]
- Skriver, H.L.; Rosengaard, N.M. Surface energy and work function of elemental metals. Phys. Rev. B 1992, 46, 7157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Ansari, K.R.; Quraishi, M.A.; Lin, Y. Investigation of corrosion inhibitors adsorption on metals using density functional theory and molecular dynamics simulation. In Corrosion Inhibitors; IntechOpen: London, UK, 2019. [Google Scholar]
- Obot, I.B.; Haruna, K.; Saleh, T.A. Atomistic Simulation: A Unique and Powerful Computational Tool for Corrosion Inhibition Research. Arab. J. Sci. Eng. 2019, 44, 1–32. [Google Scholar] [CrossRef]
- Kirkpatrick, S.; Gelatt, C.D.; Vecchi, M.P. Optimization by Simulated Annealing. Science 1983, 220, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Fabian, V. Simulated annealing simulated. Computers Math. Applic. 1997, 33, 81–94. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Dong, Y. Crystal morphology study of N, N′-diacetylchitobiose by molecular dynamics simulation. Carbohydr. Res. 2011, 346, 2457–2462. [Google Scholar] [CrossRef]
- El Ibrahimi, B.; Jmiai, A.; El Mouaden, K.; Oukhrib, R.; Soumoue, A.; El Issami, S.; Bazzi, L. Theoretical evaluation of some α-amino acids for corrosion inhibition of copper in acidic medium: DFT calculations, Monte Carlo simulations and QSPR studies. J. King Saud. Univ. Sci. 2020, 32, 163–171. [Google Scholar] [CrossRef]
- Kabanda, M.M.; Obot, I.B.; Ebenso, E.E. Computational Study of Some Amino Acid Derivatives as Potential Corrosion Inhibitors for Different Metal Surfaces and in Different Media. Int. J. Electrochem. Sci. 2013, 8, 10839–10850. [Google Scholar]
- Albrecht, M.; Yi, H.; Köksal, O.; Raabe, G.; Pan, F.; Valkonen, A.; Rissanen, K. CF3: An Electron-Withdrawing Substituent for Aromatic Anion Acceptors? “side-On” versus “on-Top” Binding of Halides. Chem. Eur. J. 2016, 22, 6956–6963. [Google Scholar] [CrossRef]
- Fukui, K.; Yonezawa, T.; Shingu, H. A molecular orbital theory of reactivity in aromatic hydrocarbons. J. Chem. Phys. 1952, 20, 722–725. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Wang, H.; Wang, L.; Liu, A. DFT study of new bipyrazole derivatives and their potential activity as corrosion inhibitors. J. Mol. Model. 2007, 13, 147–153. [Google Scholar] [CrossRef]
- Stoyanova, A.E.; Peyerimhoff, S.D. On the relationship between corrosion inhibiting effect and molecular structure. Electrochim. Acta 2002, 47, 1365–1371. [Google Scholar] [CrossRef]
- Senet, P. Chemical hardnesses of atoms and molecules from frontier orbitals. Chem. Phys. Lett. 1997, 275, 527–532. [Google Scholar] [CrossRef]
- Salman, T.A.; Zinad, D.S.; Jaber, S.H.; Al-Ghezi, M.; Mahal, A.; Takriff, M.S.; Al-Amiery, A.A. Effect of 1,3,4-thiadiazole scaffold on the corrosion inhibition of mild steel in acidic medium: An experimental and computational study. J. Bio. Tribo. Corros. 2019, 5, 1–11. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Notation | Structure/Name | Function |
|---|---|---|
| FF1 |  furan-2-carbaldehyde | Aldehyde |
| FF2 |  5-(hydroxymethyl)furfural | Aldehyde Alcohol |
| FF3 |  5-(hydroxymethyl)furoic acid | Carboxylic acid Alcohol |
| ELUMO | EHOMO | ∆E | Energy | I | A | η | χ | ω | ε | ∆N | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HF/6-31G++(2d,p) | |||||||||||
| FF1 | 1.095 | −9.673 | 10.768 | −9284.422 | 9.673 | −1.095 | 5.384 | 4.289 | 1.708 | 0.585 | 0.018 |
| FF2 | 1.060 | −9.322 | 10.382 | −12,382.22 | 9.322 | −1.060 | 5.191 | 4.131 | 1.643 | 0.608 | 0.034 |
| FF3 | 1.031 | −8.680 | 9.711 | −23,703.52 | 8.680 | −1.031 | 4.855 | 3.824 | 1.506 | 0.664 | 0.067 |
| MP2/6-31G++(2d,p) | |||||||||||
| FF1 | 0.976 | −9.396 | 10.372 | −9289.265 | 9.396 | −0.976 | 5.186 | 4.210 | 1.709 | 0.585 | 0.026 |
| FF2 | 0.950 | −8.932 | 9.882 | −12,388.66 | 8.932 | −0.950 | 4.941 | 3.991 | 1.612 | 0.620 | 0.049 |
| FF3 | 0.911 | −8.513 | 9.423 | −23,714.94 | 8.513 | −0.910 | 4.712 | 3.801 | 1.533 | 0.652 | 0.072 |
| B3LYP/6-31G++(2d,p) | |||||||||||
| FF1 | −1.751 | −6.911 | 5.160 | −9289.608 | 6.911 | 1.751 | 2.580 | 4.331 | 3.635 | 0.275 | 0.029 |
| FF2 | −1.649 | −6.775 | 5.126 | −12,389.13 | 6.775 | 1.649 | 2.563 | 4.212 | 3.461 | 0.289 | 0.052 |
| FF3 | −1.533 | −6.638 | 5.104 | −23,715.59 | 6.638 | 1.533 | 2.552 | 4.085 | 3.270 | 0.306 | 0.077 |
| ELUMO | EHOMO | ∆E | Energy | I | A | η | χ | ω | ε | ∆N | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HF/6-31G++(2d,p) | |||||||||||
| FF1 | 0.983 | −8.952 | 9.935 | −9343.37 | 8.952 | −0.983 | 4.967 | 3.984 | 1.598 | 0.626 | 0.050 |
| FF2 | 1.006 | −8.857 | 9.863 | −12,460 | 8.857 | −1.006 | 4.932 | 3.925 | 1.562 | 0.640 | 0.056 |
| FF3 | 1.031 | −8.680 | 9.711 | −23,861 | 8.680 | −1.031 | 4.855 | 3.824 | 1.506 | 0.664 | 0.067 |
| MP2/6-31G++(2d,p) | |||||||||||
| FF1 | 0.868 | −8.829 | 9.697 | −9343.87 | 8.829 | −0.868 | 4,.848 | 3.980 | 1.634 | 0.612 | 0.051 |
| FF2 | 0.886 | −8.680 | 9.567 | −12,460.8 | 8.681 | −0.886 | 4.783 | 3.897 | 1.588 | 0.630 | 0.061 |
| FF3 | 0.910 | −8.513 | 9.423 | −23,861.9 | 8.513 | −0.910 | 4.712 | 3.801 | 1.533 | 0.652 | 0.072 |
| B3LYP/6-31G++(2d,p) | |||||||||||
| FF1 | −1.704 | −6.698 | 4.994 | −9343.87 | 6.698 | 1.704 | 2.497 | 4.201 | 3.534 | 0.283 | 0.056 |
| FF2 | −1.852 | −6.479 | 4.627 | −12,460.8 | 6.479 | 1.852 | 2.314 | 4.165 | 3.749 | 0.267 | 0.068 |
| FF3 | −1.992 | −6.210 | 4.217 | −23,861.9 | 6.210 | 1.992 | 2.109 | 4.101 | 3.988 | 0.251 | 0.090 |
| Atom | C1 | C2 | C3 | C4 | O5 | C8 | O9 | C10 | O13 | O15 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| FF1 | aq | 0.001 | −0.201 | 0.014 | 0.259 | −0.170 | −0.343 | −0.296 | |||
| gas | −0.003 | −0.184 | −0.014 | 0.257 | −0.139 | −0.331 | −0.212 | ||||
| FF2 | aq | −0.020 | −0.020 | −0.440 | 0.434 | −0.177 | −0.182 | −0.280 | −0.098 | −0.517 | |
| gas | −0.043 | 0.011 | −0.418 | 0.383 | −0.153 | −0.151 | −0.200 | −0.128 | −0.467 | ||
| FF3 | aq | −0.125 | 0.042 | −0.402 | 0.149 | −0.212 | 0.597 | −0.357 | −0.075 | −0.534 | −0.551 |
| gas | −0.130 | 0.074 | −0.386 | 0.110 | −0.191 | 0.591 | −0.296 | −0.095 | −0.486 | −0.538 |
| Metal | (hkl) | Multiplicity | dhkl (Å) | % Total Facet Area |
|---|---|---|---|---|
| Copper | (111) | 8 | 2.1 | 78 |
| (200) | 6 | 1.8 | 22 | |
| Iron | (110) | 12 | 2.0 | 100 |
| (100) | 6 | 1.4 | 64 | |
| Aluminum | (111) | 8 | 2.3 | 25 |
| (200) | 6 | 2.0 | 22 | |
| Tin | (111) | 8 | 3.7 | 100 |
| (220) | 12 | 2.3 | 0 |
| Metal Surface | Cu(111) | Fe(110) | Al(111) | Sn(111) |
|---|---|---|---|---|
| FF1 | −30.689 | −57.091 | −28.414 | −22.571 |
| FF2 | −38.708 | −71.963 | −35.649 | −28.179 |
| FF3 | −41.679 | −77.539 | −38.503 | −30.138 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourzi, H.; Oukhrib, R.; El Ibrahimi, B.; Abou Oualid, H.; Abdellaoui, Y.; Balkard, B.; El Issami, S.; Hilali, M.; Bazzi, L.; Len, C. Furfural Analogs as Sustainable Corrosion Inhibitors—Predictive Efficiency Using DFT and Monte Carlo Simulations on the Cu(111), Fe(110), Al(111) and Sn(111) Surfaces in Acid Media. Sustainability 2020, 12, 3304. https://doi.org/10.3390/su12083304
Bourzi H, Oukhrib R, El Ibrahimi B, Abou Oualid H, Abdellaoui Y, Balkard B, El Issami S, Hilali M, Bazzi L, Len C. Furfural Analogs as Sustainable Corrosion Inhibitors—Predictive Efficiency Using DFT and Monte Carlo Simulations on the Cu(111), Fe(110), Al(111) and Sn(111) Surfaces in Acid Media. Sustainability. 2020; 12(8):3304. https://doi.org/10.3390/su12083304
Chicago/Turabian StyleBourzi, Hassan, Rachid Oukhrib, Brahim El Ibrahimi, Hicham Abou Oualid, Youness Abdellaoui, Bouchra Balkard, Souad El Issami, Mustapha Hilali, Lahcen Bazzi, and Christophe Len. 2020. "Furfural Analogs as Sustainable Corrosion Inhibitors—Predictive Efficiency Using DFT and Monte Carlo Simulations on the Cu(111), Fe(110), Al(111) and Sn(111) Surfaces in Acid Media" Sustainability 12, no. 8: 3304. https://doi.org/10.3390/su12083304
APA StyleBourzi, H., Oukhrib, R., El Ibrahimi, B., Abou Oualid, H., Abdellaoui, Y., Balkard, B., El Issami, S., Hilali, M., Bazzi, L., & Len, C. (2020). Furfural Analogs as Sustainable Corrosion Inhibitors—Predictive Efficiency Using DFT and Monte Carlo Simulations on the Cu(111), Fe(110), Al(111) and Sn(111) Surfaces in Acid Media. Sustainability, 12(8), 3304. https://doi.org/10.3390/su12083304