1. Introduction
Skin is the largest organ in the human body. It protects the organism against unfavorable external factors e.g., chemicals, environment pollutants, allergens, microorganisms, and it plays a crucial role in maintaining general homeostasis [
1]. Therefore, it is also an important target of oxidative stress due to the activity of reactive oxygen species (ROS), which are constantly generated in the fibroblasts and keratinocytes in response to exogenous or endogenous prooxidant agents [
2]. Furthermore, ROS produced during normal skin metabolism, constitute a part of proper skin functionality and usually cause a little damage due to the activity of intracellular mechanisms, which reduce their harmful properties. It is worth noting that intensive or prolonged oxidative stress may overwhelm antioxidant defense mechanisms of the human skin and contribute to the skin disorders development, e.g.,: skin aging, dermatitis, allergic reactions or even skin neoplasms [
3]. Main constituents of skin are fibroblasts, which arise during embryogenesis from embryonic mesenchyme. A certain amount of spindle-shaped mesenchymal cells differentiate into supporting cells—fibroblasts. They are usually elongated, spindle-shaped, with several projections lying in one plane. They have one cell nucleus, round or oval with distinct nucleoli. Sometimes several nucleoli within the nucleus can be observed. Fibroblasts are the main cells that build up the dermis. Skin consists of three layers: epidermis, dermis and subcutaneous tissue. The epidermis forms a surface layer that stays in contact with the external environment and is built principally of keratinocytes that synthesize keratin—a component of the stratum corneum of the epidermis. The skin is composed of two layers: papillary and reticular [
4,
5].
Free radicals as atoms or compounds, which have at least one unpaired electron in the outer orbit, are mainly produced in the mitochondria during electron flow through the mitochondrial respiratory chain, and they increase their reactivity through oxidation-reduction mechanisms. About 2% of oxygen consumed by the respiratory chain undergoes simultaneous reduction causing the formation of ROS. As a result of these processes, superoxide anion radicals are formed, that initiate a whole cascade of reactive oxygen species, such as hydroxyl radical, hydrogen peroxide and singlet oxygen. ROS are generated in cells in different ways, but their main source are mitochondria, which consume about 90% of the oxygen needed by the cells. Mitochondria are also most often damaged by ROS, and with age they degenerate fairly quickly. Under normal conditions ROS are permanently removed by a complex antioxidant system, but in pathological conditions there is an imbalance between prooxidants and antioxidants. This imbalance can lead to oxidative stress and subsequently overproduction of ROS, which modify cellular components such as DNA, proteins and lipids [
6,
7]. Oxidative stress largely affects the skin, which occupies the largest area of the human body and is a protective coating for the internal organs. The skin structure allows it to perform physical and biochemical protection functions. Due to its functions and localization in the human body, skin is the most exposed for oxidative stress tissue. It is most susceptible to oxidative damage, which results from the presence of potential biological targets for free radical reactions in its components.
Air pollution, naturally occurring airborne gases such as ozone or high concentrations of oxygen, ionizing and non-ionizing radiation, bacteria, viruses and various exogenous toxins and chemicals can be considered as exogenous sources of factors that promote oxidative stress in the skin. Oxidative stress can also be caused by physical damage to the skin, such as burns or injuries [
8,
9]. On the other hand, endogenous sources of oxidative damage are generated by enzymes involved in the autoxidation of endo- and exogenous compounds, as well as by lipoxygenases and cyclooxygenases during eicosanoid metabolism. Nitric oxide synthase is an example of an enzyme that produces NO (nitric oxide) radicals directly in the skin. The source of ROS may also be neutrophils activated by a bacterial invasion in which a respiratory burst is observed.
One of the classes of compounds, which activity in the human organism is based on the generation of oxidative stress, mainly in cancer cells, are anticancer drugs. An example of such compound with proved prooxidant activity is Doxorubicin (DOX). DOX is an effective anticancer agent from anthracycline antibiotic group, which has a large spectrum of activity and it is being used alone or in combination to treat a variety of tumors, both haematological and solid, such as breast cancer [
10,
11]. It inhibits cell proliferation, induces oxidative stress, inhibits topoisomerase II, and finally leads to the cell death mainly through apoptosis [
12,
13,
14]. DOX tends to accumulate in mitochondria, causing changes in their structure and function. However, the main cause of DOX side effects observed in human is an extremely high level of oxidative stress within the cell, eventually causing apoptotic cell death [
15,
16]. A generation of free radicals caused by DOX can be described in two ways. The first one is an enzymatic mechanism consisting of the formation of semiquinone free radicals by the activity of NADPH-dependent reductases, which cause a one—electron reduction of DOX to DOX-semiquinone. Redox transformations of DOX-derived quinone/semiquinone provide superoxide radicals in the presence of oxygen [
17,
18]. The second mechanism has non-enzymatic background and it involves reaction with an iron (Fe
3+), which produces Fe
2+-doxorubicin free radical complex. This Fe-DOX complex is able to reduce oxygen to hydrogen peroxide and other free radicals. In our previous experiment regarding DOX influence on breast cancer cells we revealed that DOX-metal complexes are even more efficient than a parent drug [
10,
17].
Increasingly frequent implementation of various strategies to reduce undesirable DOX side effects was observed. Besides dose regulation, combined therapies with antioxidants, especially plant origin substances, are being used. Very promising results come from the combination of DOX with dietary antioxidants from the polyphenol group of compounds in order to lower oxidative stress level [
19]. One of the antioxidative agents with promising properties is cichoric acid. Cichoric acid (CA), a dicaffeyltartaric acid, was identified in a variety of edible plants and vegetables, such as: iceberg lettuce, basil,
Cichorium intybus L., dandelion,
Echinacea purpurea and
Orthosiphon stamineus. Many of above-mentioned plants are being used in folk medicine. CA exhibits many biological properties including anti-hyaluronidase activity, protection of collagen from possible free radical damage, antiviral activity, promoting phagocyte activity and free radical scavenger properties [
20,
21]. According to the literature, CA also inhibits HIV (human immunodeficiency virus) integrase, which is responsible for the HIV DNA copy integration with the DNA of the host cell [
22]. It should be also mentioned that CA has the ability to influence an immune response in chronically stressed mice, causing a significant decrease in stress level [
23,
24]. In addition recent research revealed that CA enhances glucose uptake in muscle cells and subsequently stimulates Langerhans islets to the secretion of the insulin [
25]. Despite so many beneficial activities of CA described in literature, no studies exist regarding its antioxidative properties against DOX-induced oxidative stress in normal human fibroblasts.
Several studies have shown that CA reveals its general antioxidative properties, which may counteract doxorubicin-induced cytotoxicity, but they were conducted on cancer cell lines and they have not clarified the mechanisms of its action. Therefore, with our study, we wanted to determine if CA acts as an antioxidant against doxorubicin—induced oxidative stress and if it acts via antioxidative and/or antiapoptotic pathway. We tested it in human normal fibroblasts cell line, because fibroblasts are very responsive to chemical signals and changes made to this signals can affect other cells. Fibroblasts constitute also main cell type of the skin, which is exposed for anticancer drugs side effects. Therefore strategies to attenuate the toxic effects of doxorubicin, such as combined therapy with dietary antioxidants (e.g., CA) are implemented.
2. Materials and Methods
2.1. Reagents
Dulbecco’s modified Eagle’s medium (DMEM), containing glucose at 4.5 mg/mL (25 mM), penicillin, streptomycin, trypsin–EDTA (ethylenediaminetetraacetic acid), FBS (Fetal Bovine Serum) and PBS (Phosphate-buffered saline) (without Ca and Mg) were provided by Gibco (San Diego, CA, USA), GSH/GSSG-Glo™ Assay kit, Caspase-Glo® 3/7 Assay kit, Caspase-Glo® 9 Assay kit were provided by Promega, Madison, WI, USA. MTT reagent was purchased from Sigma-Aldrich (St. Louis, MO, USA). Acridine orange and ethidium bromide were obtained from Sigma-Aldrich (St. Louis, MO, USA). Dichlorodihydrofluorescein diacetate assay (DCFH-DA) was provided by Sigma-Aldrich, St. Louis, MO, USA. SDS (Sodium dodecyl sulfate), TCA (Trichloroacetic acid), TBA (Thiobarbituric acid), Folin-Ciocalteu reagent were provided by Sigma-Aldrich and DTNB (Ellman’s Reagent) (5,5-dithio-bis-(2-nitrobenzoic acid)) by Serva. Cichoric acid, chlorogenic acid, caffeic acid, quercetin, DPPH (2,2-diphenyl-1-picrylhydrazyl), trolox, H2O2, horseradish peroxidase, phosphate buffer pH = 7 were purchased from Sigma-Aldrich Co. and used without purification. Methanol was purchased from Merck (Darmstadt, Germany).
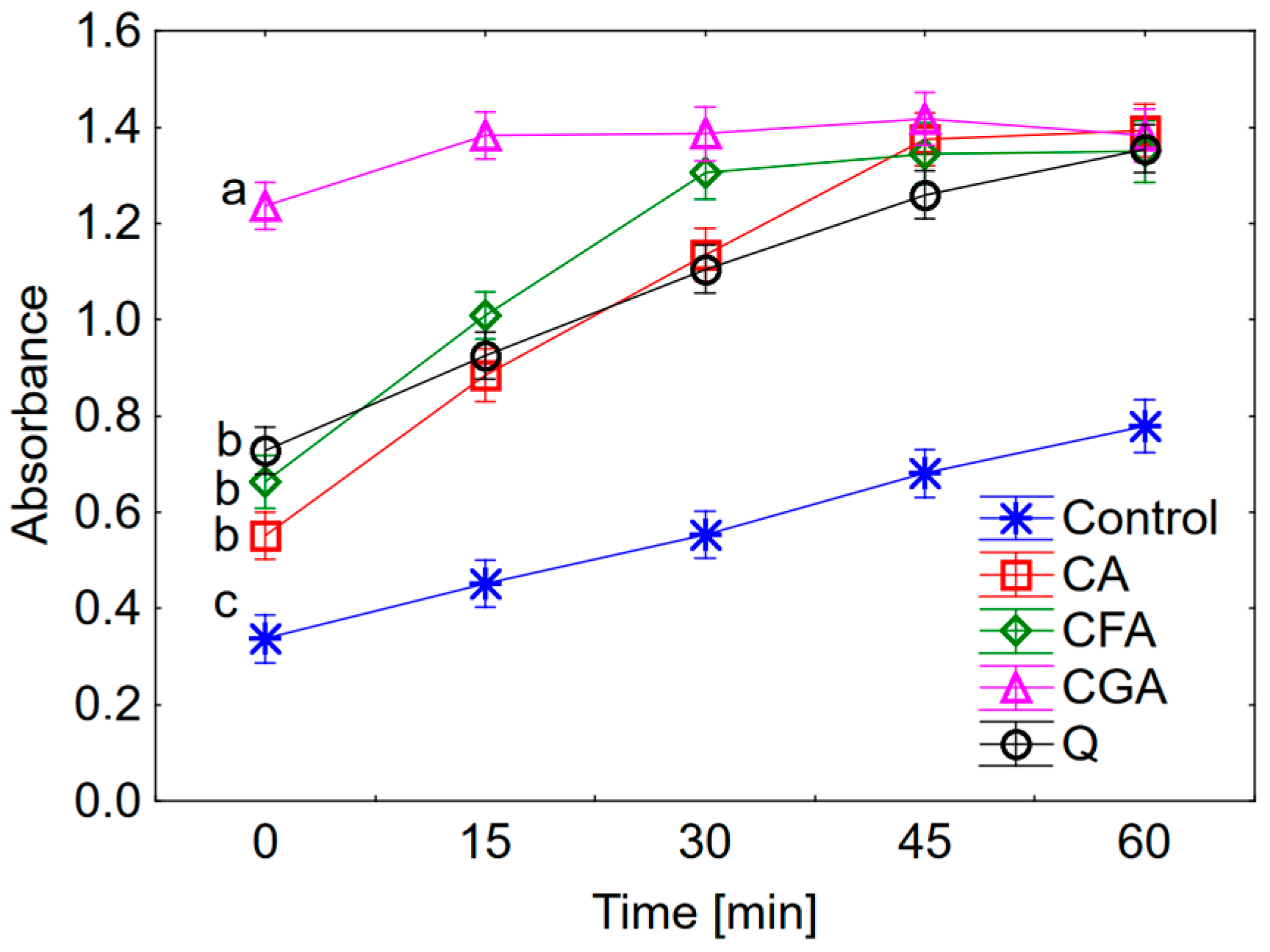
2.2. Anti-/Pro-Oxidant Activity
Antiradical activity was determined according to the DPPH assay described by [
26]. The methanolic solutions of cichoric, chlorogenic and caffeic acids, quercetin and DPPH were prepared just before analysis. 1 mL of tested compound and 2 mL of DPPH was added to each tube, vortexed and incubated in the darkness for 1 h at 23 °C. The final concentrations of phenolic compounds were 8, 5, 1 and 0.5 μM, and DPPH was 40 μM. The absorbance of solutions was measured at 516 nm against methanol as the blank using Agilent Carry 5000 spectrophotometer (Agilent, Santa Clara, CA, USA). The control sample consisted of 2 mL of DPPH solution and 1 mL of methanol. The antiradical activity of phenolic compounds was calculated as the percent of DPPH inhibition according to the equation:
where: %I—% inhibition of DPPH radical,
—absorbance of the control,
—absorbance of the sample.
The pro-oxidant activity of tested compounds was determined as the rate of oxidation of trolox according to the procedure described by Zeraik et al. [
27]. 100 μM trolox, 50 μM H
2O
2, 0.01 μM horseradish peroxidase in phosphate buffer (pH = 7) and 0.25 μM tested substances were incubated at 25 °C. The measurement was conducted at 272 nm using Agilent Carry 5000 spectrophotometer.
2.3. Cell Culture
The effect of CA, DOX and CA-DOX was examined in normal human skin fibroblast cell line (CRL1474), which was obtained from American Type Culture Collection (ATCC). Cells were maintained in DMEM (Gibco) supplemented with 10% FBS (Gibco), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37 °C in a humidified atmosphere of 5% CO2 in air. The viability of cells was estimated at CA concentrations of: 0.5 µM, 1 µM, 10 µM, 50 µM, 100 µM, 200 µM and 300 µM; DOX concentrations of: 0.09 µM, 0.18 µM, 0.38 µM, 0.75 µM, 1.5 µM, 3 µM and 6 µM; mix of CA and DOX in concentrations: 0.5 µM CA + 3 µM DOX, 1 µM CA + 3 µM DOX, 10 µM CA + 3 µM DOX, 50 µM CA + 3 µM DOX, 100 µM CA + 3 µM DOX, 200 µM CA + 3 µM DOX and 300 µM CA + 3 µM DOX. Caspases 3/7 activity, caspase 9 activity, SH (thiol) group content, TBARS (Thiobarbituric acid reactive substances) content, GSH/GSSG (Glutathione/Oxidized Glutathione) ratio, ROS content and apoptosis were examined at CA concentrations of 300 µM, DOX concentration of 3 µM and the mixture of these two compounds (CA + DOX, concentrations: 300 µM + 3 µM, respectively). Fibroblasts were seeded in 96-well plates at a density 2 × 104 cells/well. The cells were cultured for 24 h and 48 h and were treated with DOX in a concentration of 3 μM, CA in a concentration of 300 μM and with a mixture of DOX and CA (3 μM + 300 μM, respectively). Cells were seeded in: (a) transparent plates for cytotoxicity tests; (b) white plates for determination of caspase 3/7 and 9 activity, determination of GSH/GSSG ratio; (c) black plates for intracellular ROS detection.
2.4. Chemical Treatment of Cells
CA was stored in a refrigerator at temperature 4 °C. CA was dissolved in TrisHCl buffer. The compound was added to the cultured cells for a final concentration in the range of 0.5 µM to 300 µM. DOX was stored in a freezer at temperature −20 °C. The compound was added to the culture cells for a final concentration in the range of 0.09 µM to 6 µM. CA and DOX were both added to the cultured cells in selected concentrations. The control cells were incubated without the tested compounds.
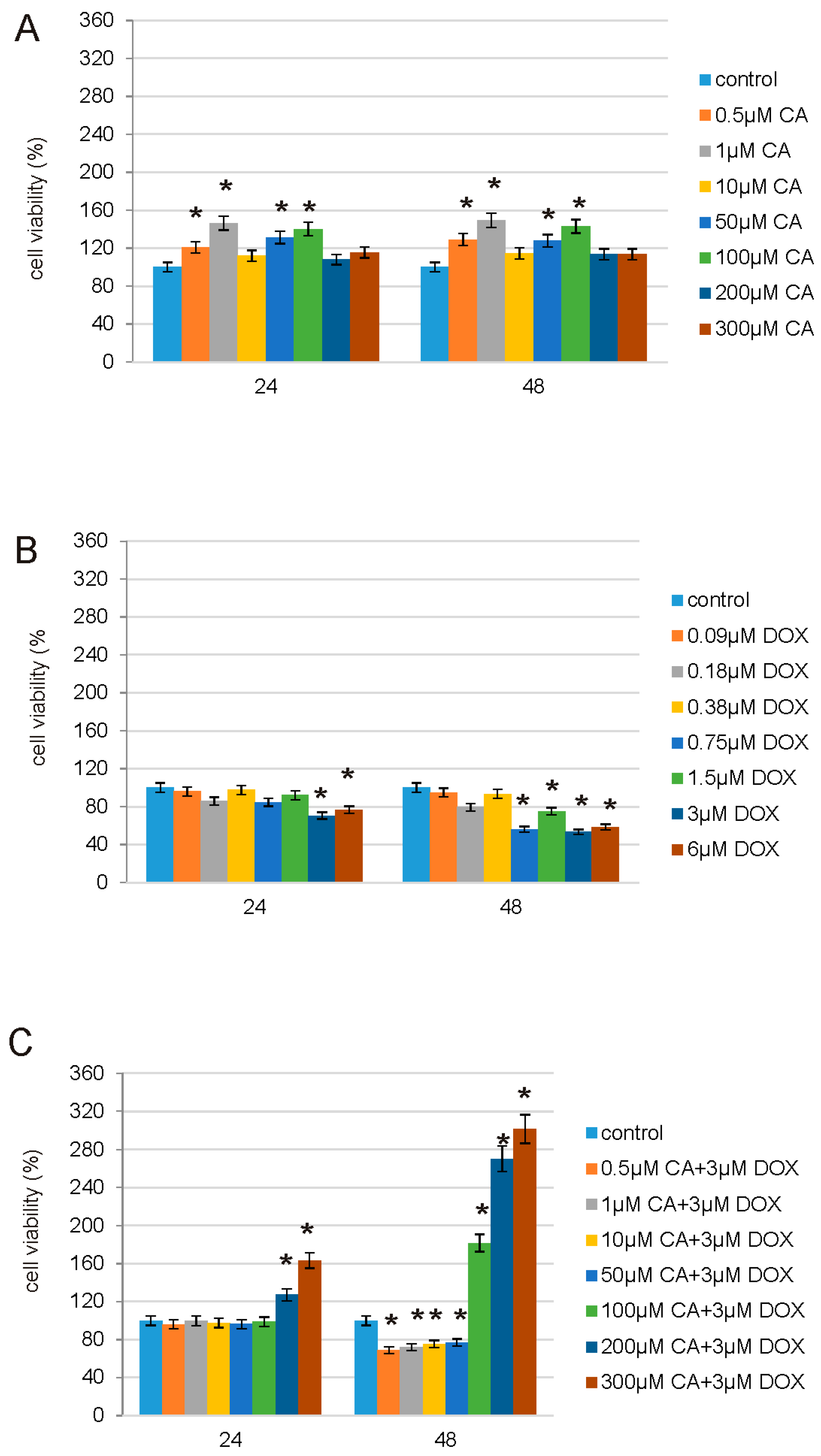
2.5. CA, DOX and CA-DOX Cytotoxicity
CA, DOX and CA-DOX cytotoxicity were measured according to the method of Carmichael using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) [
28]. The cells cultured for 24 h and 48 h were treated with: 1st—CA in the concentration range from 0.5 µM to 300 µM; 2nd—DOX in the concentration range from 0.09 µM to 6 µM; 3rd—CA combined with DOX: CA in the concentration range from 0.5 µM to 300 µM combined with DOX in the 3 µM concentration. After 24 h and 48 h cells were washed 3 times with PBS and subsequently incubated with 10 µL of MTT solution (5 mg/mL in PBS) for 2 h at 37 °C in 5% CO
2 in an incubator. Subsequently, 100 µL of DMSO was added and cells were incubated in the dark for the next 2 h. The absorbance was measured at 570 nm in a microplate reader GloMax
®-Multi Microplate Multimode Reader (Promega Corporation, Madison, WI, USA). The viability of fibroblast cells was calculated as a percentage of control cells, incubated without tested compounds. All the experiments were done in triplicates.
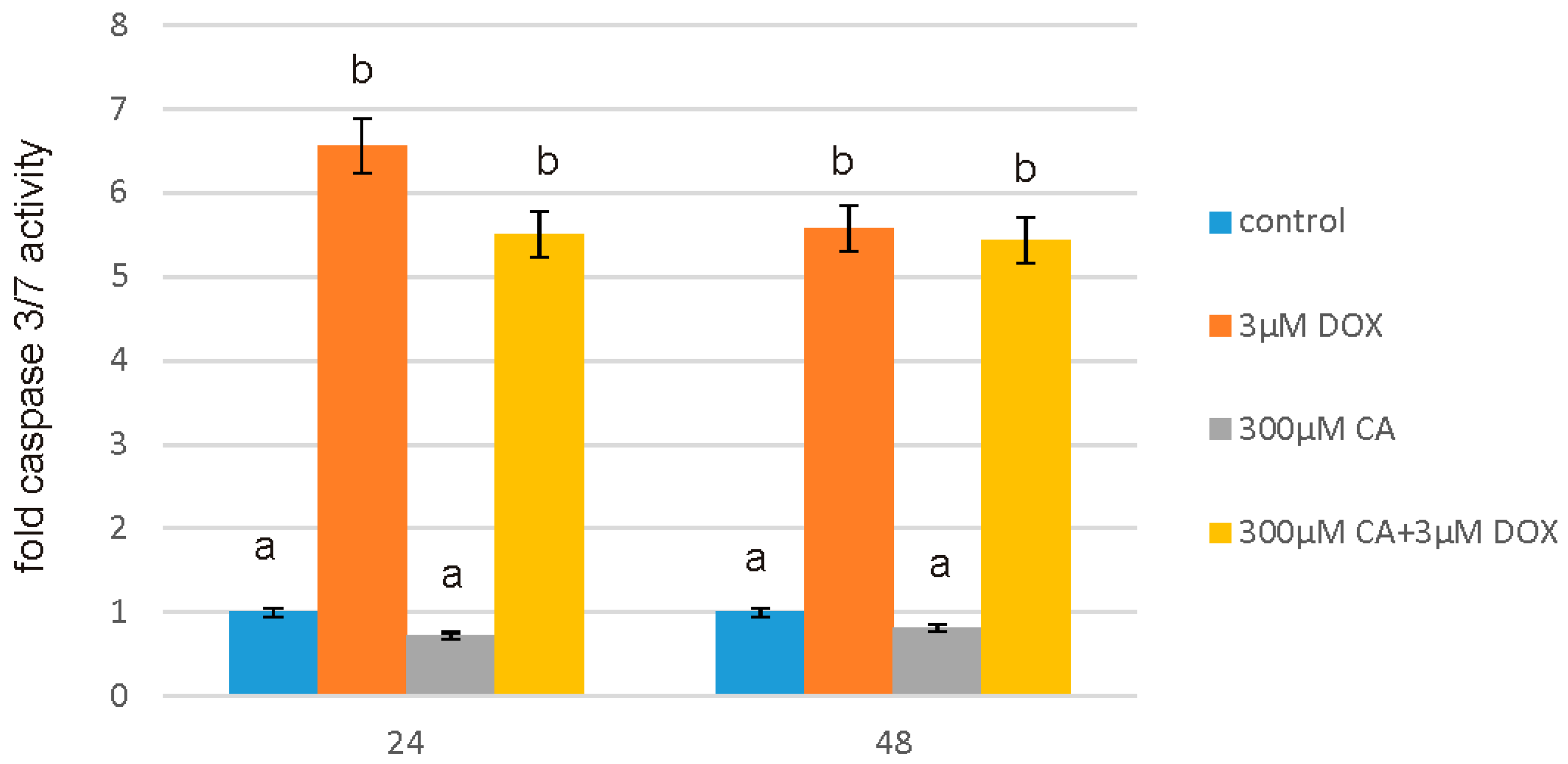
2.6. Determination of Caspase 3/7 Activity
After intended time of incubation medium was removed and Assay reagents were added to the wells. The activity of caspases 3/7 was measured using luminescent assay based on the substrate which contains the tetrapeptide sequence DEVD in a reagent optimized for caspase activity, luciferase activity and cell lysis. The cell lysis was followed by the caspase cleavage of the substrate and generation of the luminescent signal. The luminescence is proportional to the amount of caspase activity present in the sample. Assay is based on the thermostable luciferase activity, which generates stable, “glow-type” luminescent signal. The luminescence was measured in a microplate reader GloMax®-Multi Microplate Multimode Reader. All the experiments were done in triplicates.
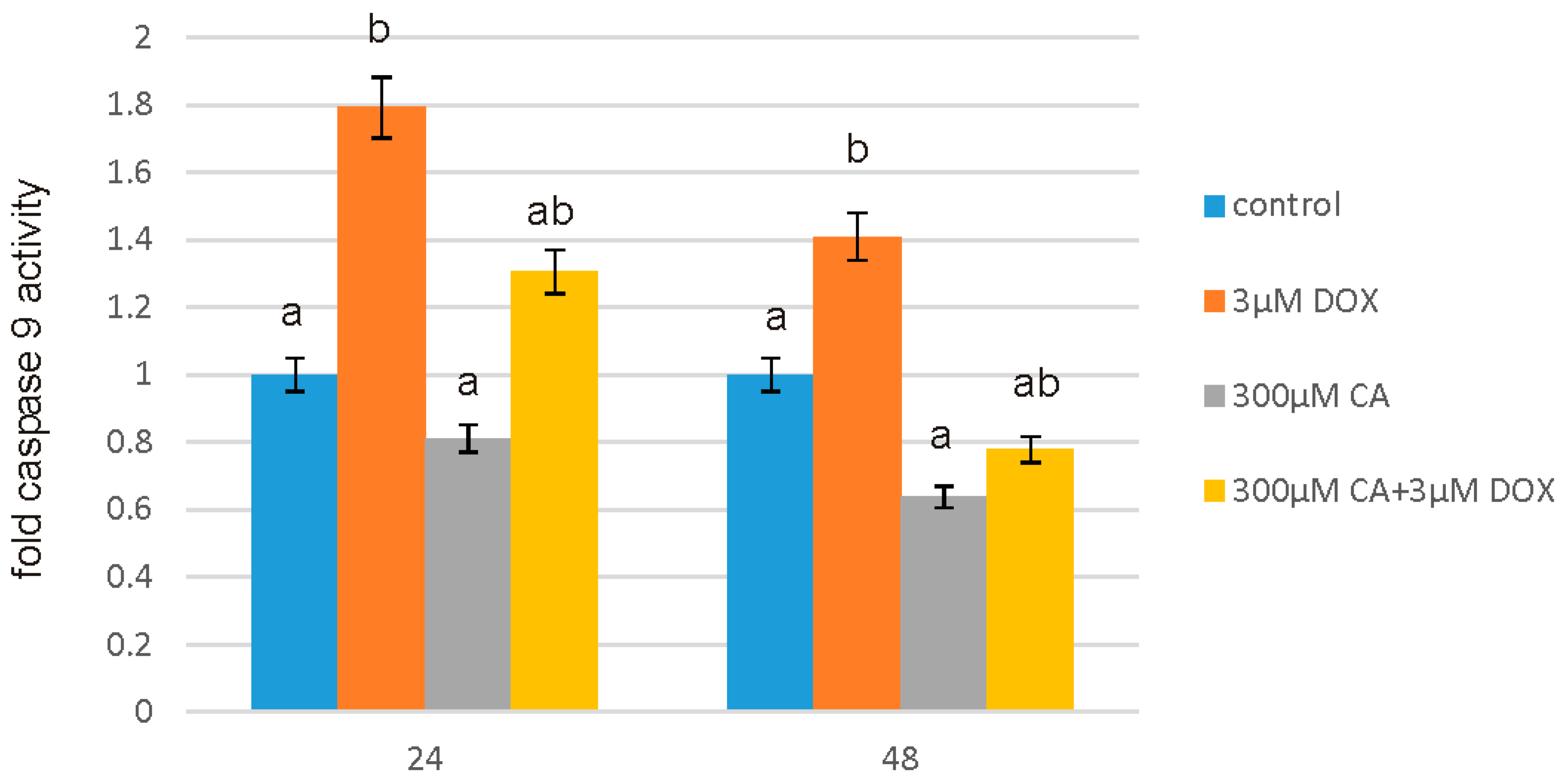
2.7. Determination of Caspase 9 Activity
After intended time of incubation medium was removed and Assay reagents were added to the wells. The activity of caspase 9 was measured by using luminescent assay based on a luminogenic caspase-9 substrate in a buffer system provided for caspase activity, luciferase activity and cell lysis. The addition of a special Caspase-Glo® 9 Reagent results in cell lysis, followed by caspase cleavage of the substrate, and generation of a “glow-type” luminescent signal. The signal generated is proportional to the amount of caspase activity present in the sample. The assay is based on thermostable luciferase, which generates the stable “glow-type” luminescent signal. The luminescence was measured in a microplate reader GloMax®-Multi Microplate Multimode Reader (Promega Corporation, Madison, WI, USA). All the experiments were done in triplicates.
2.8. Total Protein Content in Cells
Fibroblasts (1 × 10
5 cells/mL) were incubated in 2 mL of culture medium with tested compounds in tissue culture 6-well plates. The determination of protein concentration was performed spectrophotometrically as per Lowry et al. (1951) as described previously [
29]. All the experiments were done in triplicates.
2.9. Determination of SH Groups
SH-groups were measured using the method of Rice-Evans (1991) as described previously [
29]. Fibroblasts (1 × 10
5 cells/mL) were incubated in 2 mL of culture medium with tested compounds in tissue culture 6-well plates. All the experiments were done in triplicates.
2.10. Determination of TBA Reactive Species (TBARS) Levels
The method of Rice-Evans (1991) was applied in order to measure the level of membrane lipid-peroxidation products (TBARS), as described previously [
29]. Fibroblasts (1 × 10
5 cells/mL) were incubated in 2 mL of culture medium with test compounds in tissue culture 6-well plates. All the experiments were done in triplicates.
2.11. Determination of GSH/GSSG
Total glutathione and GSH/GSSG ratio were each assayed in triplicate via GSH/GSSG-Glo™ kit (Promega, Madison, WI, USA) following manufacturer’s instructions. Prior to the assay growth media were removed and cells washed with PBS. Assay is based on a luminescence measurement and detects and quantifies total glutathione (GSH + GSSG), GSSG and GSH/GSSG ratios in cultured cells. Stable luminescent signals are correlated with either the GSH or GSSG concentration of a sample. In this method GSH-dependent conversion of a GSH probe, Luciferin-NT, to luciferin by a glutathione S-transferase enzyme is coupled to a firefly luciferase reaction. Light from luciferase depends on the amount of luciferin formed, which in turn depends on the amount of GSH present. Thus, the luminescent signal is proportional to the amount of GSH. GSH/GSSG ratios are calculated directly from luminescence measurements.
2.12. Intracellular ROS Detection
The intracellular ROS level was measured according to Krętowski R. et al. [
30]. After diffusion through the cell membrane, DCFH-DA is deacetylated by cellular esterases to a non-fluorescent compound, which is later oxidized by intracellular ROS into a fluorescent 2′,7′–dichlorofluorescein (DCF). After 24 h, the medium was removed, the cells were stained with 10 μM of DCFH-DA in 200 µL PBS at 37 °C, 5% CO
2 incubator, for 45 min. Next, the dye was removed and replaced with DOX in a concentration of 3 µM, CA in a concentration of 300 µM and with a mixture of DOX + CA (3 µM + 300 µM, respectively) and incubated for 24 h and 48 h. Then, the DCF fluorescence intensity was measured using the GloMax
®-Multi Detection System (Promega Corporation, Madison, WI, USA) at the excitation wavelength of 485 nm and the emission wavelength of 535 nm. The intracellular ROS generation in fibroblasts cells was shown as the intensity of fluorescence of the DCF. All the experiments were done in triplicates.
2.13. Intracellular ROS Detection by Flow Cytometry
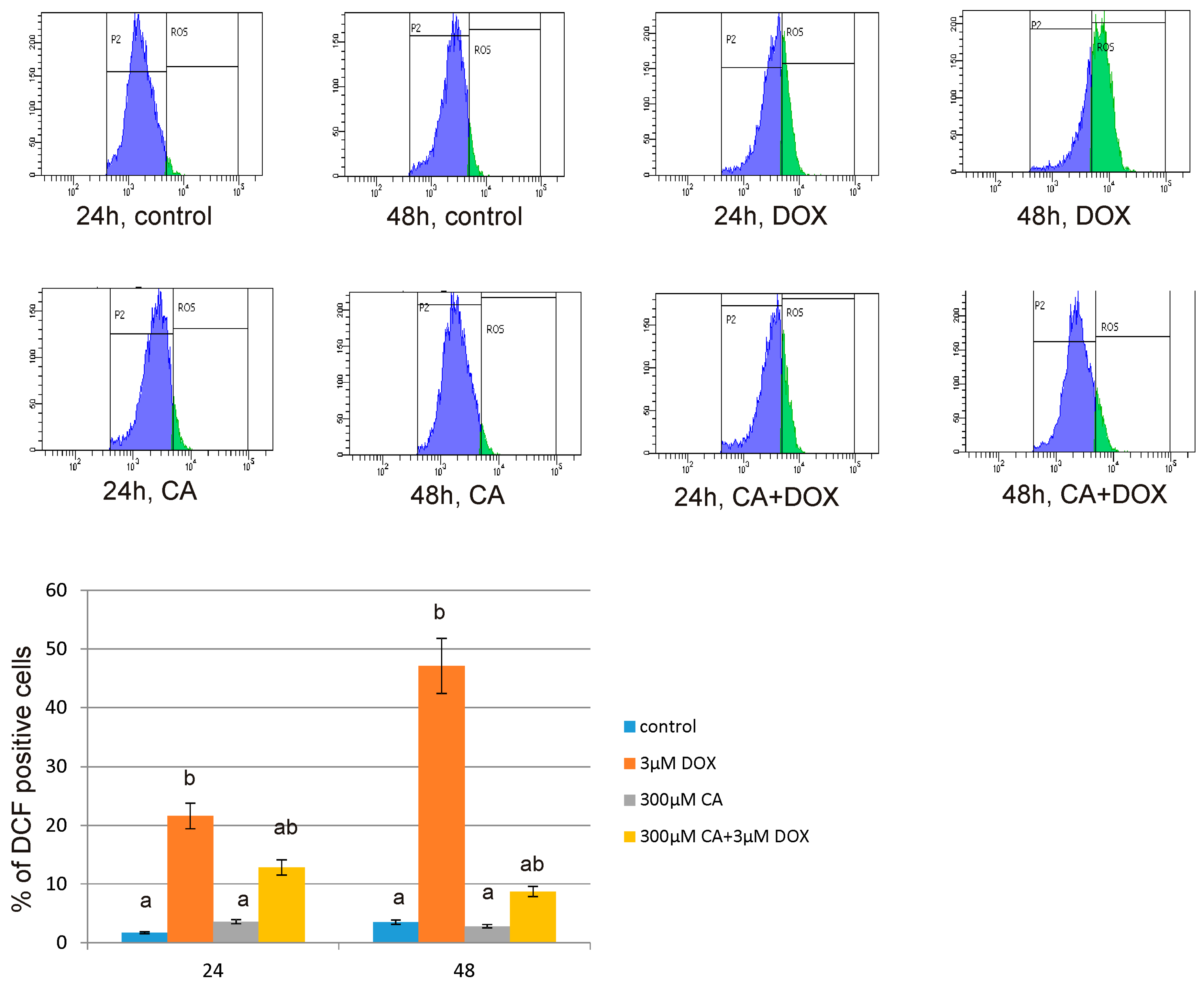
The level of intracellular reactive oxygen species (ROS) was determined using dichlorodihydrofluorescein diacetate (DCFH-DA), (Sigma, St. Louis, MO, USA). After diffusion through the cell membrane, DCFH-DA is deacetylated by cellular esterases to a non-fluorescent compound, which is later oxidized by intracellular ROS into a fluorescent 2′,7′–dichlorofluorescein (DCF). The cells (2.5 × 105) were seeded in 2 mL of growth medium in 6-well plates. After 24 h, the medium was removed, the cells were stained with 10 μM of DCFH-DA in 2 mL PBS at 37 °C, 5% CO2 incubator, for 30 min. Next, the dye was removed and replaced with DOX in a concentration of 3 µM, CA in a concentration of 300 µM and with a mixture of DOX and CA (3 µM + 300 µM, respectively) and incubated for 24 h and 48 h. Then, the cells were trypsinized, resuspended in DMEM and then in PBS. The DCF fluorescence intensity was measured according to flow cytometry method, by using FACSCanto II cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). Data were analysed using FACSDiva software (version 6, Becton Dickinson, Franklin Lakes, NJ, USA). The intracellular ROS generation in fibroblasts was depicted as the % of DCF positive cells.
2.14. Fluorescent Microscopy Assay
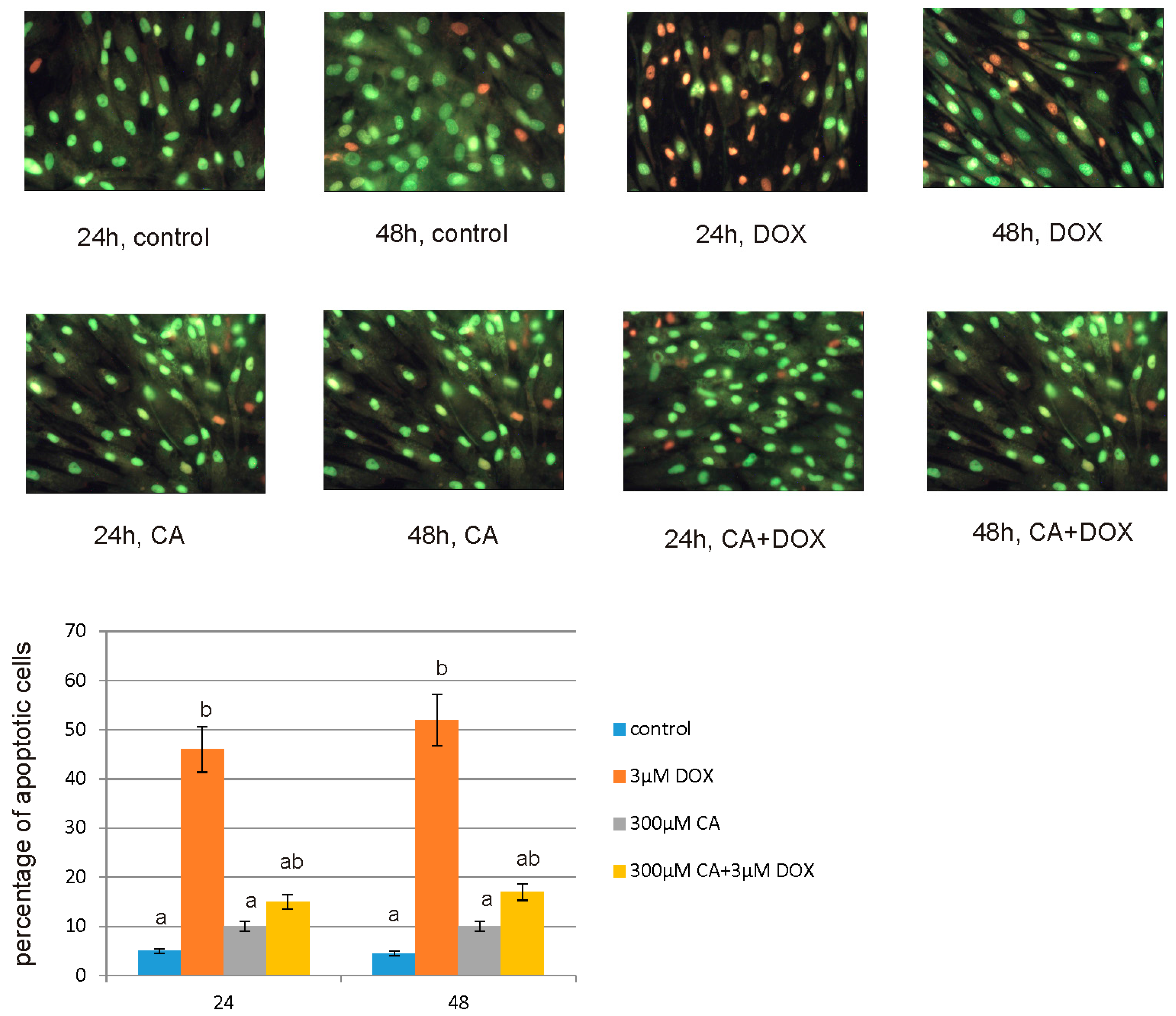
For apoptotic and necrotic cells nuclear morphology evaluation fluorescent dyes, such as ethidium bromide (10 µM) and acridine orange (10 µM) were used. The MCF-7 cells were seeded on cell imaging dishes with coverglass bottom with DOX in a concentration of 3 µM, CA in a concentration of 300 µM and with a mixture of DOX and CA (3 µM + 300 µM, respectively) and without tested compound as a control, for 24 h and 48 h. After incubation the cells were washed twice with PBS and then stained with dyes solution in the dark in room temperature, for 10 min. After incubation, staining mixture was removed and the cells were washed with PBS in order to analyze under fluorescent microscope (200× magnification). Ethidium bromide stains only cells that have lost their membrane integrity, while acridine orange stains live and dead cells. The cells were analyzed and photographed via using Leica DM IL fluorescent microscope (Leica Microsystems, Wetzlar, Germany). The following criteria were used: living cells—have regularly distributed green chromatin nucleus, early apoptotic cells characterized by bright green nucleus with condensed or often fragmented chromatin, late apoptotic cells—orange nuclei with chromatin condensation or fragmentation, while necrotic cells showed orange-stained cell nuclei. The cells were counted and percentage of apoptotic cells was the sum of early apoptotic and late apoptotic cells percent.
2.15. Statistical Analysis
For parametric data one-way analysis of variance (ANOVA) followed Tukey’s test was applied. Results from five independent experiments were expressed as mean ± standard deviation (SD) of mean for parametric data. Significance was considered when p ≤ 0.05. Statistica 13.0 was used.
4. Discussion
The selected natural phenolic compounds are common components of human diet and their antioxidant and as well as anti-inflammatory and anticancer properties are widely described, e.g., CA could be beneficial for preventing dermal injury in the setting of DOX treatment [
31,
32,
33,
34]. Cichoric acid is a compound with many potentially beneficial properties. It can be obtained from isolated and purified plant and vegetables and it has been described as a one daily nutraceutical which enhances antioxidant activity [
35]. The consumption of CA, its derivatives and CA containing plants may significantly influence redox balance in human organism and provide excellent health benefits. CA has been reported as potential anti-diabetic and anti-obesity compound [
36]. However, what is important in the context of nutrition in pathological conditions, its oral intake alleviates severe side effects of an excessive alcohol consumption, which was studied in case of acute alcohol-induced hepatic steatosis in mice. Mechanism of CA action in above described study resulted from oxidative stress inhibition and CA anti-inflammation properties [
37].
The anti- and pro-oxidant activity of cichoric acid was determined and compared with the activity of caffeic acid, chlorogenic acid and quercetin. Cichoric acid is the derivative of caffeic acid and tartaric acid. The presence of the two caffeic acid moieties in the cichoric acid molecule determines its high antioxidant properties. Therefore the caffeic acid was chosen in this comparative study. Chlorogenic acid is another chosen phenolic compound which is an ester of caffeic and quinic acids. It was selected in this study in order to estimate whether the number of caffeic acid moieties or the present of additional structure (e.g., quinic acid fragment) affect the anti-/pro-oxidant properties of molecules. Quercetin was chosen as one of the most intensively studied antioxidants. Obtained results indicate that according to the increasing antioxidant activity the studied compounds may be ordered as follows: cichoric acid > caffeic acid~quercetin > chlorogenic acid. The selected compounds were studied for their pro-oxidative effect on trolox oxidation. In this assay the radicals of polyphenolics are produced in their reaction with H
2O
2 catalysed by horseradish peroxidase. Then the phenoxyl radicals react with trolox and cause its oxidation to trolox radicals and trolox quinones. In the same time the phenoxyl radicals are transformed to phenolic compounds. The maximum absorption for trolox quinone is 272 nm. This is a general procedure for study the pro-oxidant activity of phenolic compounds [
27]. Obtained results showed that the pro-oxidant activity increases in the series: cichoric acid < quercetin < caffeic acid < chlorogenic acid. Cichoric acid was selected for this study because of its high antioxidant properties and relatively lower pro-oxidant activity compared to compounds which display structural similarities.
An increasing interest in the evaluation of clinical effectiveness of natural extracts, dietary supplements, plant origin antioxidants, especially from the polyphenol group of compounds is being observed. The understanding of the molecular mechanisms underlying dietary antioxidants action in diseases prevention and treatment is important to effectively complete these improvements with healthy lifestyle changes. The aim of this actions is mainly to improve human health, prevent diseases and eliminate some severe side effects of the commonly used chemotherapeutics, especially in the cancer treatment. An example of such chemotherapeutic, widely used in the treatment of a variety of cancers, both hematological malignancies and solid tumors is DOX. However, the clinical application of DOX is limited, which results from its high, cumulative dose-dependent toxicity, especially cardiotoxicity, leading to cardiomyopathy. An observed DOX-induced toxicity may be a result of DOX-induced mitochondrial dysfunction and accumulation of oxidative stress products, such as lipid peroxidation products and/or protein oxidation products [
17,
38].
In order to study possible protective properties of CA on DOX-treated normal human fibroblasts we decided to choose, based on prior experiments, a range of DOX [
39] and CA [
40] concentrations and estimate the most effective concentrations for both tested compounds. MTT assay revealed that in normal conditions, without any stress factors, CA is efficient in the stimulation of fibroblasts proliferation especially in 0.5 µM, 1 µM, 50 µM and 100 µM concentration. On the other hand, DOX inhibits fibroblasts viability most effectively at the concentration of 3 µM. Therefore, this DOX concentration was chosen for further analysis. The third part of MTT assay showed that pretreatment of fibroblasts with CA (0.5–300 µM) for 2 h before stimulate with DOX was the most effective at 300 µM of CA. Because of that reason, this combination of DOX with CA was applied for the evaluation of the anti-apoptotic and antioxidative effect. Literature data indicate the fact that CA has positive influence on normal cells, e.g., human dermal fibroblasts, where it induces Hepatocyte Growth Factor production on hepatocytes and where it may act as an anti-viral agent [
41,
42].
Reactive oxygen species (ROS) occurring due to too high level of oxidative stress are chemically reactive molecules that may damage main cellular macromolecules, such as DNA, lipids and proteins, subsequently causing genetic mutations. An excessive exposure to ROS may be seriously deleterious because of oxidative damage to important molecules in cell [
43]. To assess the effect of CA on proteins in this work we used one of the most widely studied markers of protein oxidation—protein thiol groups content. We observed the oxidative action of DOX, which conforms that DOX exhibits pro-oxidant properties against proteins and this action is connected with its cytotoxic activity. On the other hand stimulation with CA caused a significant increase in thiol group content, which means that tested compound act as an antioxidant. Especially after 48 h of treatment we can observe that prooxidative activity of DOX was effectively reversed by CA treatment. Study by Tsai KL et al. confirmed that pretreatment with CA may inhibit oxidative stress caused by external factors [
44].
Many natural compounds from the polyphenol group, which also include CA, are characterized by high antioxidant capacities and have been considered as a potential protectors against DOX-induced toxicity. This protective effect may be also connected with the reduction of lipid peroxidation process [
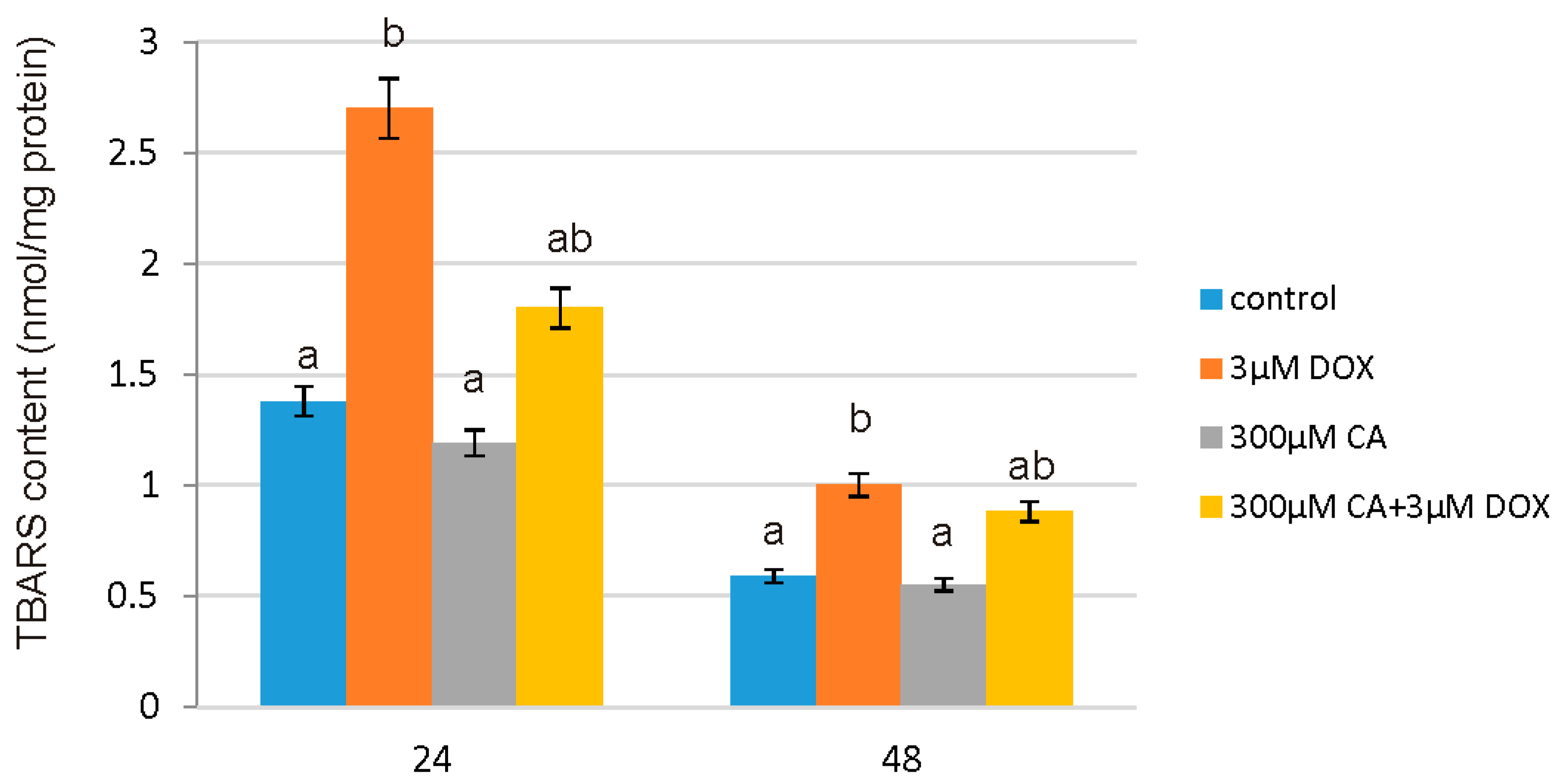
19]. In support of this report, we observed that CA does not cause oxidative alterations of membrane lipids and protects against DOX-induced TBARS production, which is probably related with antioxidant properties of CA. Already after 24 h we noticed significant increase in TBARS content in DOX-treated cells and a decrease in cells pretreated with 300 µM of CA. CA activity as a compound that prevents lipid peroxidation process was confirmed by several studies [
45,
46,
47]. Polyphenols exhibit antioxidative properties at several levels, among others they prevent the lipid peroxidation of biological membranes and inhibit the oxidation of lipoproteins and polyunsaturated fatty acids, which is in consistence with our results [
48].
Low ROS concentration within the cell is essential for keeping redox balance and for the stimulation of cell proliferation rate. However, an excessive level of accumulated in cell ROS may cause serious damage in protein, lipid and DNA structures through their oxidation and subsequently apoptosis and cell death [
49,
50]. According to the literature CA, which belongs to the polyphenols group of compounds, may provoke ROS generation in cancer cells and also in preadipocytes, but in normal cells CA was a potent ROS scavenger, reducing ROS accumulation under basal as well as oxidative stress conditions [
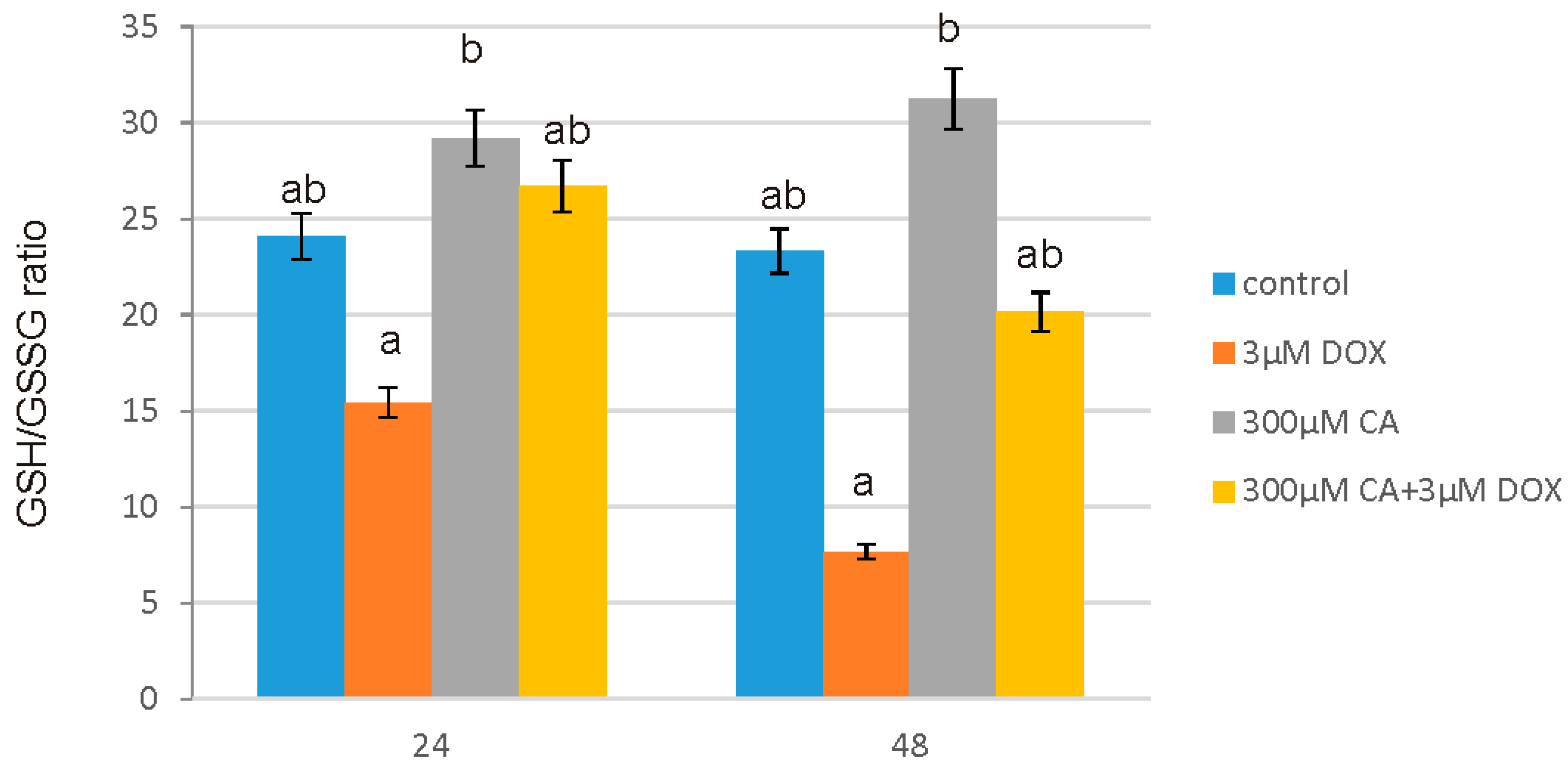
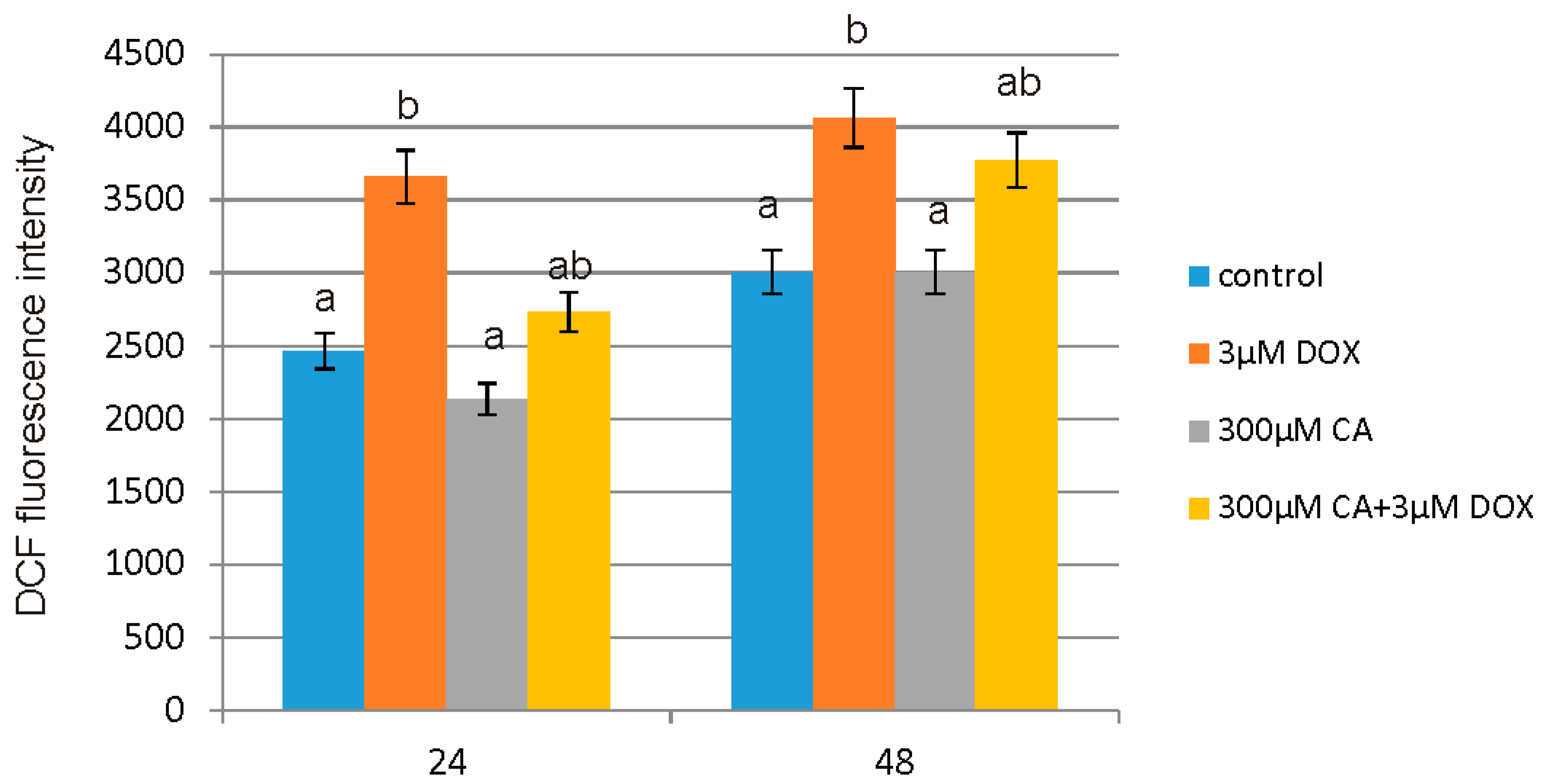
51]. It is in accordance with our results regarding ROS generation in fibroblast cells. We observed a significant increase in ROS production under the influence of DOX and a decrease in ROS content caused by CA. Pretreatment of fibroblast cells with CA (300 μM) for 2 h before stimulate with DOX significantly inhibited the formation of ROS. Flow cytometry results confirm that intervention with CA mitigated DOX-caused oxidative stress and reduced ROS formation in fibroblasts stimulated with DOX. Therefore we presume that the primary mechanism through which CA alleviates DOX-facilitated cell death of fibroblast cells is its antioxidant function. An observed, DOX-induced significant increase in ROS content was accompanied by a decrease in GSH/GSSG ratio, especially after 48 h of treatment. Similarly as in other tested oxidative stress parameters, GSH/GSSG ratio was significantly reduced after exposure to DOX and these results were effectively reversed by CA treatment. Our results are in accordance with literature data indicating positive influence of selected polyphenols on GSH content. One of the main phenolic compounds of curry spice turmeric is curcumin, the consumption of which brings beneficial effects. It was shown that curcumin plays a protective role against adriamycin-induced toxicity through three main mechanisms: first—it inhibits lipid peroxidation, second – it stabilizes cell membranes and third—it raises glutathione level [
52]. Therefore we presume that CA likewise influences GSH level in fibroblast cells. According to Fauser JK et al. a reduced level of the main GSH antioxidant may result in apoptosis and it also may be connected with a reduction of G0/G1 and arrest of the S and M cell cycle phases, together with increased ROS generation [
53]. Apoptosis is a programmed cell death that is extremely important for human organism homeostasis maintaining, for tissue development and malignancies treatment. Regulation of this process is main target for anticancer drugs, however chemotherapeutics, such as anthracycline antibiotics, e.g., DOX, may also induce apoptosis in normal, non-cancerous cells. At least two pathways have been described by which an apoptosis occurs: first is an extrinsic death-receptor dependent apoptosis and the second—intrinsic mitochondrial dependent apoptosis. In both pathways selected caspases activation occurs, which means activation of initiator caspases (e.g., caspase-8 and -9) and effector caspases (e.g., caspase-3, -6, and -7) [
54,
55]. A variety of evidence support also the role of reactive oxygen species in apoptotic cell death. It has been shown that oxygen free radicals are directly involved in the pathogenesis of apoptosis. Caspase 3/7 assay, caspase 9 assay and fluorescent microscopy assay showed that at 3 µM of DOX concentration, numerous apoptotic cells were seen, while pretreatment with 300 µM of CA reduced the number of positive fluorescent cells and decreased the activity of studied caspases significantly. We observed an increase in caspase 3, 7 and 9 activity under the influence of DOX as compared to control non-treated cells, which was accompanied by a decrease in GSH/GSSG ratio and a significant increase in ROS content. Apoptosis is particularly related to the cascade of caspases. Literature data indicated that the release of cytochrome c from the mitochondria into the cytosol, may result in the caspase-9 activation, which subsequently activates effector caspase, such as caspase-3, which causes DNA fragmentation and eventually—cell death [
56]. In our study, we found that CA significantly prevented DOX-induced activation of caspase 3 and 7, which is in accordance with literature data that confirms such CA activity. It may thus be hypothesized that this anti-apoptotic effect of CA might be attributed to observed increase in the levels of antioxidant glutathione (reduced GSH) and subsequently to a decrease in oxidative stress level. Tsai KL et al. revealed that CA precluded the activation of caspase 3 and subsequent DNA strand breaks, through the inhibition of the translocation of NF-κB from the cytosol to the nucleus, suppression of Bax and promotion the Bcl-2 expression [
44]. In our experiment pretreatment with CA was associated with the inhibition of the downstream apoptotic signaling pathways, finally preventing activation of caspase-3, caspase-7 and caspase -9 induced by DOX. It is suggested that CA is a natural food-derived compound that inhibits apoptosis [
57]. Our findings confirm this hypothesis, because under the influence of CA we showed a significant increase in cells proliferation and an decrease in caspase 3/7 and 9 level, which is accompanied by an increase in GSH content and a decrease in ROS content. Anti-apoptotic effect of CA in DOX-treated fibroblasts was confirmed by fluorescence microscopy assay, which indicated that tested compound was efficient both in time and dose-dependent manner.

 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










