Molecular Targets of Epigallocatechin—Gallate (EGCG): A Special Focus on Signal Transduction and Cancer



Abstract
:1. Introduction
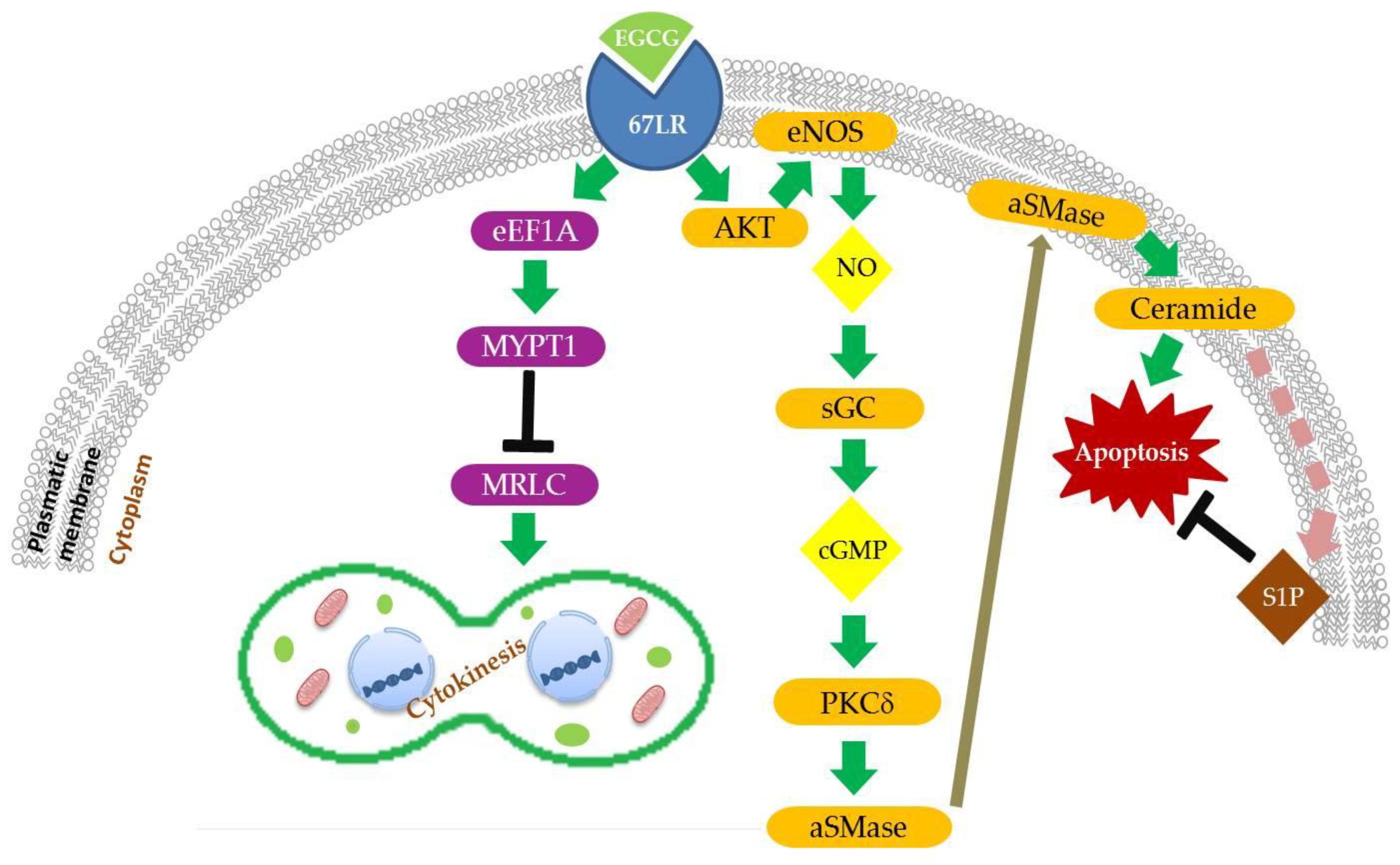
2. 67-kDa Laminin Receptor Signalling Pathways
2.1. Lipid Rafts-Mediated Apoptosis
2.1.1. EGCG/67LR/Akt/eNOS/NO/cGMP/PKCδ/aSMase Pathway
2.1.2. EGCG/67LR/Ceramide/SphK1/S1P Pathway
2.2. Cancer Cell Growth Inhibition
2.2.1. EGCG/67LR/eEF1a/MYPT1/MRLC Pathway
2.2.2. EGCG/67LR/cAMP/PKA/PP2A Pathway
2.3. Modulation of Cancer Stem Cells Properties
3. Other EGCG-Interacting Proteins
4. EGCG Epigenetic Regulation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Graham, H.N. Green tea composition, consumption, and polyphenol chemistry. Prev. Med. 1992, 21, 334–350. [Google Scholar] [CrossRef]
- Lowe, G.M.; Gana, K.; Rahman, K. Dietary supplementation with green tea extract promotes enhanced human leukocyte activity. J. Complement. Integr. Med. 2015, 12, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Boschmann, M.; Thielecke, F. The effects of epigallocatechin-3-gallate on thermogenesis and fat oxidation in obese men: A pilot study. J. Am. Coll. Nutr. 2007, 26, 389S–395S. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Sanchez, K.; Leyva, M.J.; Wu, M.; Betts, N.M.; Aston, C.E.; Lyons, T.J. Green tea supplementation affects body weight, lipids, and lipid peroxidation in obese subjects with metabolic syndrome. J. Am. Coll. Nutr. 2010, 29, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Ide, K.; Yamada, H.; Takuma, N.; Park, M.; Wakamiya, N.; Nakase, J.; Ukawa, Y.; Sagesaka, Y.M. Green tea consumption affects cognitive dysfunction in the elderly: A pilot study. Nutrients 2014, 6, 4032–4042. [Google Scholar] [CrossRef] [PubMed]
- Bettuzzi, S.; Brausi, M.; Rizzi, F.; Castagnetti, G.; Peracchia, G.; Corti, A. Chemoprevention of human prostate cancer by oral administration of green tea catechins in volunteers with high-grade prostate intraepithelial neoplasia: A preliminary report from a one-year proof-of-principle study. Cancer Res. 2006, 66, 1234–1240. [Google Scholar] [CrossRef]
- Khan, N.; Afaq, F.; Saleem, M.; Ahmad, N.; Mukhtar, H. Targeting multiple signaling pathways by green tea polyphenol (−)-epigallocatechin-3-gallate. Cancer Res. 2006, 66, 2500–2505. [Google Scholar] [CrossRef]
- Yang, C.S.; Sang, S.; Lambert, J.D.; Lee, M.J. Bioavailability issues in studying the health effects of plant polyphenolic compounds. Mol. Nutr. Food Res. 2008, 52 (Suppl. 1), S139–S151. [Google Scholar] [CrossRef]
- Lee, M.J.; Wang, Z.Y.; Li, H.; Chen, L.; Sun, Y.; Gobbo, S.; Balentine, D.A.; Yang, C.S. Analysis of plasma and urinary tea polyphenols in human subjects. Cancer Epidemiol. Biomarkers Prev. 1995, 4, 393–399. [Google Scholar]
- Gan, R.Y.; Li, H.B.; Sui, Z.Q.; Corke, H. Absorption, metabolism, anti-cancer effect and molecular targets of epigallocatechin gallate (egcg): An updated review. Crit. Rev. Food Sci. Nutr. 2018, 58, 924–941. [Google Scholar] [CrossRef]
- Zubair, H.; Azim, S.; Ahmad, A.; Khan, M.A.; Patel, G.K.; Singh, S.; Singh, A.P. Cancer chemoprevention by phytochemicals: Nature’s healing touch. Molecules 2017, 22, 395. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Yang, Y.; Liu, K.; Zhao, J.; Chen, X.; Liu, H.; Bai, R.; Li, X.; Jiang, Y.; Zhang, X.; et al. Combination curcumin and (−)-epigallocatechin-3-gallate inhibits colorectal carcinoma microenvironment-induced angiogenesis by jak/stat3/il-8 pathway. Oncogenesis 2017, 6, e384. [Google Scholar] [PubMed]
- Chan, M.M.; Chen, R.; Fong, D. Targeting cancer stem cells with dietary phytochemical—Repositioned drug combinations. Cancer Lett. 2018, 433, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, D.; Zhu, K. Sox2ot variant 7 contributes to the synergistic interaction between egcg and doxorubicin to kill osteosarcoma via autophagy and stemness inhibition. J. Exp. Clin. Cancer Res. 2018, 37, 37. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.; Wagner, A.; Neureiter, D.; Pichler, M.; Jakab, M.; Illig, R.; Berr, F.; Kiesslich, T. The green tea catechin epigallocatechin gallate induces cell cycle arrest and shows potential synergism with cisplatin in biliary tract cancer cells. BMC Complement. Altern. Med. 2015, 15, 194. [Google Scholar] [CrossRef]
- Zhou, Y.; Tang, J.; Du, Y.; Ding, J.; Liu, J.Y. The green tea polyphenol egcg potentiates the antiproliferative activity of sunitinib in human cancer cells. Tumour. Biol. 2016, 37, 8555–8566. [Google Scholar] [CrossRef]
- Yuan, X.; He, Y.; Zhou, G.; Li, X.; Feng, A.; Zheng, W. Target challenging-cancer drug delivery to gastric cancer tissues with a fucose graft epigallocatechin-3-gallate-gold particles nanocomposite approach. J. Photochem. Photobiol. B 2018, 183, 147–153. [Google Scholar] [CrossRef]
- Hajipour, H.; Hamishehkar, H.; Nazari Soltan Ahmad, S.; Barghi, S.; Maroufi, N.F.; Taheri, R.A. Improved anticancer effects of epigallocatechin gallate using rgd-containing nanostructured lipid carriers. Artif. Cells Nanomed. Biotechnol. 2018. [Google Scholar] [CrossRef]
- Sanna, V.; Singh, C.K.; Jashari, R.; Adhami, V.M.; Chamcheu, J.C.; Rady, I.; Sechi, M.; Mukhtar, H.; Siddiqui, I.A. Targeted nanoparticles encapsulating (−)-epigallocatechin-3-gallate for prostate cancer prevention and therapy. Sci. Rep. 2017, 7, 41573. [Google Scholar] [CrossRef] [Green Version]
- Krupkova, O.; Ferguson, S.J.; Wuertz-Kozak, K. Stability of (−)-epigallocatechin gallate and its activity in liquid formulations and delivery systems. J. Nutr. Biochem. 2016, 37, 1–12. [Google Scholar] [CrossRef]
- Rizzi, F.; Naponelli, V.; Silva, A.; Modernelli, A.; Ramazzina, I.; Bonacini, M.; Tardito, S.; Gatti, R.; Uggeri, J.; Bettuzzi, S. Polyphenon e(r), a standardized green tea extract, induces endoplasmic reticulum stress, leading to death of immortalized pnt1a cells by anoikis and tumorigenic pc3 by necroptosis. Carcinogenesis 2014, 35, 828–839. [Google Scholar] [CrossRef] [PubMed]
- Modernelli, A.; Naponelli, V.; Giovanna Troglio, M.; Bonacini, M.; Ramazzina, I.; Bettuzzi, S.; Rizzi, F. Egcg antagonizes bortezomib cytotoxicity in prostate cancer cells by an autophagic mechanism. Sci. Rep. 2015, 5, 15270. [Google Scholar] [CrossRef] [PubMed]
- Thawonsuwan, J.; Kiron, V.; Satoh, S.; Panigrahi, A.; Verlhac, V. Epigallocatechin-3-gallate (egcg) affects the antioxidant and immune defense of the rainbow trout, oncorhynchus mykiss. Fish. Physiol. Biochem. 2010, 36, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.D.; Elias, R.J. The antioxidant and pro-oxidant activities of green tea polyphenols: A role in cancer prevention. Arch. Biochem. Biophys. 2010, 501, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Hastak, K.; Afaq, F.; Ahmad, N.; Mukhtar, H. Essential role of caspases in epigallocatechin-3-gallate-mediated inhibition of nuclear factor kappa b and induction of apoptosis. Oncogene 2004, 23, 2507–2522. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Adachi, S.; Masuda, M.; Kozawa, O.; Moriwaki, H. Cancer chemoprevention with green tea catechins by targeting receptor tyrosine kinases. Mol. Nutr. Food Res. 2011, 55, 832–843. [Google Scholar] [CrossRef]
- Singh, B.N.; Shankar, S.; Srivastava, R.K. Green tea catechin, epigallocatechin-3-gallate (egcg): Mechanisms, perspectives and clinical applications. Biochem. Pharmacol. 2011, 82, 1807–1821. [Google Scholar] [CrossRef]
- Lin, C.H.; Shen, Y.A.; Hung, P.H.; Yu, Y.B.; Chen, Y.J. Epigallocathechin gallate, polyphenol present in green tea, inhibits stem-like characteristics and epithelial-mesenchymal transition in nasopharyngeal cancer cell lines. BMC Complement. Altern. Med. 2012, 12, 201. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Shim, J.Y.; Zhu, B.T. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol. Pharmacol. 2005, 68, 1018–1030. [Google Scholar] [CrossRef]
- Shirakami, Y.; Shimizu, M. Possible mechanisms of green tea and its constituents against cancer. Molecules 2018, 23, 2284. [Google Scholar] [CrossRef]
- Rahmani, A.H.; Al Shabrmi, F.M.; Allemailem, K.S.; Aly, S.M.; Khan, M.A. Implications of green tea and its constituents in the prevention of cancer via the modulation of cell signalling pathway. BioMed Res. Int. 2015, 2015, 925640. [Google Scholar] [CrossRef] [PubMed]
- Naponelli, V.; Ramazzina, I.; Lenzi, C.; Bettuzzi, S.; Rizzi, F. Green tea catechins for prostate cancer prevention: Present achievements and future challenges. Antioxidants 2017, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Ellinger, S.; Muller, N.; Stehle, P.; Ulrich-Merzenich, G. Consumption of green tea or green tea products: Is there an evidence for antioxidant effects from controlled interventional studies? Phytomedicine 2011, 18, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Ganapathy, S.; Hingorani, S.R.; Srivastava, R.K. Egcg inhibits growth, invasion, angiogenesis and metastasis of pancreatic cancer. Front. Biosci. 2008, 13, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Hussain, T.; Mukhtar, H. Molecular pathway for (−)-epigallocatechin-3-gallate-induced cell cycle arrest and apoptosis of human prostate carcinoma cells. Arch. Biochem. Biophys. 2003, 410, 177–185. [Google Scholar] [CrossRef]
- Shimizu, M.; Shirakami, Y.; Moriwaki, H. Targeting receptor tyrosine kinases for chemoprevention by green tea catechin, egcg. Int. J. Mol. Sci. 2008, 9, 1034–1049. [Google Scholar] [CrossRef]
- Shimizu, M.; Weinstein, I.B. Modulation of signal transduction by tea catechins and related phytochemicals. Mutat. Res. 2005, 591, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.; Shukla, S.; Gupta, S. Promoter demethylation and chromatin remodeling by green tea polyphenols leads to re-expression of gstp1 in human prostate cancer cells. Int. J. Cancer 2010, 126, 2520–2533. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Hussain, A.; Sundaram, M.K.; Alalami, U.; Gunasekera, D.; Ramesh, L.; Hamza, A.; Quraishi, U. (−)-epigallocatechin-3-gallate reverses the expression of various tumor-suppressor genes by inhibiting DNA methyltransferases and histone deacetylases in human cervical cancer cells. Oncol. Rep. 2015, 33, 1976–1984. [Google Scholar] [CrossRef] [PubMed]
- Rouzer, C.A.; Marnett, L.J. Green tea gets molecular. Cancer Prev. Res. 2011, 4, 1343–1345. [Google Scholar] [CrossRef]
- Saeki, K.; Hayakawa, S.; Nakano, S.; Ito, S.; Oishi, Y.; Suzuki, Y.; Isemura, M. In vitro and in silico studies of the molecular interactions of epigallocatechin-3-o-gallate (egcg) with proteins that explain the health benefits of green tea. Molecules 2018, 23, 1295. [Google Scholar] [CrossRef] [PubMed]
- Ermakova, S.; Choi, B.Y.; Choi, H.S.; Kang, B.S.; Bode, A.M.; Dong, Z. The intermediate filament protein vimentin is a new target for epigallocatechin gallate. J. Biol. Chem. 2005, 280, 16882–16890. [Google Scholar] [CrossRef]
- He, Z.; Tang, F.; Ermakova, S.; Li, M.; Zhao, Q.; Cho, Y.Y.; Ma, W.Y.; Choi, H.S.; Bode, A.M.; Yang, C.S.; et al. Fyn is a novel target of (−)-epigallocatechin gallate in the inhibition of jb6 cl41 cell transformation. Mol. Carcinog. 2008, 47, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.H.; Choi, H.S.; Pugliese, A.; Lee, S.Y.; Chae, J.I.; Choi, B.Y.; Bode, A.M.; Dong, Z. (−)-epigallocatechin gallate regulates cd3-mediated t cell receptor signaling in leukemia through the inhibition of zap-70 kinase. J. Biol. Chem. 2008, 283, 28370–28379. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; He, Z.; Ermakova, S.; Zheng, D.; Tang, F.; Cho, Y.Y.; Zhu, F.; Ma, W.Y.; Sham, Y.; Rogozin, E.A.; et al. Direct inhibition of insulin-like growth factor-i receptor kinase activity by (−)-epigallocatechin-3-gallate regulates cell transformation. Cancer Epidemiol. Biomarkers Prev. 2007, 16, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Ermakova, S.P.; Kang, B.S.; Choi, B.Y.; Choi, H.S.; Schuster, T.F.; Ma, W.Y.; Bode, A.M.; Dong, Z. (−)-epigallocatechin gallate overcomes resistance to etoposide-induced cell death by targeting the molecular chaperone glucose-regulated protein 78. Cancer Res. 2006, 66, 9260–9269. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, Y. Small molecule-sensing strategy and techniques for understanding the functionality of green tea. Biosci. Biotechnol. Biochem. 2015, 79, 687–699. [Google Scholar] [CrossRef] [Green Version]
- Nandakumar, V.; Vaid, M.; Katiyar, S.K. (−)-epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, cip1/p21 and p16ink4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis 2011, 32, 537–544. [Google Scholar] [CrossRef]
- Hu, Q.; Chang, X.; Yan, R.; Rong, C.; Yang, C.; Cheng, S.; Gu, X.; Yao, H.; Hou, X.; Mo, Y.; et al. (−)-epigallocatechin-3-gallate induces cancer cell apoptosis via acetylation of amyloid precursor protein. Med. Oncol. 2015, 32, 390. [Google Scholar] [CrossRef]
- Sazuka, M.; Imazawa, H.; Shoji, Y.; Mita, T.; Hara, Y.; Isemura, M. Inhibition of collagenases from mouse lung carcinoma cells by green tea catechins and black tea theaflavins. Biosci. Biotechnol. Biochem. 1997, 61, 1504–1506. [Google Scholar] [CrossRef]
- Fujimura, Y.; Tachibana, H.; Yamada, K. Lipid raft-associated catechin suppresses the fcepsilonri expression by inhibiting phosphorylation of the extracellular signal-regulated kinase1/2. FEBS Lett. 2004, 556, 204–210. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Huang, Y.; Kumazoe, M.; Lesnick, C.; Yamada, S.; Ueda, N.; Suzuki, T.; Yamashita, S.; Kim, Y.H.; Fujimura, Y.; et al. Sphingosine kinase-1 protects multiple myeloma from apoptosis driven by cancer-specific inhibition of rtks. Mol. Cancer Ther. 2015, 14, 2303–2312. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.P.; Kuo, S.C.; Huang, W.W.; Yang, J.S.; Lai, K.C.; Chen, H.J.; Lin, K.L.; Chiu, Y.J.; Huang, L.J.; Chung, J.G. (−)-epigallocatechin gallate induced apoptosis in human adrenal cancer nci-h295 cells through caspase-dependent and caspase-independent pathway. Anticancer Res. 2009, 29, 1435–1442. [Google Scholar] [PubMed]
- Zhu, J.; Jiang, Y.; Yang, X.; Wang, S.; Xie, C.; Li, X.; Li, Y.; Chen, Y.; Wang, X.; Meng, Y.; et al. Wnt/beta-catenin pathway mediates (−)-epigallocatechin-3-gallate (egcg) inhibition of lung cancer stem cells. Biochem. Biophys. Res. Commun. 2017, 482, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Umeda, D.; Tachibana, H.; Yamada, K. Epigallocatechin-3-o-gallate disrupts stress fibers and the contractile ring by reducing myosin regulatory light chain phosphorylation mediated through the target molecule 67 kda laminin receptor. Biochem. Biophys. Res. Commun. 2005, 333, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Suthakar, G.; Srivastava, R.K. Epigallocatechin-3-gallate inhibits cell cycle and induces apoptosis in pancreatic cancer. Front. Biosci. 2007, 12, 5039–5051. [Google Scholar] [CrossRef]
- Hwang, Y.S.; Park, K.K.; Chung, W.Y. Epigallocatechin-3 gallate inhibits cancer invasion by repressing functional invadopodia formation in oral squamous cell carcinoma. Eur. J. Pharmacol. 2013, 715, 286–295. [Google Scholar] [CrossRef]
- Olotu, F.A.; Agoni, C.; Adeniji, E.; Abdullahi, M.; Soliman, M.E. Probing gallate-mediated selectivity and high-affinity binding of epigallocatechin gallate: A way-forward in the design of selective inhibitors for anti-apoptotic bcl-2 proteins. Appl. Biochem. Biotechnol. 2018, 29, 1–20. [Google Scholar] [CrossRef]
- Shin, S.; Kim, M.K.; Jung, W.; Chong, Y. (−)-epigallocatechin gallate derivatives reduce the expression of both urokinase plasminogen activator and plasminogen activator inhibitor-1 to inhibit migration, adhesion, and invasion of mda-mb-231 cells. Phytother. Res. 2018, 32, 2086–2096. [Google Scholar] [CrossRef]
- Shi, J.; Liu, F.; Zhang, W.; Liu, X.; Lin, B.; Tang, X. Epigallocatechin-3-gallate inhibits nicotine-induced migration and invasion by the suppression of angiogenesis and epithelial-mesenchymal transition in non-small cell lung cancer cells. Oncol. Rep. 2015, 33, 2972–2980. [Google Scholar] [CrossRef]
- Umeda, D.; Yano, S.; Yamada, K.; Tachibana, H. Green tea polyphenol epigallocatechin-3-gallate signaling pathway through 67-kda laminin receptor. J. Biol. Chem. 2008, 283, 3050–3058. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.Y.; Lee, J.K.; Jeon, Y.K.; Kim, C.W. Exosome derived from epigallocatechin gallate treated breast cancer cells suppresses tumor growth by inhibiting tumor-associated macrophage infiltration and m2 polarization. BMC Cancer 2013, 13, 421. [Google Scholar] [CrossRef] [PubMed]
- Borutinskaite, V.; Virksaite, A.; Gudelyte, G.; Navakauskiene, R. Green tea polyphenol egcg causes anti-cancerous epigenetic modulations in acute promyelocytic leukemia cells. Leuk. Lymphoma 2018, 59, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Shi, M.; Li, G.; Wang, N.; Wei, J.; Wang, T.; Wang, Y. Regulation of id1 expression by epigallocatechin3gallate and its effect on the proliferation and apoptosis of poorly differentiated ags gastric cancer cells. Int. J. Oncol. 2013, 43, 1052–1058. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kwak, J.; Choi, H.K.; Choi, K.C.; Kim, S.; Lee, J.; Jun, W.; Park, H.J.; Yoon, H.G. Egcg suppresses prostate cancer cell growth modulating acetylation of androgen receptor by anti-histone acetyltransferase activity. Int. J. Mol. Med. 2012, 30, 69–74. [Google Scholar] [PubMed]
- Balasubramanian, S.; Adhikary, G.; Eckert, R.L. The bmi-1 polycomb protein antagonizes the (−)-epigallocatechin-3-gallate-dependent suppression of skin cancer cell survival. Carcinogenesis 2010, 31, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Watanabe, T.; Mondal, A.; Suzuki, K.; Kurusu-Kanno, M.; Li, Z.; Yamazaki, T.; Fujiki, H.; Suganuma, M. Mechanism-based inhibition of cancer metastasis with (−)-epigallocatechin gallate. Biochem. Biophys. Res. Commun. 2014, 443, 1–6. [Google Scholar] [CrossRef]
- Chen, H.; Landen, C.N.; Li, Y.; Alvarez, R.D.; Tollefsbol, T.O. Epigallocatechin gallate and sulforaphane combination treatment induce apoptosis in paclitaxel-resistant ovarian cancer cells through htert and bcl-2 down-regulation. Exp. Cell Res. 2013, 319, 697–706. [Google Scholar] [CrossRef]
- Basu, A.; Haldar, S. Combinatorial effect of epigallocatechin-3-gallate and trail on pancreatic cancer cell death. Int. J. Oncol. 2009, 34, 281–286. [Google Scholar] [CrossRef]
- Belguise, K.; Guo, S.; Sonenshein, G.E. Activation of foxo3a by the green tea polyphenol epigallocatechin-3-gallate induces estrogen receptor alpha expression reversing invasive phenotype of breast cancer cells. Cancer Res. 2007, 67, 5763–5770. [Google Scholar] [CrossRef]
- Harper, C.E.; Patel, B.B.; Wang, J.; Eltoum, I.A.; Lamartiniere, C.A. Epigallocatechin-3-gallate suppresses early stage, but not late stage prostate cancer in tramp mice: Mechanisms of action. Prostate 2007, 67, 1576–1589. [Google Scholar] [CrossRef]
- Deb, G.; Thakur, V.S.; Limaye, A.M.; Gupta, S. Epigenetic induction of tissue inhibitor of matrix metalloproteinase-3 by green tea polyphenols in breast cancer cells. Mol. Carcinog. 2015, 54, 485–499. [Google Scholar] [CrossRef]
- Moses, M.A.; Henry, E.C.; Ricke, W.A.; Gasiewicz, T.A. The heat shock protein 90 inhibitor, (−)-epigallocatechin gallate, has anticancer activity in a novel human prostate cancer progression model. Cancer Prev. Res. 2015, 8, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Kumazoe, M.; Sugihara, K.; Tsukamoto, S.; Huang, Y.; Tsurudome, Y.; Suzuki, T.; Suemasu, Y.; Ueda, N.; Yamashita, S.; Kim, Y.; et al. 67-kda laminin receptor increases cgmp to induce cancer-selective apoptosis. J. Clin. Investig. 2013, 123, 787–799. [Google Scholar] [CrossRef]
- Vahora, H.; Khan, M.A.; Alalami, U.; Hussain, A. The potential role of nitric oxide in halting cancer progression through chemoprevention. J. Cancer Prev. 2016, 21, 1–12. [Google Scholar] [CrossRef]
- Surh, Y.J.; Chun, K.S.; Cha, H.H.; Han, S.S.; Keum, Y.S.; Park, K.K.; Lee, S.S. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: Down-regulation of cox-2 and inos through suppression of nf-kappa b activation. Mutat. Res. 2001, 480–481, 243–268. [Google Scholar] [CrossRef]
- Hayakawa, S.; Saito, K.; Miyoshi, N.; Ohishi, T.; Oishi, Y.; Miyoshi, M.; Nakamura, Y. Anti-cancer effects of green tea by either anti- or pro- oxidative mechanisms. Asian Pac. J. Cancer Prev. 2016, 17, 1649–1654. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, S.; Porter, A.G. Nitric oxide-induced transcriptional up-regulation of protective genes by nrf2 via the antioxidant response element counteracts apoptosis of neuroblastoma cells. J. Biol. Chem. 2004, 279, 20096–20107. [Google Scholar] [CrossRef]
- Li, Y.J.; Wu, S.L.; Lu, S.M.; Chen, F.; Guo, Y.; Gan, S.M.; Shi, Y.L.; Liu, S.; Li, S.L. (−)-epigallocatechin-3-gallate inhibits nasopharyngeal cancer stem cell self-renewal and migration and reverses the epithelial-mesenchymal transition via nf-kappab p65 inactivation. Tumour. Biol. 2015, 36, 2747–2761. [Google Scholar] [CrossRef]
- He, L.; Zhang, E.; Shi, J.; Li, X.; Zhou, K.; Zhang, Q.; Le, A.D.; Tang, X. (−)-epigallocatechin-3-gallate inhibits human papillomavirus (hpv)-16 oncoprotein-induced angiogenesis in non-small cell lung cancer cells by targeting hif-1alpha. Cancer Chemother. Pharmacol. 2013, 71, 713–725. [Google Scholar] [CrossRef]
- Yamada, S.; Tsukamoto, S.; Huang, Y.; Makio, A.; Kumazoe, M.; Yamashita, S.; Tachibana, H. Epigallocatechin-3-O-gallate up-regulates microrna-let-7b expression by activating 67-kda laminin receptor signaling in melanoma cells. Sci. Rep. 2016, 6, 19225. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bian, S.; Yang, C.S. Green tea polyphenol egcg suppresses lung cancer cell growth through upregulating mir-210 expression caused by stabilizing hif-1alpha. Carcinogenesis 2011, 32, 1881–1889. [Google Scholar] [CrossRef] [PubMed]
- Urusova, D.V.; Shim, J.H.; Kim, D.J.; Jung, S.K.; Zykova, T.A.; Carper, A.; Bode, A.M.; Dong, Z. Epigallocatechin-gallate suppresses tumorigenesis by directly targeting pin1. Cancer Prev. Res. 2011, 4, 1366–1377. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Chen, H.G.; Yan, Q.; Deng, M.; Liu, J.; Doerge, S.; Ma, W.; Dong, Z.; Li, D.W. Protein phosphatase-2a is a target of epigallocatechin-3-gallate and modulates p53-bak apoptotic pathway. Cancer Res. 2008, 68, 4150–4162. [Google Scholar] [CrossRef] [PubMed]
- Toden, S.; Tran, H.M.; Tovar-Camargo, O.A.; Okugawa, Y.; Goel, A. Epigallocatechin-3-gallate targets cancer stem-like cells and enhances 5-fluorouracil chemosensitivity in colorectal cancer. Oncotarget 2016, 7, 16158–16171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukamoto, S.; Huang, Y.; Umeda, D.; Yamada, S.; Yamashita, S.; Kumazoe, M.; Kim, Y.; Murata, M.; Yamada, K.; Tachibana, H. 67-kda laminin receptor-dependent protein phosphatase 2a (pp2a) activation elicits melanoma-specific antitumor activity overcoming drug resistance. J. Biol. Chem. 2014, 289, 32671–32681. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, S.N.; Kala, R.; Tollefsbol, T.O. Molecular mechanisms for inhibition of colon cancer cells by combined epigenetic-modulating epigallocatechin gallate and sodium butyrate. Exp. Cell Res. 2014, 324, 40–53. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, S.X.; Ma, J.W.; Li, H.Y.; Ye, J.C.; Xie, S.M.; Du, B.; Zhong, X.Y. Egcg inhibits properties of glioma stem-like cells and synergizes with temozolomide through downregulation of p-glycoprotein inhibition. J. Neurooncol. 2015, 121, 41–52. [Google Scholar] [CrossRef]
- Suzuki, Y.; Isemura, M. Binding interaction between (−)-epigallocatechin gallate causes impaired spreading of cancer cells on fibrinogen. Biomed. Res. 2013, 34, 301–308. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, H.; Koga, K.; Fujimura, Y.; Yamada, K. A receptor for green tea polyphenol egcg. Nat. Struct. Mol. Biol. 2004, 11, 380–381. [Google Scholar] [CrossRef]
- Lee, S.H.; Nam, H.J.; Kang, H.J.; Kwon, H.W.; Lim, Y.C. Epigallocatechin-3-gallate attenuates head and neck cancer stem cell traits through suppression of notch pathway. Eur. J. Cancer 2013, 49, 3210–3218. [Google Scholar] [CrossRef] [PubMed]
- Adachi, S.; Nagao, T.; Ingolfsson, H.I.; Maxfield, F.R.; Andersen, O.S.; Kopelovich, L.; Weinstein, I.B. The inhibitory effect of (−)-epigallocatechin gallate on activation of the epidermal growth factor receptor is associated with altered lipid order in ht29 colon cancer cells. Cancer Res. 2007, 67, 6493–6501. [Google Scholar] [CrossRef] [PubMed]
- Duhon, D.; Bigelow, R.L.; Coleman, D.T.; Steffan, J.J.; Yu, C.; Langston, W.; Kevil, C.G.; Cardelli, J.A. The polyphenol epigallocatechin-3-gallate affects lipid rafts to block activation of the c-met receptor in prostate cancer cells. Mol. Carcinog. 2010, 49, 739–749. [Google Scholar] [CrossRef]
- Tabuchi, M.; Hayakawa, S.; Honda, E.; Ooshima, K.; Itoh, T.; Yoshida, K.; Park, A.M.; Higashino, H.; Isemura, M.; Munakata, H. Epigallocatechin-3-gallate suppresses transforming growth factor-beta signaling by interacting with the transforming growth factor-beta type ii receptor. World J. Exp. Med. 2013, 3, 100–107. [Google Scholar] [CrossRef]
- Wubetu, G.Y.; Shimada, M.; Morine, Y.; Ikemoto, T.; Ishikawa, D.; Iwahashi, S.; Yamada, S.; Saito, Y.; Arakawa, Y.; Imura, S. Epigallocatechin gallate hinders human hepatoma and colon cancer sphere formation. J. Gastroenterol. Hepatol. 2016, 31, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Punathil, T.; Tollefsbol, T.O.; Katiyar, S.K. Egcg inhibits mammary cancer cell migration through inhibition of nitric oxide synthase and guanylate cyclase. Biochem. Biophys. Res. Commun. 2008, 375, 162–167. [Google Scholar] [CrossRef]
- Sen, T.; Dutta, A.; Chatterjee, A. Epigallocatechin-3-gallate (egcg) downregulates gelatinase-b (mmp-9) by involvement of fak/erk/nfkappab and ap-1 in the human breast cancer cell line mda-mb-231. Anticancer Drugs 2010, 21, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; Katiyar, S.K. Green tea polyphenol, (−)-epigallocatechin-3-gallate, induces toxicity in human skin cancer cells by targeting beta-catenin signaling. Toxicol. Appl. Pharmacol. 2013, 273, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Marsh, L.; Srivastava, R.K. Egcg inhibits growth of human pancreatic tumors orthotopically implanted in balb c nude mice through modulation of fkhrl1/foxo3a and neuropilin. Mol. Cell. Biochem. 2013, 372, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Moradzadeh, M.; Hosseini, A.; Erfanian, S.; Rezaei, H. Epigallocatechin-3-gallate promotes apoptosis in human breast cancer t47d cells through down-regulation of pi3k/akt and telomerase. Pharmacol. Rep. 2017, 69, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Ketchart, W.; Smith, K.M.; Krupka, T.; Wittmann, B.M.; Hu, Y.; Rayman, P.A.; Doughman, Y.Q.; Albert, J.M.; Bai, X.; Finke, J.H.; et al. Inhibition of metastasis by hexim1 through effects on cell invasion and angiogenesis. Oncogene 2013, 32, 3829–3839. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.L.; Xu, J.; Yao, H.J.; Luo, K.L.; Li, J.M.; Wu, T.; Wu, G.Z. Inhibition of human 67-kda laminin receptor sensitizes multidrug resistance colon cancer cell line sw480 for apoptosis induction. Tumour. Biol. 2016, 37, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Montuori, N.; Selleri, C.; Risitano, A.M.; Raiola, A.M.; Ragno, P.; Del Vecchio, L.; Rotoli, B.; Rossi, G. Expression of the 67-kda laminin receptor in acute myeloid leukemia cells mediates adhesion to laminin and is frequently associated with monocytic differentiation. Clin. Cancer Res. 1999, 5, 1465–1472. [Google Scholar] [PubMed]
- Yu, H.N.; Zhang, L.C.; Yang, J.G.; Das, U.N.; Shen, S.R. Effect of laminin tyrosine-isoleucine-glycine-serine-arginine peptide on the growth of human prostate cancer (pc-3) cells in vitro. Eur. J. Pharmacol. 2009, 616, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Sun, L.; Zhang, H.; Li, Z.; Ning, X.; Shi, Y.; Guo, C.; Han, S.; Wu, K.; Fan, D. Hypoxia-mediated up-regulation of mgr1-ag/37lrp in gastric cancers occurs via hypoxia-inducible-factor 1-dependent mechanism and contributes to drug resistance. Int. J. Cancer 2009, 124, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Pesapane, A.; Ragno, P.; Selleri, C.; Montuori, N. Recent advances in the function of the 67 kda laminin receptor and its targeting for personalized therapy in cancer. Curr. Pharm. Des. 2017, 23, 4745–4757. [Google Scholar]
- Pesapane, A.; Di Giovanni, C.; Rossi, F.W.; Alfano, D.; Formisano, L.; Ragno, P.; Selleri, C.; Montuori, N.; Lavecchia, A. Discovery of new small molecules inhibiting 67 kda laminin receptor interaction with laminin and cancer cell invasion. Oncotarget 2015, 6, 18116–18133. [Google Scholar] [CrossRef]
- Li, Y.; Li, D.; Chen, J.; Wang, S. A polysaccharide from pinellia ternata inhibits cell proliferation and metastasis in human cholangiocarcinoma cells by targeting of cdc42 and 67kda laminin receptor (lr). Int. J. Biol. Macromol. 2016, 93, 520–525. [Google Scholar] [CrossRef]
- Fujimura, Y.; Sumida, M.; Sugihara, K.; Tsukamoto, S.; Yamada, K.; Tachibana, H. Green tea polyphenol egcg sensing motif on the 67-kda laminin receptor. PLoS ONE 2012, 7, e37942. [Google Scholar] [CrossRef]
- Fujimura, Y.; Yamada, K.; Tachibana, H. A lipid raft-associated 67kda laminin receptor mediates suppressive effect of epigallocatechin-3-o-gallate on fcepsilonri expression. Biochem. Biophys. Res. Commun. 2005, 336, 674–681. [Google Scholar] [CrossRef]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Auzins, A.; Sun, X.; Xu, Y.; Harnischfeger, F.; Lu, Y.; Li, Z.; Chen, Y.H.; Zheng, W.; Liu, W. The synaptic recruitment of lipid rafts is dependent on cd19-pi3k module and cytoskeleton remodeling molecules. J. Leukoc. Biol. 2015, 98, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Varshney, P.; Yadav, V.; Saini, N. Lipid rafts in immune signalling: Current progress and future perspective. Immunology 2016, 149, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell. Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.J. Growth factor receptors, lipid rafts and caveolae: An evolving story. Biochim. Biophys. Acta 2005, 1746, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Diluvio, G.; Del Gaudio, F.; Giuli, M.V.; Franciosa, G.; Giuliani, E.; Palermo, R.; Besharat, Z.M.; Pignataro, M.G.; Vacca, A.; d’Amati, G.; et al. Notch3 inactivation increases triple negative breast cancer sensitivity to gefitinib by promoting egfr tyrosine dephosphorylation and its intracellular arrest. Oncogenesis 2018, 7, 42. [Google Scholar] [CrossRef]
- Pike, L.J.; Han, X.; Gross, R.W. Epidermal growth factor receptors are localized to lipid rafts that contain a balance of inner and outer leaflet lipids: A shotgun lipidomics study. J. Biol. Chem. 2005, 280, 26796–26804. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Wakasaki, T.; Toh, S.; Shimizu, M.; Adachi, S. Chemoprevention of head and neck cancer by green tea extract: Egcg-the role of egfr signaling and “lipid raft”. J. Oncol. 2011, 2011, 540148. [Google Scholar] [CrossRef]
- Guo, T.; Xu, L.; Che, X.; Zhang, S.; Li, C.; Wang, J.; Gong, J.; Ma, R.; Fan, Y.; Hou, K.; et al. Formation of the igf1r/cav1/src tri-complex antagonizes trail-induced apoptosis in gastric cancer cells. Cell. Biol. Int. 2017, 41, 749–760. [Google Scholar] [CrossRef]
- Alawin, O.A.; Ahmed, R.A.; Ibrahim, B.A.; Briski, K.P.; Sylvester, P.W. Antiproliferative effects of gamma-tocotrienol are associated with lipid raft disruption in her2-positive human breast cancer cells. J. Nutr. Biochem. 2016, 27, 266–277. [Google Scholar] [CrossRef]
- Sur, S.; Pal, D.; Roy, R.; Barua, A.; Roy, A.; Saha, P.; Panda, C.K. Tea polyphenols egcg and tf restrict tongue and liver carcinogenesis simultaneously induced by n-nitrosodiethylamine in mice. Toxicol. Appl. Pharmacol. 2016, 300, 34–46. [Google Scholar] [CrossRef]
- Ma, Y.C.; Li, C.; Gao, F.; Xu, Y.; Jiang, Z.B.; Liu, J.X.; Jin, L.Y. Epigallocatechin gallate inhibits the growth of human lung cancer by directly targeting the egfr signaling pathway. Oncol. Rep. 2014, 31, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Filippi, A.; Picot, T.; Aanei, C.M.; Nagy, P.; Szollosi, J.; Campos, L.; Ganea, C.; Mocanu, M.M. Epigallocatechin-3-o-gallate alleviates the malignant phenotype in a-431 epidermoid and sk-br-3 breast cancer cell lines. Int. J. Food Sci. Nutr. 2018, 69, 584–597. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.J. Rafts defined: A report on the keystone symposium on lipid rafts and cell function. J. Lipid. Res. 2006, 47, 1597–1598. [Google Scholar] [CrossRef]
- Kim, J.Y.; Wang, L.; Lee, J.; Ou, J.J. Hepatitis c virus induces the localization of lipid rafts to autophagosomes for its rna replication. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Rosenberger, C.M.; Brumell, J.H.; Finlay, B.B. Microbial pathogenesis: Lipid rafts as pathogen portals. Curr. Biol. 2000, 10, R823–R825. [Google Scholar] [CrossRef]
- Guimaraes, A.J.; de Cerqueira, M.D.; Zamith-Miranda, D.; Lopez, P.H.; Rodrigues, M.L.; Pontes, B.; Viana, N.B.; DeLeon-Rodriguez, C.M.; Rossi, D.C.P.; Casadevall, A.; et al. Host membrane glycosphingolipids and lipid microdomains facilitate histoplasma capsulatum internalization by macrophages. Cell. Microbiol. 2018, e12976. [Google Scholar] [CrossRef] [PubMed]
- Smart, E.J.; Graf, G.A.; McNiven, M.A.; Sessa, W.C.; Engelman, J.A.; Scherer, P.E.; Okamoto, T.; Lisanti, M.P. Caveolins, liquid-ordered domains, and signal transduction. Mol. Cell. Biol. 1999, 19, 7289–7304. [Google Scholar] [CrossRef]
- Hwangbo, C.; Tae, N.; Lee, S.; Kim, O.; Park, O.K.; Kim, J.; Kwon, S.H.; Lee, J.H. Syntenin regulates tgf-beta1-induced smad activation and the epithelial-to-mesenchymal transition by inhibiting caveolin-mediated tgf-beta type i receptor internalization. Oncogene 2016, 35, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Laurenzana, A.; Fibbi, G.; Chilla, A.; Margheri, G.; Del Rosso, T.; Rovida, E.; Del Rosso, M.; Margheri, F. Lipid rafts: Integrated platforms for vascular organization offering therapeutic opportunities. Cell. Mol. Life Sci. 2015, 72, 1537–1557. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Lipid rafts as major platforms for signaling regulation in cancer. Adv. Biol. Regul. 2015, 57, 130–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, Y.T.; Podar, K.; Catley, L.; Tseng, Y.H.; Akiyama, M.; Shringarpure, R.; Burger, R.; Hideshima, T.; Chauhan, D.; Mitsiades, N.; et al. Insulin-like growth factor-1 induces adhesion and migration in human multiple myeloma cells via activation of beta1-integrin and phosphatidylinositol 3’-kinase/akt signaling. Cancer Res. 2003, 63, 5850–5858. [Google Scholar]
- Raghu, H.; Sodadasu, P.K.; Malla, R.R.; Gondi, C.S.; Estes, N.; Rao, J.S. Localization of upar and mmp-9 in lipid rafts is critical for migration, invasion and angiogenesis in human breast cancer cells. BMC Cancer 2010, 10, 647. [Google Scholar] [CrossRef] [PubMed]
- Lacour, S.; Hammann, A.; Grazide, S.; Lagadic-Gossmann, D.; Athias, A.; Sergent, O.; Laurent, G.; Gambert, P.; Solary, E.; Dimanche-Boitrel, M.T. Cisplatin-induced cd95 redistribution into membrane lipid rafts of ht29 human colon cancer cells. Cancer Res. 2004, 64, 3593–3598. [Google Scholar] [PubMed]
- George, K.S.; Wu, S. Lipid raft: A floating island of death or survival. Toxicol. Appl. Pharmacol. 2012, 259, 311–319. [Google Scholar] [Green Version]
- Alves, A.C.S.; Dias, R.A.; Kagami, L.P.; das Neves, G.M.; Torres, F.C.; Eifler-Lima, V.L.; Carvalho, I.; de Miranda Silva, C.; Kawano, D.F. Beyond the “lock and key” paradigm: Targeting lipid rafts to induce the selective apoptosis of cancer cells. Curr. Med. Chem. 2018, 25, 2082–2104. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Hirotsu, K.; Kumazoe, M.; Goto, Y.; Sugihara, K.; Suda, T.; Tsurudome, Y.; Suzuki, T.; Yamashita, S.; Kim, Y.; et al. Green tea polyphenol egcg induces lipid-raft clustering and apoptotic cell death by activating protein kinase cdelta and acid sphingomyelinase through a 67 kda laminin receptor in multiple myeloma cells. Biochem. J. 2012, 443, 525–534. [Google Scholar] [CrossRef]
- Mocanu, M.M.; Ganea, C.; Georgescu, L.; Varadi, T.; Shrestha, D.; Baran, I.; Katona, E.; Nagy, P.; Szollosi, J. Epigallocatechin 3-o-gallate induces 67 kda laminin receptor-mediated cell death accompanied by downregulation of erbb proteins and altered lipid raft clustering in mammary and epidermoid carcinoma cells. J. Nat. Prod. 2014, 77, 250–257. [Google Scholar] [CrossRef]
- Huang, Y.; Kumazoe, M.; Bae, J.; Yamada, S.; Takai, M.; Hidaka, S.; Yamashita, S.; Kim, Y.; Won, Y.; Murata, M.; et al. Green tea polyphenol epigallocatechin-o-gallate induces cell death by acid sphingomyelinase activation in chronic myeloid leukemia cells. Oncol. Rep. 2015, 34, 1162–1168. [Google Scholar] [CrossRef]
- Yang, C.S.; Wang, H. Cancer preventive activities of tea catechins. Molecules 2016, 21, 1679. [Google Scholar] [CrossRef]
- Luo, K.W.; Lung, W.Y.; Chun, X.; Luo, X.L.; Huang, W.R. Egcg inhibited bladder cancer t24 and 5637 cell proliferation and migration via pi3k/akt pathway. Oncotarget 2018, 9, 12261–12272. [Google Scholar] [CrossRef]
- Velavan, B.; Divya, T.; Sureshkumar, A.; Sudhandiran, G. Nano-chemotherapeutic efficacy of (−) -epigallocatechin 3-gallate mediating apoptosis in a549cells: Involvement of reactive oxygen species mediated nrf2/keap1signaling. Biochem. Biophys. Res. Commun. 2018, 503, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.J.; Qiao, K.S.; Sun, P.; Chen, P.; Li, Q. Study of egcg induced apoptosis in lung cancer cells by inhibiting pi3k/akt signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 4557–4563. [Google Scholar]
- Wang, Y.Q.; Lu, J.L.; Liang, Y.R.; Li, Q.S. Suppressive effects of egcg on cervical cancer. Molecules 2018, 23, 2334. [Google Scholar] [CrossRef] [PubMed]
- Britschgi, A.; Simon, H.U.; Tobler, A.; Fey, M.F.; Tschan, M.P. Epigallocatechin-3-gallate induces cell death in acute myeloid leukaemia cells and supports all-trans retinoic acid-induced neutrophil differentiation via death-associated protein kinase 2. Br. J. Haematol. 2010, 149, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Shammas, M.A.; Neri, P.; Koley, H.; Batchu, R.B.; Bertheau, R.C.; Munshi, V.; Prabhala, R.; Fulciniti, M.; Tai, Y.T.; Treon, S.P.; et al. Specific killing of multiple myeloma cells by (−)-epigallocatechin-3-gallate extracted from green tea: Biologic activity and therapeutic implications. Blood 2006, 108, 2804–2810. [Google Scholar] [CrossRef]
- Kirschnek, S.; Paris, F.; Weller, M.; Grassme, H.; Ferlinz, K.; Riehle, A.; Fuks, Z.; Kolesnick, R.; Gulbins, E. Cd95-mediated apoptosis in vivo involves acid sphingomyelinase. J. Biol. Chem. 2000, 275, 27316–27323. [Google Scholar] [PubMed]
- Garcia-Barros, M.; Paris, F.; Cordon-Cardo, C.; Lyden, D.; Rafii, S.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 2003, 300, 1155–1159. [Google Scholar] [CrossRef]
- Zhang, Y.; Mattjus, P.; Schmid, P.C.; Dong, Z.; Zhong, S.; Ma, W.Y.; Brown, R.E.; Bode, A.M.; Schmid, H.H. Involvement of the acid sphingomyelinase pathway in uva-induced apoptosis. J. Biol. Chem. 2001, 276, 11775–11782. [Google Scholar] [CrossRef]
- Goni, F.M.; Alonso, A. Sphingomyelinases: Enzymology and membrane activity. FEBS Lett. 2002, 531, 38–46. [Google Scholar] [CrossRef]
- Morita, Y.; Perez, G.I.; Paris, F.; Miranda, S.R.; Ehleiter, D.; Haimovitz-Friedman, A.; Fuks, Z.; Xie, Z.; Reed, J.C.; Schuchman, E.H.; et al. Oocyte apoptosis is suppressed by disruption of the acid sphingomyelinase gene or by sphingosine-1-phosphate therapy. Nat. Med. 2000, 6, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Gulbins, E. Regulation of death receptor signaling and apoptosis by ceramide. Pharmacol. Res. 2003, 47, 393–399. [Google Scholar] [CrossRef]
- Hueber, A.O.; Bernard, A.M.; Herincs, Z.; Couzinet, A.; He, H.T. An essential role for membrane rafts in the initiation of fas/cd95-triggered cell death in mouse thymocytes. EMBO Rep. 2002, 3, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Kischkel, F.C.; Hellbardt, S.; Behrmann, I.; Germer, M.; Pawlita, M.; Krammer, P.H.; Peter, M.E. Cytotoxicity-dependent apo-1 (fas/cd95)-associated proteins form a death-inducing signaling complex (disc) with the receptor. EMBO J. 1995, 14, 5579–5588. [Google Scholar] [CrossRef] [PubMed]
- Grassme, H.; Cremesti, A.; Kolesnick, R.; Gulbins, E. Ceramide-mediated clustering is required for cd95-disc formation. Oncogene 2003, 22, 5457–5470. [Google Scholar] [CrossRef] [PubMed]
- Gajate, C.; Mollinedo, F. The antitumor ether lipid et-18-och(3) induces apoptosis through translocation and capping of fas/cd95 into membrane rafts in human leukemic cells. Blood 2001, 98, 3860–3863. [Google Scholar] [CrossRef] [PubMed]
- London, E. Ceramide selectively displaces cholesterol from ordered lipid domains (rafts): Implications for lipid raft structure and function. J. Biol. Chem. 2004, 279, 9997–10004. [Google Scholar]
- Cremesti, A.; Paris, F.; Grassme, H.; Holler, N.; Tschopp, J.; Fuks, Z.; Gulbins, E.; Kolesnick, R. Ceramide enables fas to cap and kill. J. Biol. Chem. 2001, 276, 23954–23961. [Google Scholar] [CrossRef]
- Fanzo, J.C.; Lynch, M.P.; Phee, H.; Hyer, M.; Cremesti, A.; Grassme, H.; Norris, J.S.; Coggeshall, K.M.; Rueda, B.R.; Pernis, A.B.; et al. Cd95 rapidly clusters in cells of diverse origins. Cancer Biol. Ther. 2003, 2, 392–395. [Google Scholar] [CrossRef]
- Wu, L.Y.; De Luca, T.; Watanabe, T.; Morre, D.M.; Morre, D.J. Metabolite modulation of hela cell response to enox2 inhibitors egcg and phenoxodiol. Biochim. Biophys. Acta 2011, 1810, 784–789. [Google Scholar] [CrossRef]
- Kim, M.H.; Chung, J. Synergistic cell death by egcg and ibuprofen in du-145 prostate cancer cell line. Anticancer Res. 2007, 27, 3947–3956. [Google Scholar] [PubMed]
- Tan, X.; Zhang, Y.; Jiang, B.; Zhou, D. Changes in ceramide levels upon catechins-induced apoptosis in lovo cells. Life Sci. 2002, 70, 2023–2029. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, F.; Li, G.; Huang, J.; Liu, Y.; Zhang, Q.; Tang, Q.; Hu, C.; Zhang, R. Coptisine induces apoptosis in human hepatoma cells through activating 67-kda laminin receptor/cgmp signaling. Front. Pharmacol. 2018, 9, 517. [Google Scholar] [CrossRef] [PubMed]
- Kumazoe, M.; Kim, Y.; Bae, J.; Takai, M.; Murata, M.; Suemasu, Y.; Sugihara, K.; Yamashita, S.; Tsukamoto, S.; Huang, Y.; et al. Phosphodiesterase 5 inhibitor acts as a potent agent sensitizing acute myeloid leukemia cells to 67-kda laminin receptor-dependent apoptosis. FEBS Lett. 2013, 587, 3052–3057. [Google Scholar] [CrossRef] [PubMed]
- Kumazoe, M.; Tsukamoto, S.; Lesnick, C.; Kay, N.E.; Yamada, K.; Shanafelt, T.D.; Tachibana, H. Vardenafil, a clinically available phosphodiesterase inhibitor, potentiates the killing effect of egcg on cll cells. Br. J. Haematol. 2015, 168, 610–613. [Google Scholar] [CrossRef] [PubMed]
- Casaletto, J.B.; McClatchey, A.I. Spatial regulation of receptor tyrosine kinases in development and cancer. Nat. Rev. Cancer 2012, 12, 387–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brizuela, L.; Dayon, A.; Doumerc, N.; Ader, I.; Golzio, M.; Izard, J.C.; Hara, Y.; Malavaud, B.; Cuvillier, O. The sphingosine kinase-1 survival pathway is a molecular target for the tumor-suppressive tea and wine polyphenols in prostate cancer. FASEB J. 2010, 24, 3882–3894. [Google Scholar] [CrossRef] [PubMed]
- Olivera, A.; Spiegel, S. Sphingosine-1-phosphate as second messenger in cell proliferation induced by pdgf and fcs mitogens. Nature 1993, 365, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Cuvillier, O.; Pirianov, G.; Kleuser, B.; Vanek, P.G.; Coso, O.A.; Gutkind, S.; Spiegel, S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 1996, 381, 800–803. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, S.; Kumazoe, M.; Huang, Y.; Lesnick, C.; Kay, N.E.; Shanafelt, T.D.; Tachibana, H. Sphk1 inhibitor potentiates the anti-cancer effect of egcg on leukaemia cells. Br. J. Haematol. 2017, 178, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, S.; Ganguli, A.; Das, A.; Nag, D.; Chakrabarti, G. Epigallocatechin-3-gallate shows anti-proliferative activity in hela cells targeting tubulin-microtubule equilibrium. Chem. Biol. Interact. 2015, 242, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Shenouda, N.S.; Zhou, C.; Browning, J.D.; Ansell, P.J.; Sakla, M.S.; Lubahn, D.B.; Macdonald, R.S. Phytoestrogens in common herbs regulate prostate cancer cell growth in vitro. Nutr. Cancer 2004, 49, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Umeda, D.; Yano, S.; Yamada, K.; Tachibana, H. Involvement of 67-kda laminin receptor-mediated myosin phosphatase activation in antiproliferative effect of epigallocatechin-3-O-gallate at a physiological concentration on caco-2 colon cancer cells. Biochem. Biophys. Res. Commun. 2008, 371, 172–176. [Google Scholar] [CrossRef] [PubMed]
- D’Avino, P.P.; Giansanti, M.G.; Petronczki, M. Cytokinesis in animal cells. Cold Spring Harb. Perspect. Biol. 2015, 7, a015834. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.L. The mechanism of cortical ingression during early cytokinesis: Thinking beyond the contractile ring hypothesis. Trends Cell. Biol. 2005, 15, 581–588. [Google Scholar] [CrossRef]
- Matsumura, F. Regulation of myosin ii during cytokinesis in higher eukaryotes. Trends Cell. Biol. 2005, 15, 371–377. [Google Scholar] [CrossRef]
- Moussavi, R.S.; Kelley, C.A.; Adelstein, R.S. Phosphorylation of vertebrate nonmuscle and smooth muscle myosin heavy chains and light chains. Mol. Cell. Biochem. 1993, 127–128, 219–227. [Google Scholar] [CrossRef]
- Scholey, J.M.; Taylor, K.A.; Kendrick-Jones, J. Regulation of non-muscle myosin assembly by calmodulin-dependent light chain kinase. Nature 1980, 287, 233–235. [Google Scholar] [CrossRef]
- Ikebe, M.; Koretz, J.; Hartshorne, D.J. Effects of phosphorylation of light chain residues threonine 18 and serine 19 on the properties and conformation of smooth muscle myosin. J. Biol. Chem. 1988, 263, 6432–6437. [Google Scholar]
- Kawano, Y.; Fukata, Y.; Oshiro, N.; Amano, M.; Nakamura, T.; Ito, M.; Matsumura, F.; Inagaki, M.; Kaibuchi, K. Phosphorylation of myosin-binding subunit (mbs) of myosin phosphatase by rho-kinase in vivo. J. Cell. Biol. 1999, 147, 1023–1038. [Google Scholar] [CrossRef]
- Negrutskii, B.S.; El’skaya, A.V. Eukaryotic translation elongation factor 1 alpha: Structure, expression, functions, and possible role in aminoacyl-trna channeling. Prog. Nucleic Acid Res. Mol. Biol. 1998, 60, 47–78. [Google Scholar] [PubMed]
- Gangwani, L.; Mikrut, M.; Galcheva-Gargova, Z.; Davis, R.J. Interaction of zpr1 with translation elongation factor-1alpha in proliferating cells. J. Cell. Biol. 1998, 143, 1471–1484. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, A.; Caraglia, M.; Longo, O.; Marra, M.; Abbruzzese, A.; Arcari, P. The translation elongation factor 1a in tumorigenesis, signal transduction and apoptosis: Review article. Amino Acids 2004, 26, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Izawa, T.; Fukata, Y.; Kimura, T.; Iwamatsu, A.; Dohi, K.; Kaibuchi, K. Elongation factor-1 alpha is a novel substrate of rho-associated kinase. Biochem. Biophys. Res. Commun. 2000, 278, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Peterson, R.T.; Desai, B.N.; Hardwick, J.S.; Schreiber, S.L. Protein phosphatase 2a interacts with the 70-kda s6 kinase and is activated by inhibition of fkbp12-rapamycinassociated protein. Proc. Natl. Acad. Sci. USA 1999, 96, 4438–4442. [Google Scholar] [CrossRef]
- Zhang, Q.; Claret, F.X. Phosphatases: The new brakes for cancer development? Enzym. Res. 2012, 2012, 659649. [Google Scholar] [CrossRef] [PubMed]
- Kiss, A.; Becsi, B.; Kolozsvari, B.; Komaromi, I.; Kover, K.E.; Erdodi, F. Epigallocatechin-3-gallate and penta-o-galloyl-beta-d-glucose inhibit protein phosphatase-1. FEBS J. 2013, 280, 612–626. [Google Scholar] [CrossRef]
- Kitano, K.; Nam, K.Y.; Kimura, S.; Fujiki, H.; Imanishi, Y. Sealing effects of (−)-epigallocatechin gallate on protein kinase c and protein phosphatase 2a. Biophys. Chem. 1997, 65, 157–164. [Google Scholar] [CrossRef]
- Janssens, V.; Goris, J.; Van Hoof, C. Pp2a: The expected tumor suppressor. Curr. Opin. Genet. Dev. 2005, 15, 34–41. [Google Scholar] [CrossRef]
- Stamenkovic, I.; Yu, Q. Merlin, a “magic” linker between extracellular cues and intracellular signaling pathways that regulate cell motility, proliferation, and survival. Curr. Protein Pept. Sci. 2010, 11, 471–484. [Google Scholar] [CrossRef]
- Horiguchi, A.; Zheng, R.; Shen, R.; Nanus, D.M. Inactivation of the nf2 tumor suppressor protein merlin in du145 prostate cancer cells. Prostate 2008, 68, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, A.; Shibata, Y.; Hall, I.M.; Dutta, A. Chromosomal structural variations during progression of a prostate epithelial cell line to a malignant metastatic state inactivate the nf2, nipsnap1, ugt2b17, and lpin2 genes. Cancer Biol. Ther. 2013, 14, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, A.M.; Fernandez-Valle, C. Role of merlin/nf2 inactivation in tumor biology. Oncogene 2016, 35, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. Micrornas: Tiny regulators with great potential. Cell 2001, 107, 823–826. [Google Scholar] [CrossRef]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. Ras is regulated by the let-7 microrna family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.; Lorenz, P.; Gross, G.; Ibrahim, S.; Kunz, M. Microrna let-7b targets important cell cycle molecules in malignant melanoma cells and interferes with anchorage-independent growth. Cell Res. 2008, 18, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Zedan, A.H.; Hansen, T.F.; Assenholt, J.; Pleckaitis, M.; Madsen, J.S.; Osther, P.J.S. Microrna expression in tumour tissue and plasma in patients with newly diagnosed metastatic prostate cancer. Tumour. Biol. 2018, 40, 1010428318775864. [Google Scholar] [CrossRef]
- Zhou, H.; Chen, J.X.; Yang, C.S.; Yang, M.Q.; Deng, Y.; Wang, H. Gene regulation mediated by micrornas in response to green tea polyphenol egcg in mouse lung cancer. BMC Genom. 2014, 15 (Suppl. 11), S3. [Google Scholar] [CrossRef]
- Pan, X.; Zhao, B.; Song, Z.; Han, S.; Wang, M. Estrogen receptor-alpha36 is involved in epigallocatechin-3-gallate induced growth inhibition of er-negative breast cancer stem/progenitor cells. J. Pharmacol. Sci. 2016, 130, 85–93. [Google Scholar] [CrossRef]
- Jiang, P.; Xu, C.; Chen, L.; Chen, A.; Wu, X.; Zhou, M.; Haq, I.U.; Mariyam, Z.; Feng, Q. Egcg inhibits csc-like properties through targeting mir-485/cd44 axis in a549-cisplatin resistant cells. Mol. Carcinog. 2018, 57, 1835–1844. [Google Scholar] [CrossRef]
- Nishimura, N.; Hartomo, T.B.; Pham, T.V.; Lee, M.J.; Yamamoto, T.; Morikawa, S.; Hasegawa, D.; Takeda, H.; Kawasaki, K.; Kosaka, Y.; et al. Epigallocatechin gallate inhibits sphere formation of neuroblastoma be(2)-c cells. Environ. Health Prev. Med. 2012, 17, 246–251. [Google Scholar] [CrossRef]
- Kumazoe, M.; Takai, M.; Hiroi, S.; Takeuchi, C.; Yamanouchi, M.; Nojiri, T.; Onda, H.; Bae, J.; Huang, Y.; Takamatsu, K.; et al. Pde3 inhibitor and egcg combination treatment suppress cancer stem cell properties in pancreatic ductal adenocarcinoma. Sci. Rep. 2017, 7, 1917. [Google Scholar] [CrossRef] [PubMed]
- Kumazoe, M.; Takai, M.; Bae, J.; Hiroi, S.; Huang, Y.; Takamatsu, K.; Won, Y.; Yamashita, M.; Hidaka, S.; Yamashita, S.; et al. Foxo3 is essential for cd44 expression in pancreatic cancer cells. Oncogene 2017, 36, 2643–2654. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.F.; Kane, S.E.; Sonenshein, G.E. Trastuzumab-resistant her2-driven breast cancer cells are sensitive to epigallocatechin-3 gallate. Cancer Res. 2007, 67, 9018–9023. [Google Scholar] [CrossRef] [PubMed]
- Farabegoli, F.; Govoni, M.; Ciavarella, C.; Orlandi, M.; Papi, A. A rxr ligand 6-oh-11-o-hydroxyphenanthrene with antitumour properties enhances (−)-epigallocatechin-3-gallate activity in three human breast carcinoma cell lines. BioMed Res. Int. 2014, 2014, 853086. [Google Scholar] [CrossRef]
- Lugnier, C. Cyclic nucleotide phosphodiesterase (pde) superfamily: A new target for the development of specific therapeutic agents. Pharmacol. Ther. 2006, 109, 366–398. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.P.; Zhou, X.Z. The prolyl isomerase pin1: A pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Sola, D.; Dalla-Favera, R. Pinning down the c-myc oncoprotein. Nat. Cell Biol. 2004, 6, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Sears, R.C. The life cycle of c-myc: From synthesis to degradation. Cell Cycle 2004, 3, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Ryo, A.; Nakamura, M.; Wulf, G.; Liou, Y.C.; Lu, K.P. Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with apc. Nat. Cell Biol. 2001, 3, 793–801. [Google Scholar] [CrossRef]
- Bao, L.; Kimzey, A.; Sauter, G.; Sowadski, J.M.; Lu, K.P.; Wang, D.G. Prevalent overexpression of prolyl isomerase pin1 in human cancers. Am. J. Pathol. 2004, 164, 1727–1737. [Google Scholar] [CrossRef]
- Ayala, G.; Wang, D.; Wulf, G.; Frolov, A.; Li, R.; Sowadski, J.; Wheeler, T.M.; Lu, K.P.; Bao, L. The prolyl isomerase pin1 is a novel prognostic marker in human prostate cancer. Cancer Res. 2003, 63, 6244–6251. [Google Scholar]
- Moore, J.D.; Potter, A. Pin1 inhibitors: Pitfalls, progress and cellular pharmacology. Bioorg. Med. Chem. Lett. 2013, 23, 4283–4291. [Google Scholar] [CrossRef]
- Hidaka, M.; Kosaka, K.; Tsushima, S.; Uchida, C.; Takahashi, K.; Takahashi, N.; Tsubuki, M.; Hara, Y.; Uchida, T. Food polyphenols targeting peptidyl prolyl cis/trans isomerase pin1. Biochem. Biophys. Res. Commun. 2018, 499, 681–687. [Google Scholar] [CrossRef]
- Xi, L.; Wang, Y.; He, Q.; Zhang, Q.; Du, L. Interaction between pin1 and its natural product inhibitor epigallocatechin-3-gallate by spectroscopy and molecular dynamics simulations. Spectrochim. Acta A Mol. Biomol. Spectrosc 2016, 169, 134–143. [Google Scholar] [CrossRef]
- Katsuno, Y.; Lamouille, S.; Derynck, R. Tgf-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Diamond, M.E.; Ottaviano, A.J.; Joseph, M.J.; Ananthanarayan, V.; Munshi, H.G. Transforming growth factor-beta 1 promotes matrix metalloproteinase-9-mediated oral cancer invasion through snail expression. Mol. Cancer Res. 2008, 6, 10–20. [Google Scholar] [CrossRef]
- Joseph, M.J.; Dangi-Garimella, S.; Shields, M.A.; Diamond, M.E.; Sun, L.; Koblinski, J.E.; Munshi, H.G. Slug is a downstream mediator of transforming growth factor-beta1-induced matrix metalloproteinase-9 expression and invasion of oral cancer cells. J. Cell. Biochem. 2009, 108, 726–736. [Google Scholar] [CrossRef]
- Sinpitaksakul, S.N.; Pimkhaokham, A.; Sanchavanakit, N.; Pavasant, P. Tgf-beta1 induced mmp-9 expression in hnscc cell lines via smad/mlck pathway. Biochem. Biophys. Res. Commun. 2008, 371, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.; Nandy, S.K.; Sarkar, J.; Chakraborti, T.; Chakraborti, S. Inhibition of pro-/active mmp-2 by green tea catechins and prediction of their interaction by molecular docking studies. Mol. Cell. Biochem. 2017, 427, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, J.; Nandy, S.K.; Chowdhury, A.; Chakraborti, T.; Chakraborti, S. Inhibition of mmp-9 by green tea catechins and prediction of their interaction by molecular docking analysis. Biomed. Pharmacother. 2016, 84, 340–347. [Google Scholar] [CrossRef]
- Schramm, L. Going green: The role of the green tea component egcg in chemoprevention. J. Carcinog. Mutagen. 2013, 4, 1000142. [Google Scholar] [CrossRef] [PubMed]
- Riley, P.A. Epimutation and cancer: Carcinogenesis viewed as error-prone inheritance of epigenetic information. J. Oncol. 2018, 2018, 2645095. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Huang, Q.; Ji, L.; Wang, Y.; Qi, X.; Liu, L.; Liu, Z.; Lu, L. Epigenetic regulation of active Chinese herbal components for cancer prevention and treatment: A follow-up review. Pharmacol. Res. 2016, 114, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.Z.; Wang, Y.; Ai, N.; Hou, Z.; Sun, Y.; Lu, H.; Welsh, W.; Yang, C.S. Tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003, 63, 7563–7570. [Google Scholar]
- Pal, D.; Sur, S.; Roy, R.; Mandal, S.; Kumar Panda, C. Epigallocatechin gallate in combination with eugenol or amarogentin shows synergistic chemotherapeutic potential in cervical cancer cell line. J. Cell. Physiol. 2018, 234, 825–836. [Google Scholar] [CrossRef]
- Oya, Y.; Mondal, A.; Rawangkan, A.; Umsumarng, S.; Iida, K.; Watanabe, T.; Kanno, M.; Suzuki, K.; Li, Z.; Kagechika, H.; et al. Down-regulation of histone deacetylase 4, -5 and -6 as a mechanism of synergistic enhancement of apoptosis in human lung cancer cells treated with the combination of a synthetic retinoid, am80 and green tea catechin. J. Nutr. Biochem. 2017, 42, 7–16. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Han, L.; Zhou, Y.; Sun, S. Green tea polyphenol egcg reverse cisplatin resistance of a549/ddp cell line through candidate genes demethylation. Biomed. Pharmacother. 2015, 69, 285–290. [Google Scholar] [CrossRef]
- Jin, H.; Chen, J.X.; Wang, H.; Lu, G.; Liu, A.; Li, G.; Tu, S.; Lin, Y.; Yang, C.S. Nnk-induced DNA methyltransferase 1 in lung tumorigenesis in a/j mice and inhibitory effects of (−)-epigallocatechin-3-gallate. Nutr. Cancer 2015, 67, 167–176. [Google Scholar] [CrossRef]
- Liu, L.; Zuo, J.; Wang, G. Epigallocatechin-3-gallate suppresses cell proliferation and promotes apoptosis in ec9706 and eca109 esophageal carcinoma cells. Oncol. Lett. 2017, 14, 4391–4395. [Google Scholar] [CrossRef]
- Le, C.T.; Leenders, W.P.J.; Molenaar, R.J.; van Noorden, C.J.F. Effects of the green tea polyphenol epigallocatechin-3-gallate on glioma: A critical evaluation of the literature. Nutr. Cancer 2018, 70, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Li, W.G.; Li, Q.H.; Tan, Z. Epigallocatechin gallate induces telomere fragmentation in hela and 293 but not in mrc-5 cells. Life Sci. 2005, 76, 1735–1746. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, P.; Gao, F.; Yang, J.; Yao, K. Effects of epigallocatechin gallate on the proliferation and apoptosis of the nasopharyngeal carcinoma cell line cne2. Exp. Ther. Med. 2014, 8, 1783–1788. [Google Scholar] [CrossRef]
- Wang, X.; Hao, M.W.; Dong, K.; Lin, F.; Ren, J.H.; Zhang, H.Z. Apoptosis induction effects of egcg in laryngeal squamous cell carcinoma cells through telomerase repression. Arch. Pharm. Res. 2009, 32, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Sadava, D.; Whitlock, E.; Kane, S.E. The green tea polyphenol, epigallocatechin-3-gallate inhibits telomerase and induces apoptosis in drug-resistant lung cancer cells. Biochem. Biophys. Res. Commun. 2007, 360, 233–237. [Google Scholar] [CrossRef]
- Kuzuhara, T.; Sei, Y.; Yamaguchi, K.; Suganuma, M.; Fujiki, H. DNA and rna as new binding targets of green tea catechins. J. Biol. Chem. 2006, 281, 17446–17456. [Google Scholar] [CrossRef] [PubMed]
- Kuzuhara, T.; Tanabe, A.; Sei, Y.; Yamaguchi, K.; Suganuma, M.; Fujiki, H. Synergistic effects of multiple treatments, and both DNA and rna direct bindings on, green tea catechins. Mol. Carcinog. 2007, 46, 640–645. [Google Scholar] [CrossRef]


{kind=link}
| Cell Cycle, Proliferation & Survival | Apoptosis & Cell Death | Motility, Invasion and Metastatization | Inflammation | Epigenetic Control | Others |
|---|---|---|---|---|---|
| p16 [48] | Bax [49] | MMP-2 * [50] | FcεRI [51] | DNMT1 * [39] | DAPK1 [52] |
| p18 [35] | Bad [53] | MMP-9 * [50] | IL-8 [54] | DNMT3A [48] | MRLC [55] |
| p21 [48] | Bak [56] | MMP-14 [57] | IGF-1R * [45] | DNMT3B * [39] | MYPT1 [55] |
| p27 [56] | Bcl-2 * [58] | uPA [59] | VEGF [60] | HDAC1 * [39] | eEF1a [61] |
| Cyclin D [56] | Bcl-xl [53] | PAI-1 [59] | CSF-1 [62] | HDAC2 [63] | ID1 [64] |
| Cyclin E [35] | Bcl-xs [56] | E-cadherine [39] | CCL-2 [62] | HAT [65] | RAR-β [39] |
| Cyclin A [66] | Caspase3 [56] | SLUG [67] | COX-2 [60] | hTERT [68] | HSP70 [53] |
| Cyclin B [66] | Caspase8 [69] | SNAIL1 [70] | iNOS [71] | EZH2 [72] | HSP90 * [73] |
| CDK4 [56] | Caspase9 [56] | Vimentin * [42] | eNOS [74,75,76,77,78] | GRP78 * [46] | |
| CDK6 [56] | Apaf-1 [53] | Twist [79] | PECAM-1 [80] | ||
| CDK2 [35] | Puma [56] | N-cadherine [79] | miR-16 [62] | ||
| CDK1 [66] | XIAP [53] | HIF-1α [60] | let-7b miRNA [81] | ||
| Erk1/2 [56] | Cytochrome C [56] | β-catenin [54] | miR-210 [82] | ||
| Pin * [83] | p53 [84] | Wnt [54] | miR34a [85] | ||
| PPA2 [86] | Survivin [87] | TIMP-3 [72] | miR145 [85] | ||
| PKA [86] | Fas [69] | miR200c [85] | |||
| STAT [12] | DR5 [69] | ZAP70 * [44] | |||
| AR [65] | PARP [88] | TRAF-6 * [89] | |||
| 67LR * [90] | Oct4 [85] | ||||
| FcεRI [51] | Sox2 [91] | ||||
| EGFR [92] | Notch1 [85] | ||||
| HGFR [93] | Nanog [85] | ||||
| TGFR-II * [94] | CD133 [95] | ||||
| cGMP [74] [96] | |||||
| cAMP [86] | |||||
| P-glycoprotein [88] | |||||
| NF-kB [97] | |||||
| c-Myc [98] | |||||
| FOXO3a [99] | |||||
| GSK-3β [98] | |||||
| PI3K [100] | |||||
| AKT [100] | |||||
| PKC-δ [74] | |||||
| JAK-1/2 [12] | |||||
| Src [57] | |||||
| CK1α [98] | |||||
| p38 MAPK [56] | |||||
| JNK [56] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Negri, A.; Naponelli, V.; Rizzi, F.; Bettuzzi, S. Molecular Targets of Epigallocatechin—Gallate (EGCG): A Special Focus on Signal Transduction and Cancer. Nutrients 2018, 10, 1936. https://doi.org/10.3390/nu10121936
Negri A, Naponelli V, Rizzi F, Bettuzzi S. Molecular Targets of Epigallocatechin—Gallate (EGCG): A Special Focus on Signal Transduction and Cancer. Nutrients. 2018; 10(12):1936. https://doi.org/10.3390/nu10121936
Chicago/Turabian StyleNegri, Aide, Valeria Naponelli, Federica Rizzi, and Saverio Bettuzzi. 2018. "Molecular Targets of Epigallocatechin—Gallate (EGCG): A Special Focus on Signal Transduction and Cancer" Nutrients 10, no. 12: 1936. https://doi.org/10.3390/nu10121936
APA StyleNegri, A., Naponelli, V., Rizzi, F., & Bettuzzi, S. (2018). Molecular Targets of Epigallocatechin—Gallate (EGCG): A Special Focus on Signal Transduction and Cancer. Nutrients, 10(12), 1936. https://doi.org/10.3390/nu10121936