Effect of Genetically Low 25-Hydroxyvitamin D on Mortality Risk: Mendelian Randomization Analysis in 3 Large European Cohorts

 , , , , , , , , ,
, , , , , , , , ,  add
Show full author list
add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cohorts and Data Collection
2.2. Statistical Methods
3. Results
4. Discussion
5. Strengths and Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Disclaimer
References
- Pludowski, P.; Holick, M.F.; Pilz, S.; Wagner, C.L.; Hollis, B.W.; Grant, W.B.; Shoenfeld, Y.; Lerchbaum, E.; Llewellyn, D.J.; Kienreich, K.; et al. Vitamin D effects on musculoskeletal health, immunity, autoimmunity, cardiovascular disease, cancer, fertility, pregnancy, dementia and mortality-a review of recent evidence. Autoimmun. Rev. 2013, 12, 976–989. [Google Scholar] [CrossRef] [PubMed]
- Autier, P.; Boniol, M.; Pizot, C.; Mullie, P. Vitamin D status and ill health: A systematic review. Lancet Diabetes Endocrinol. 2014, 2, 76–89. [Google Scholar] [CrossRef]
- Kestenbaum, B.; Katz, R.; de Boer, I.; Hoofnagle, A.; Sarnak, M.J.; Shlipak, M.G.; Jenny, N.S.; Siscovick, D.S. Vitamin D, parathyroid hormone, and cardiovascular events among older adults. J. Am. Coll. Cardiol. 2011, 58, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Schottker, B.; Jorde, R.; Peasey, A.; Thorand, B.; Jansen, E.H.; Groot, L.; Streppel, M.; Gardiner, J.; Ordonez-Mena, J.M.; Perna, L.; et al. Vitamin D and mortality: Meta-analysis of individual participant data from a large consortium of cohort studies from Europe and the United States. BMJ 2014, 348, g3656. [Google Scholar] [CrossRef]
- Daraghmeh, A.H.; Bertoia, M.L.; Al-Qadi, M.O.; Abdulbaki, A.M.; Roberts, M.B.; Eaton, C.B. Evidence for the vitamin D hypothesis: The NHANES III extended mortality follow-up. Atherosclerosis 2016, 255, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.F.; Kim, J.J.; Mohr, S.B.; Gorham, E.D.; Grant, W.B.; Giovannucci, E.L.; Baggerly, L.; Hofflich, H.; Ramsdell, J.W.; Zeng, K.; et al. Meta-analysis of all-cause mortality according to serum 25-hydroxyvitamin D. Am. J. Public Health 2014, 104, e43–e50. [Google Scholar] [CrossRef] [PubMed]
- Grober, U.; Reichrath, J.; Holick, M.F. Live longer with vitamin D? Nutrients 2015, 7, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Gaksch, M.; Jorde, R.; Grimnes, G.; Joakimsen, R.; Schirmer, H.; Wilsgaard, T.; Mathiesen, E.B.; Njolstad, I.; Lochen, M.L.; Marz, W.; et al. Vitamin D and mortality: Individual participant data meta-analysis of standardized 25-hydroxyvitamin D in 26916 individuals from a European consortium. PLoS ONE 2017, 12, e0170791. [Google Scholar] [CrossRef]
- Zhu, K.; Knuiman, M.; Divitini, M.; Hung, J.; Lim, E.M.; Cooke, B.R.; Walsh, J.P. Serum 25-hydroxyvitamin D as a predictor of mortality and cardiovascular events: A 20-year study of a community-based cohort. Clin. Endocrinol. 2017. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Gluud, L.L.; Nikolova, D.; Whitfield, K.; Wetterslev, J.; Simonetti, R.G.; Bjelakovic, M.; Gluud, C. Vitamin D supplementation for prevention of mortality in adults. Cochrane Database Syst. Rev. 2014, CD007470. [Google Scholar] [CrossRef]
- Rejnmark, L.; Avenell, A.; Masud, T.; Anderson, F.; Meyer, H.E.; Sanders, K.M.; Salovaara, K.; Cooper, C.; Smith, H.E.; Jacobs, E.T.; et al. Vitamin D with calcium reduces mortality: Patient level pooled analysis of 70,528 patients from eight major vitamin D trials. J. Clin. Endocrinol. Metab. 2012, 97, 2670–2681. [Google Scholar] [CrossRef] [PubMed]
- Bolland, M.J.; Grey, A.; Gamble, G.D.; Reid, I.R. The effect of vitamin D supplementation on skeletal, vascular, or cancer outcomes: A trial sequential meta-analysis. Lancet Diabetes Endocrinol. 2014, 2, 307–320. [Google Scholar] [CrossRef]
- Chowdhury, R.; Kunutsor, S.; Vitezova, A.; Oliver-Williams, C.; Chowdhury, S.; Kiefte-de-Jong, J.C.; Khan, H.; Baena, C.P.; Prabhakaran, D.; Hoshen, M.B.; et al. Vitamin D and risk of cause specific death: Systematic review and meta-analysis of observational cohort and randomised intervention studies. BMJ 2014, 348, g1903. [Google Scholar] [CrossRef] [PubMed]
- Cashman, K.D.; Dowling, K.G.; Skrabakova, Z.; Gonzalez-Gross, M.; Valtuena, J.; De Henauw, S.; Moreno, L.; Damsgaard, C.T.; Michaelsen, K.F.; Molgaard, C.; et al. Vitamin D deficiency in Europe: Pandemic? Am. J. Clin. Nutr. 2016, 103, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Thompson, S.G. Mendelian Randomization: Methods for Using Genetic Variants in Causal Estimation; Chapman and Hall/CRC: New York, NY, USA, 2015. [Google Scholar]
- Afzal, S.; Brondum-Jacobsen, P.; Bojesen, S.E.; Nordestgaard, B.G. Genetically low vitamin D concentrations and increased mortality: Mendelian randomisation analysis in three large cohorts. BMJ 2014, 349, g6330. [Google Scholar] [CrossRef] [PubMed]
- Welsh, P.; Sattar, N. Vitamin D genes and mortality. BMJ 2014, 349, g6599. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.B.; Launer, L.J.; Eiriksdottir, G.; Kjartansson, O.; Jonsson, P.V.; Sigurdsson, G.; Thorgeirsson, G.; Aspelund, T.; Garcia, M.E.; Cotch, M.F.; et al. Age, Gene/Environment Susceptibility-Reykjavik Study: Multidisciplinary applied phenomics. Am. J. Epidemiol. 2007, 165, 1076–1087. [Google Scholar] [CrossRef]
- Winkelmann, B.R.; Marz, W.; Boehm, B.O.; Zotz, R.; Hager, J.; Hellstern, P.; Senges, J.; Group, L.S. Rationale and design of the LURIC study—A resource for functional genomics, pharmacogenomics and long-term prognosis of cardiovascular disease. Pharmacogenomics 2001, 2, S1–S73. [Google Scholar] [CrossRef]
- Jorde, R.; Schirmer, H.; Wilsgaard, T.; Joakimsen, R.M.; Mathiesen, E.B.; Njolstad, I.; Lochen, M.L.; Figenschau, Y.; Berg, J.P.; Svartberg, J.; et al. Polymorphisms related to the serum 25-hydroxyvitamin D level and risk of myocardial infarction, diabetes, cancer and mortality. The Tromso Study. PLoS ONE 2012, 7, e37295. [Google Scholar] [CrossRef]
- Wang, T.J.; Zhang, F.; Richards, J.B.; Kestenbaum, B.; van Meurs, J.B.; Berry, D.; Kiel, D.P.; Streeten, E.A.; Ohlsson, C.; Koller, D.L.; et al. Common genetic determinants of vitamin D insufficiency: A genome-wide association study. Lancet 2010, 376, 180–188. [Google Scholar] [CrossRef]
- Dastani, Z.; Berger, C.; Langsetmo, L.; Fu, L.; Wong, B.Y.; Malik, S.; Goltzman, D.; Cole, D.E.; Richards, J.B. In healthy adults, biological activity of vitamin D, as assessed by serum PTH, is largely independent of DBP concentrations. J. Bone Miner. Res. 2014, 29, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Otterbein, L.R.; Cosio, C.; Graceffa, P.; Dominguez, R. Crystal structures of the vitamin D-binding protein and its complex with actin: Structural basis of the actin-scavenger system. Proc. Natl. Acad. Sci. USA 2002, 99, 8003–8008. [Google Scholar] [CrossRef] [PubMed]
- Durazo-Arvizu, R.A.; Tian, L.; Brooks, S.P.J.; Sarafin, K.; Cashman, K.D.; Kiely, M.; Merkel, J.; Myers, G.L.; Coates, P.M.; Sempos, C.T. The Vitamin D Standardization Program (VDSP) Manual for Retrospective Laboratory Standardization of Serum 25-Hydroxyvitamin D Data. J. AOAC Int. 2017, 100, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.J.; Vimaleswaran, K.S.; Whittaker, J.C.; Hingorani, A.D.; Hypponen, E. Evaluation of genetic markers as instruments for Mendelian randomization studies on vitamin D. PLoS ONE 2012, 7, e37465. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Yu, K.; Stolzenberg-Solomon, R.; Simon, K.C.; McCullough, M.L.; Gallicchio, L.; Jacobs, E.J.; Ascherio, A.; Helzlsouer, K.; Jacobs, K.B.; et al. Genome-wide association study of circulating vitamin D levels. Hum. Mol. Genet. 2010, 19, 2739–2745. [Google Scholar] [CrossRef] [Green Version]
- Palmer, T.M.; Sterne, J.A.; Harbord, R.M.; Lawlor, D.A.; Sheehan, N.A.; Meng, S.; Granell, R.; Smith, G.D.; Didelez, V. Instrumental variable estimation of causal risk ratios and causal odds ratios in Mendelian randomization analyses. Am. J. Epidemiol. 2011, 173, 1392–1403. [Google Scholar] [CrossRef] [PubMed]
- Fieller, E.C. Some Problems in Interval Estimation. J. R. Stat. Soc. Ser. B (Methodol.) 1954, 16, 175–185. [Google Scholar] [CrossRef]
- Ross, A.C.; Manson, J.E.; Abrams, S.A.; Aloia, J.F.; Brannon, P.M.; Clinton, S.K.; Durazo-Arvizu, R.A.; Gallagher, J.C.; Gallo, R.L.; Jones, G.; et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: What clinicians need to know. J. Clin. Endocrinol. Metab. 2011, 96, 53–58. [Google Scholar] [CrossRef]
- Pierce, B.L.; Burgess, S. Efficient design for Mendelian randomization studies: Subsample and 2-sample instrumental variable estimators. Am. J. Epidemiol. 2013, 178, 1177–1184. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; O’Reilly, P.F.; Aschard, H.; Hsu, Y.H.; Richards, J.B.; Dupuis, J.; Ingelsson, E.; Karasik, D.; Pilz, S.; Berry, D.; et al. Genome-wide association study in 79,366 European-ancestry individuals informs the genetic architecture of 25-hydroxyvitamin D levels. Nat. Commun. 2018, 9, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.S.; Gao, L.H.; Zhang, X.Y.; He, J.W.; Fu, W.Z.; Liu, Y.J.; Hu, Y.Q.; Zhang, Z.L. Genetically Low Vitamin D Levels, Bone Mineral Density, and Bone Metabolism Markers: A Mendelian Randomisation Study. Sci. Rep. 2016, 6, 33202. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Sharp, S.J.; Burgess, S.; Scott, R.A.; Imamura, F.; InterAct, C.; Langenberg, C.; Wareham, N.J.; Forouhi, N.G. Association between circulating 25-hydroxyvitamin D and incident type 2 diabetes: A mendelian randomisation study. Lancet Diabetes Endocrinol. 2015, 3, 35–42. [Google Scholar] [CrossRef]
- McKay, G.J.; Young, I.S.; McGinty, A.; Bentham, G.C.; Chakravarthy, U.; Rahu, M.; Seland, J.; Soubrane, G.; Tomazzoli, L.; Topouzis, F.; et al. Associations between Serum Vitamin D and Genetic Variants in Vitamin D Pathways and Age-Related Macular Degeneration in the European Eye Study. Ophthalmology 2016, 124, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Vimaleswaran, K.S.; Cavadino, A.; Berry, D.J.; LifeLines Cohort Study Investigators; Jorde, R.; Dieffenbach, A.K.; Lu, C.; Alves, A.C.; Heerspink, H.J.; Tikkanen, E.; et al. Association of vitamin D status with arterial blood pressure and hypertension risk: A mendelian randomisation study. Lancet Diabetes Endocrinol. 2014, 2, 719–729. [Google Scholar] [CrossRef]
- Brondum-Jacobsen, P.; Benn, M.; Afzal, S.; Nordestgaard, B.G. No evidence that genetically reduced 25-hydroxyvitamin D is associated with increased risk of ischaemic heart disease or myocardial infarction: A Mendelian randomization study. Int. J. Epidemiol. 2015, 44, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Afzal, S.; Brondum-Jacobsen, P.; Bojesen, S.E.; Nordestgaard, B.G. Vitamin D concentration, obesity, and risk of diabetes: A mendelian randomisation study. Lancet Diabetes Endocrinol. 2014, 2, 298–306. [Google Scholar] [CrossRef]
- Vimaleswaran, K.S.; Berry, D.J.; Lu, C.; Tikkanen, E.; Pilz, S.; Hiraki, L.T.; Cooper, J.D.; Dastani, Z.; Li, R.; Houston, D.K.; et al. Causal relationship between obesity and vitamin D status: Bi-directional Mendelian randomization analysis of multiple cohorts. PLoS Med. 2013, 10, e1001383. [Google Scholar] [CrossRef]
- Kueider, A.M.; Tanaka, T.; An, Y.; Kitner-Triolo, M.H.; Palchamy, E.; Ferrucci, L.; Thambisetty, M. State- and trait-dependent associations of vitamin-D with brain function during aging. Neurobiol. Aging 2016, 39, 38–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitrakopoulou, V.I.; Tsilidis, K.K.; Haycock, P.C.; Dimou, N.L.; Al-Dabhani, K.; Martin, R.M.; Lewis, S.J.; Gunter, M.J.; Mondul, A.; Shui, I.M.; et al. Circulating vitamin D concentration and risk of seven cancers: Mendelian randomisation study. BMJ 2017, 359, j4761. [Google Scholar] [CrossRef] [PubMed]
- Chandler, P.D.; Tobias, D.K.; Wang, L.; Smith-Warner, S.A.; Chasman, D.I.; Rose, L.; Giovannucci, E.L.; Buring, J.E.; Ridker, P.M.; Cook, N.R.; et al. Association between Vitamin D Genetic Risk Score and Cancer Risk in a Large Cohort of U.S. Women. Nutrients 2018, 10, 55. [Google Scholar] [CrossRef] [PubMed]
- Mokry, L.E.; Ross, S.; Morris, J.A.; Manousaki, D.; Forgetta, V.; Richards, J.B. Genetically decreased vitamin D and risk of Alzheimer disease. Neurology 2016, 87. [Google Scholar] [CrossRef] [PubMed]
- Schottker, B.; Brenner, H. Vitamin D as a Resilience Factor, Helpful for Survival of Potentially Fatal Conditions: A Hypothesis Emerging from Recent Findings of the ESTHER Cohort Study and the CHANCES Consortium. Nutrients 2015, 7, 3264–3278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kupferschmidt, K. Uncertain verdict as vitamin D goes on trial. Science 2012, 337, 1476–1478. [Google Scholar] [CrossRef]
- Scragg, R.; Stewart, A.W.; Waayer, D.; Lawes, C.M.M.; Toop, L.; Sluyter, J.; Murphy, J.; Khaw, K.T.; Camargo, C.A., Jr. Effect of Monthly High-Dose Vitamin D Supplementation on Cardiovascular Disease in the Vitamin D Assessment Study: A Randomized Clinical Trial. JAMA Cardiol. 2017, 2, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Lappe, J.; Watson, P.; Travers-Gustafson, D.; Recker, R.; Garland, C.; Gorham, E.; Baggerly, K.; McDonnell, S.L. Effect of Vitamin D and Calcium Supplementation on Cancer Incidence in Older Women: A Randomized Clinical Trial. JAMA 2017, 317, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Zittermann, A.; Ernst, J.B.; Prokop, S.; Fuchs, U.; Dreier, J.; Kuhn, J.; Knabbe, C.; Birschmann, I.; Schulz, U.; Berthold, H.K.; et al. Effect of vitamin D on all-cause mortality in heart failure (EVITA): A 3-year randomized clinical trial with 4000 IU vitamin D daily. Eur. Heart J. 2017, 38, 2279–2286. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.E.; Cook, N.R.; Lee, I.M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Gordon, D.; Copeland, T.; D’Agostino, D.; et al. Vitamin D Supplements and Prevention of Cancer and Cardiovascular Disease. N. Engl. J. Med. 2018. [Google Scholar] [CrossRef]


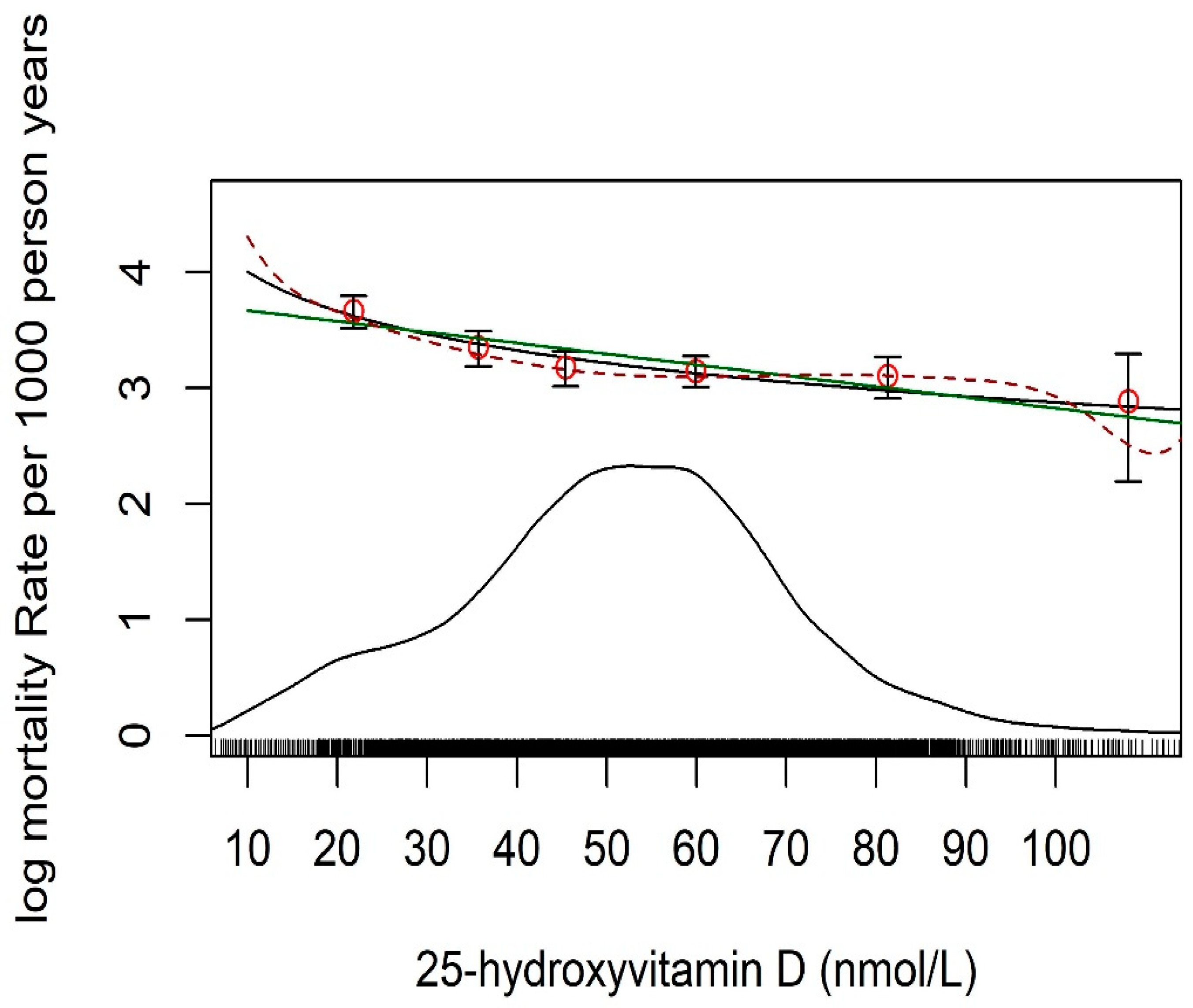{kind=link}
| Characteristic | Total Sample for Analysis | AGES | LURIC | Tromsø | ||||
|---|---|---|---|---|---|---|---|---|
| N = 10501 | N = 3172 | N = 2864 | N = 4465 | |||||
| N/Mean | %/SD | N/Mean | %/SD | N/Mean | %/SD | N/Mean | %/SD | |
| Sex (females) * | 5266 | 50.1 | 1834 | 57.8 | 866 | 30.2 | 2566 | 57.5 |
| Age | 67.1 | 10.1 | 76.4 | 5.5 | 62.9 | 10.5 | 63.2 | 7.7 |
| BMI, kg/m2 | 26.8 | 4.2 | 27.1 | 4.4 | 27.4 | 4.0 | 26.2 | 4.2 |
| Season blood drawn a | ||||||||
| spring | 3011 | 28.7 | 810 | 25.5 | 602 | 21.0 | 1599 | 35.8 |
| summer | 1328 | 12.7 | 448 | 14.1 | 711 | 24.8 | 169 | 3.8 |
| fall | 3180 | 30.3 | 1086 | 34.2 | 880 | 30.7 | 1214 | 27.2 |
| winter | 2982 | 28.4 | 828 | 26.1 | 671 | 23.4 | 1483 | 33.2 |
| 25(OH)D, nmol/Lb | 51.7 | 18.1 | 57.1 | 17.8 | 42.3 | 22.7 | 54.0 | 11.4 |
| LDL, mmol/L | 3.8 | 1.2 | 3.5 | 1.0 | 3.0 | 0.9 | 4.5 | 1.2 |
| Glucose, mmol/Lc | 5.7 | 1.5 | 5.8 | 1.1 | 5.6 | 1.8 | - | - |
| SBP, mmHg | 144.3 | 22.5 | 142.5 | 20.3 | 141.3 | 23.6 | 147.4 | 22.9 |
| Arterial hypertension *,d | 8101 | 77.2 | 2558 | 80.7 | 2677 | 93.5 | 2866 | 65.2 |
| Active smoker * | 2418 | 23.0 | 403 | 12.7 | 540 | 18.9 | 1475 | 33 |
| Diabetes *,e | 1482 | 14.3 | 365 | 11.5 | 943 | 32.9 | 174 | 4.0 |
| Previous cancer * | 1034 | 9.9 | 489 | 15.4 | 209 | 7.3 | 336 | 7.5 |
| CVD history *,f | 2400 | 22.9 | 613 | 19.3 | 1349 | 47.1 | 438 | 9.8 |
| Death * | 4003 | 38.1 | 1184 | 37.3 | 855 | 29.9 | 1964 | 44.0 |
| GENE | Activity | SNP | Beta | p-Value |
|---|---|---|---|---|
| DHCR7/NADSYN1 | Regulates vitamin D precursor | rs12785878 | −1.18 nmol/L | 1.77 × 10−6 |
| CYP2R1 | 25-hydroxylation of vitamin D | rs12794714 | −1.26 nmol/L | 1.34 × 10−7 |
| Genetic score (G1) | SUM | −1.25 nmol/L | 5.99 × 10−13 | |
| DHCR7/NADSYN1 | Regulates vitamin D precursor | rs12785878 rs11234027 | −0.63 | 8.68 × 10−6 |
| CYP2R1 | 25-hydroxylation of vitamin D | rs12794714 rs10741657 | −0.70 | 5.07 × 10−8 |
| Genetic score (G2) | SUM | −0.66 | 6.19 × 10−12 |
| Model | <30 | 30−39.9 | 40−49.9 | 50−74.9 | 75−99.9 (Ref) | 100−150 | |
|---|---|---|---|---|---|---|---|
| Basic information | Median nmol/L | 21.8 | 35.7 | 45.4 | 59.9 | 81.3 | 108.1 |
| N | 1303 | 1230 | 2174 | 4882 | 824 | 88 | |
| Deaths, n | 574 | 485 | 869 | 1808 | 250 | 17 | |
| Person years | 10592 | 12452 | 26904 | 57820 | 8434 | 837 | |
| Death Rate * | 54.2 | 38.9 | 32.3 | 31.3 | 29.6 | 20.3 | |
| Observational estimate by categories | HR | 1.76 | 1.29 | 1.11 | 1.06 | 1.00 | 0.83 |
| 95% CI | 1.50−2.07 | 1.11−1.51 | 0.95−1.28 | 0.93−1.22 | 0.51−1.36 | ||
| Observational estimate by linearity at midpoint | HR | 1.71 | 1.51 | 1.38 | 1.21 | 1.00 | 0.79 |
| 95% CI | 1.51−1.94 | 1.37−1.66 | 1.28−1.49 | 1.16−1.27 | 0.74−0.83 | ||
| MR (G1) estimate by linearity at midpoint | HR | 2.26 | 1.87 | 1.64 | 1.34 | 1.00 | 0.69 |
| 95% CI | 0.52−10.96 | 0.60-6.26 | 0.67−4.24 | 0.79−2.36 | 0.34−1.55 | ||
| MR (G2) estimate by linearity at midpoint | HR | 2.45 | 1.99 | 1.72 | 1.38 | 1.00 | 0.67 |
| 95% CI | 0.52−12.98 | 0.61−7.13 | 0.68−4.70 | 0.79−2.51 | 0.32−1.34 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aspelund, T.; Grübler, M.R.; Smith, A.V.; Gudmundsson, E.F.; Keppel, M.; Cotch, M.F.; Harris, T.B.; Jorde, R.; Grimnes, G.; Joakimsen, R.; et al. Effect of Genetically Low 25-Hydroxyvitamin D on Mortality Risk: Mendelian Randomization Analysis in 3 Large European Cohorts. Nutrients 2019, 11, 74. https://doi.org/10.3390/nu11010074
Aspelund T, Grübler MR, Smith AV, Gudmundsson EF, Keppel M, Cotch MF, Harris TB, Jorde R, Grimnes G, Joakimsen R, et al. Effect of Genetically Low 25-Hydroxyvitamin D on Mortality Risk: Mendelian Randomization Analysis in 3 Large European Cohorts. Nutrients. 2019; 11(1):74. https://doi.org/10.3390/nu11010074
Chicago/Turabian StyleAspelund, Thor, Martin R. Grübler, Albert V. Smith, Elias F. Gudmundsson, Martin Keppel, Mary Frances Cotch, Tamara B. Harris, Rolf Jorde, Guri Grimnes, Ragnar Joakimsen, and et al. 2019. "Effect of Genetically Low 25-Hydroxyvitamin D on Mortality Risk: Mendelian Randomization Analysis in 3 Large European Cohorts" Nutrients 11, no. 1: 74. https://doi.org/10.3390/nu11010074
APA StyleAspelund, T., Grübler, M. R., Smith, A. V., Gudmundsson, E. F., Keppel, M., Cotch, M. F., Harris, T. B., Jorde, R., Grimnes, G., Joakimsen, R., Schirmer, H., Wilsgaard, T., Mathiesen, E. B., Njølstad, I., Løchen, M. -L., März, W., Kleber, M. E., Tomaschitz, A., Grove-Laugesen, D., ... Eiriksdottir, G. (2019). Effect of Genetically Low 25-Hydroxyvitamin D on Mortality Risk: Mendelian Randomization Analysis in 3 Large European Cohorts. Nutrients, 11(1), 74. https://doi.org/10.3390/nu11010074