High Protein Diet and Metabolic Plasticity in Non-Alcoholic Fatty Liver Disease: Myths and Truths



Abstract
:1. Introduction
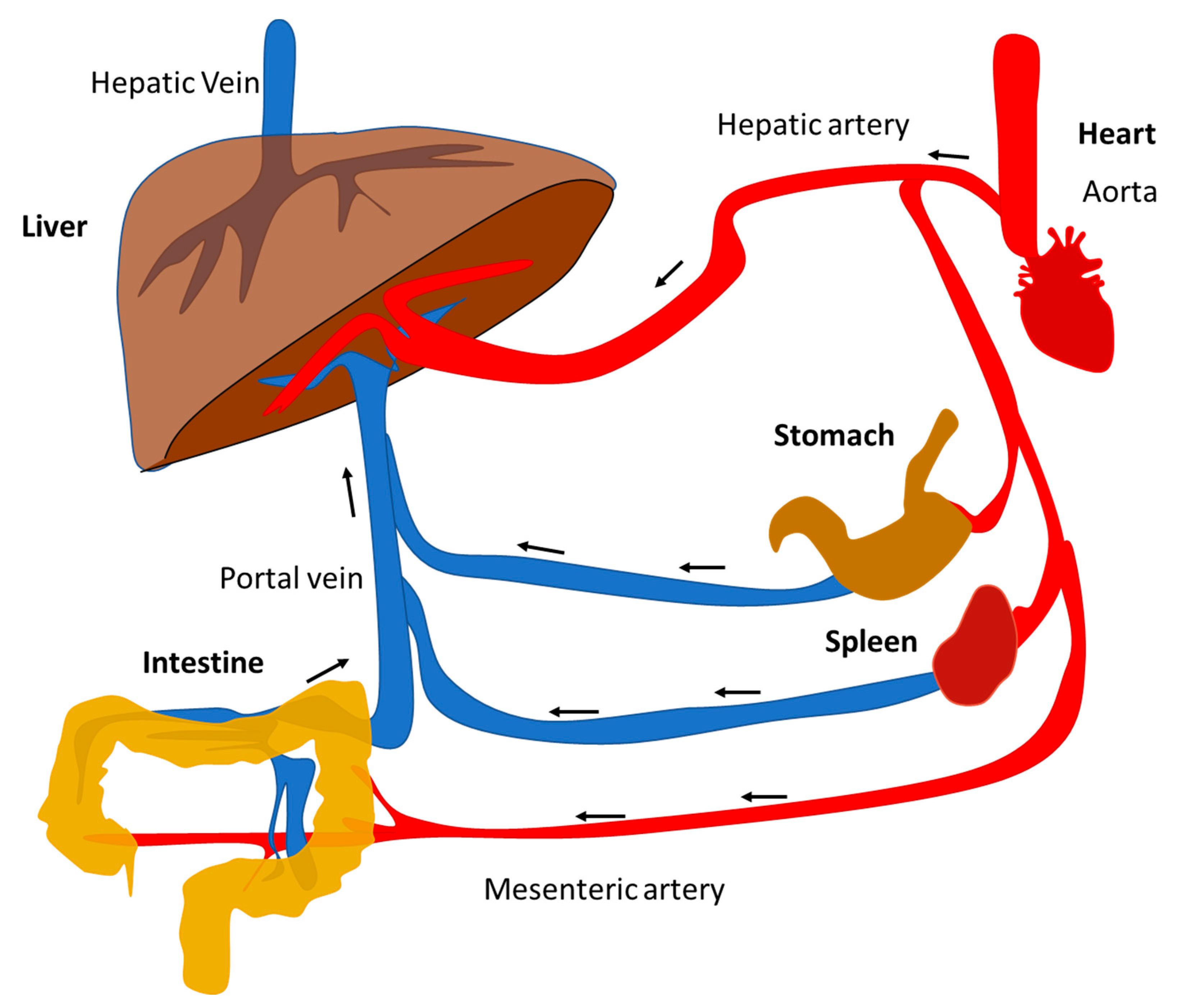
2. Pathobiology of NAFLD
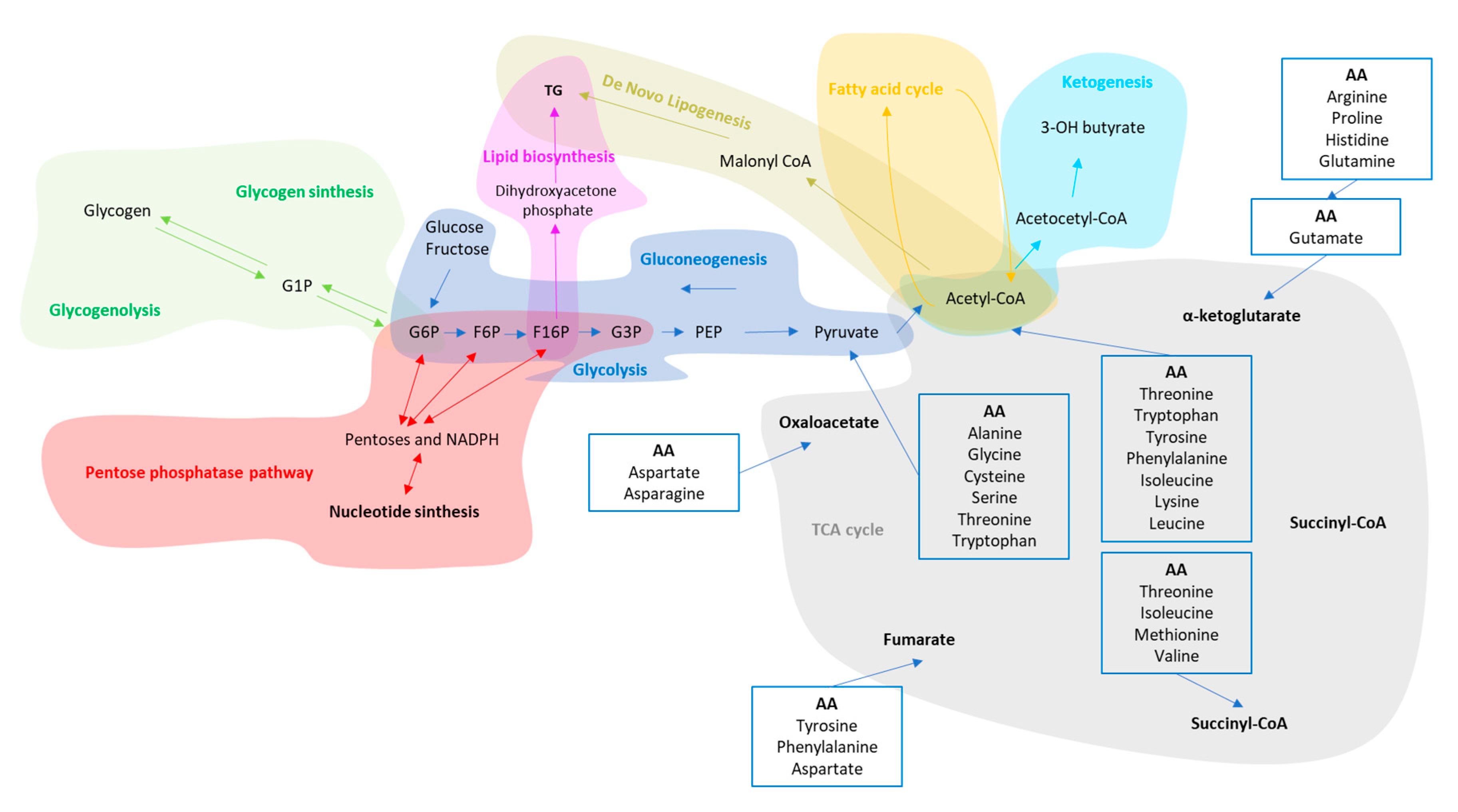
3. Liver Metabolic Plasticity for Carbohydrates, Lipids and Protein
4. Fuel Selection in NAFLD: Evidences and False Myths
5. High Protein Diet in NAFLD, Cure or Disease?
6. High Protein Diets Limitation
7. Skeletal Muscle
8. Physical Activity
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NAFLD | Non-alcoholic fatty liver disease |
| AAAs | Aromatic amino acids |
| AAs | Amino acids |
| ATP | Adenosine triphosphate |
| BCAAs | Branched-Chain amino acids |
| CHO | Carbohydrates |
| CV | Central vein |
| DNL | De novo lipogenesis |
| F1,6P | Fructose-1, 6-phosphate |
| F6P | Fructose-6-phosphate |
| fa-CoA | Fatty acyl-coenzyme |
| FAO | Fatty acid oxidation |
| FAs | Fatty acids |
| FAS | Fatty acid synthesis |
| G3P | Glyceraldehyde-3-phosphate |
| G-3-P | Glycerol 3-phosphate |
| G6P | Glucose-6-phosphate |
| HCC | Hepatocellular carcinoma |
| HDL | High-density lipoprotein |
| Heps | Hepatocytes |
| HSCs | Hepatic stellate cells |
| IMTG | Intramyocellular triacylglycerol |
| KCs | Kupffer cells |
| LDL | Low-density lipoprotein |
| LSEC | Liver sinusoidal endothelial cells |
| MetS | Metabolic syndrome |
| NASH | Non-alcoholic steatohepatitis |
| NEFA | Non-esterified fatty acids |
| PO | Proteins |
| PPP | Pentose phosphate pathway |
| SFA | Saturated fatty acid |
| SM | Skeletal muscle |
| T2DM | Diabetes mellitus type 2 |
| TG | Triglycerides |
References
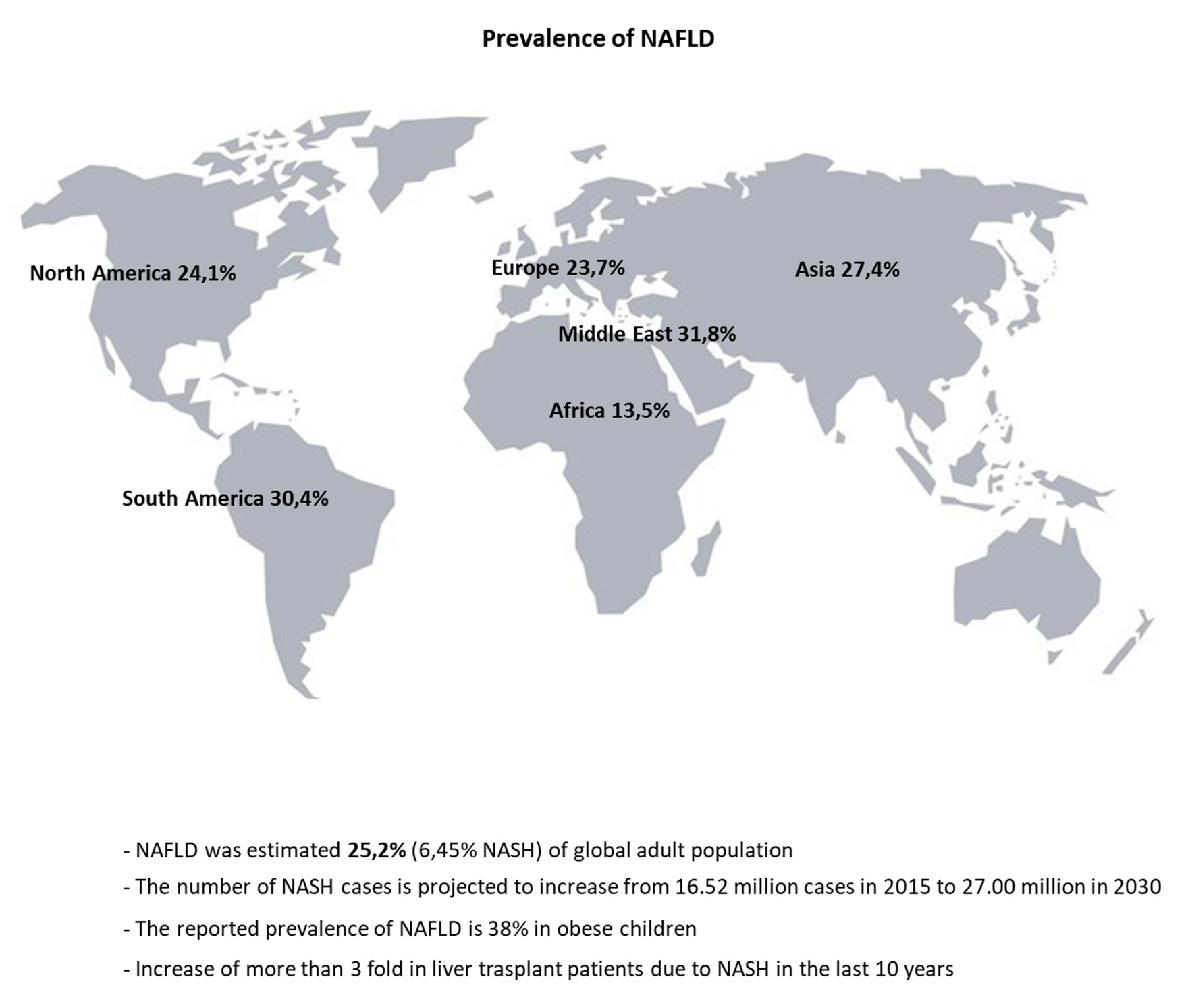
- Puri, P.; Sanyal, A.J. Nonalcoholic fatty liver disease: Definitions, risk factors, and workup. Clin. Liver Dis. 2012, 1, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xue, J.; Chen, P.; Chen, L.; Yan, S.; Liu, L. Prevalence of nonalcoholic fatty liver disease in mainland of China: A meta-analysis of published studies. J. Gastroenterol. Hepatol. 2014, 29, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef]
- Xanthakos, S.; Miles, L.; Bucuvalas, J.; Daniels, S.; Garcia, V.; Inge, T. Histologic spectrum of nonalcoholic fatty liver disease in morbidly obese adolescents. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2006, 4, 226–232. [Google Scholar] [CrossRef]
- Song, P.; Yu, J.; Wang, M.; Chang, X.; Wang, J.; An, L. Prevalence and Correlates of Suspected Nonalcoholic Fatty Liver Disease in Chinese Children. Int. J. Environ. Res. Public Health 2017, 14, 465. [Google Scholar] [CrossRef] [Green Version]
- Fraser, A.; Longnecker, M.P.; Lawlor, D.A. Prevalence of elevated alanine aminotransferase among US adolescents and associated factors: NHANES 1999–2004. Gastroenterology 2007, 133, 1814–1820. [Google Scholar] [CrossRef] [Green Version]
- Conjeevaram Selvakumar, P.; Kabbany, M.; Alkhouri, N. Nonalcoholic Fatty Liver Disease in Children: Not a Small Matter. Pediatric Drugs 2018, 20, 315–329. [Google Scholar] [CrossRef]
- Goldberg, D.; Ditah, I.C.; Saeian, K.; Lalehzari, M.; Aronsohn, A.; Gorospe, E.C.; Charlton, M. Changes in the Prevalence of Hepatitis C Virus Infection, Nonalcoholic Steatohepatitis, and Alcoholic Liver Disease Among Patients With Cirrhosis or Liver Failure on the Waitlist for Liver Transplantation. Gastroenterology 2017, 152, 1090–1099. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, J.G.; Landaverde, C.; Jennings, L.; Goldstein, R.M.; Davis, G.L. Patients with NASH and cryptogenic cirrhosis are less likely than those with hepatitis C to receive liver transplants. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2011, 9, 700–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Hagstrom, H.; Nasr, P.; Ekstedt, M.; Hammar, U.; Stal, P.; Hultcrantz, R.; Kechagias, S. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J. Hepatol. 2017, 67, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Franzen, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Pinzani, M.; Marra, F.; Carloni, V. Signal transduction in hepatic stellate cells. Liver 1998, 18, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.A.; Lee, H.C.; Choe, J.; Kim, M.J.; Lee, M.J.; Chang, H.S.; Bae, I.Y.; Kim, H.K.; An, J.; Shim, J.H.; et al. Association between non-alcoholic fatty liver disease and cancer incidence rate. J. Hepatol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Yatsuji, S.; Hashimoto, E.; Tobari, M.; Taniai, M.; Tokushige, K.; Shiratori, K. Clinical features and outcomes of cirrhosis due to non-alcoholic steatohepatitis compared with cirrhosis caused by chronic hepatitis C. J. Gastroenterol. Hepatol. 2009, 24, 248–254. [Google Scholar] [CrossRef]
- Peverill, W.; Powell, L.W.; Skoien, R. Evolving concepts in the pathogenesis of NASH: Beyond steatosis and inflammation. Int. J. Mol. Sci. 2014, 15, 8591–8638. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Brunt, E.M.; Kleiner, D.E.; Kowdley, K.V.; Chalasani, N.; Lavine, J.E.; Ratziu, V.; McCullough, A. Endpoints and clinical trial design for nonalcoholic steatohepatitis. Hepatology 2011, 54, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.Y.; Suh, K.S.; Kwon, C.H.; Yi, N.J.; Cho, S.Y.; Jang, J.J.; Kim, S.H.; Lee, K.U. The hepatic regeneration power of mild steatotic grafts is not impaired in living-donor liver transplantation. Liver Transplant. Off. Publ. Am. Assoc. Study Liver Dis. Int. Liver Transplant. Soc. 2005, 11, 210–217. [Google Scholar] [CrossRef]
- Tandra, S.; Yeh, M.M.; Brunt, E.M.; Vuppalanchi, R.; Cummings, O.W.; Ünalp-Arida, A.; Wilson, L.A.; Chalasani, N. Presence and significance of microvesicular steatosis in nonalcoholic fatty liver disease. J. Hepatol. 2011, 55, 654–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fromenty, B.; Robin, M.A.; Igoudjil, A.; Mansouri, A.; Pessayre, D. The ins and outs of mitochondrial dysfunction in NASH. Diabetes Metab. 2004, 30, 121–138. [Google Scholar] [CrossRef]
- Gluchowski, N.L.; Becuwe, M.; Walther, T.C.; Farese, R.V., Jr. Lipid droplets and liver disease: From basic biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Pitt, H.A. Hepato-pancreato-biliary fat: The good, the bad and the ugly. HPB (Oxford) 2007, 9, 92–97. [Google Scholar] [CrossRef] [Green Version]
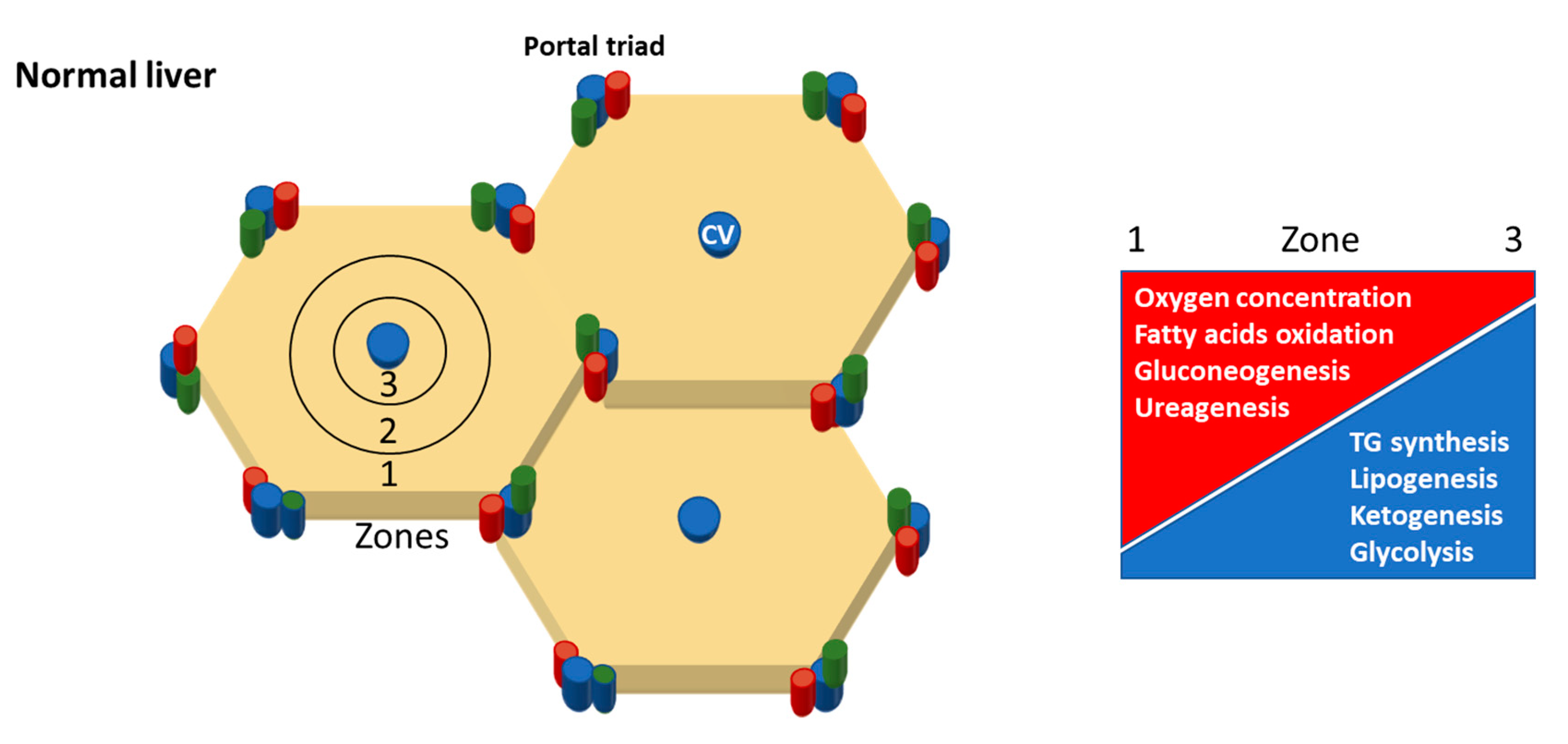
- Rappaport, A.M. The structural and functional unit in the human liver (liver acinus). Anat. Rec. 1958, 130, 673–689. [Google Scholar] [CrossRef]
- Halpern, K.B.; Shenhav, R.; Matcovitch-Natan, O.; Tóth, B.; Lemze, D.; Golan, M.; Massasa, E.E.; Baydatch, S.; Landen, S.; Moor, A.E.; et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 2017, 542, 352. [Google Scholar] [CrossRef]
- Chalasani, N.; Wilson, L.; Kleiner, D.E.; Cummings, O.W.; Brunt, E.M.; Unalp, A. Relationship of steatosis grade and zonal location to histological features of steatohepatitis in adult patients with non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 829–834. [Google Scholar] [CrossRef] [Green Version]
- Carter-Kent, C.; Brunt, E.M.; Yerian, L.M.; Alkhouri, N.; Angulo, P.; Kohli, R.; Ling, S.C.; Xanthakos, S.A.; Whitington, P.F.; Charatcharoenwitthaya, P.; et al. Relations of steatosis type, grade, and zonality to histological features in pediatric nonalcoholic fatty liver disease. J. Pediatric Gastroenterol. Nutr. 2011, 52, 190–197. [Google Scholar] [CrossRef]
- Hall, Z.; Bond, N.J.; Ashmore, T.; Sanders, F.; Ament, Z.; Wang, X.; Murray, A.J.; Bellafante, E.; Virtue, S.; Vidal-Puig, A.; et al. Lipid zonation and phospholipid remodeling in nonalcoholic fatty liver disease. Hepatology 2017, 65, 1165–1180. [Google Scholar] [CrossRef]
- Softic, S.; Meyer, J.G.; Wang, G.X.; Gupta, M.K.; Batista, T.M.; Lauritzen, H.; Fujisaka, S.; Serra, D.; Herrero, L.; Willoughby, J.; et al. Dietary Sugars Alter Hepatic Fatty Acid Oxidation via Transcriptional and Post-translational Modifications of Mitochondrial Proteins. Cell Metab. 2019, 30, 735–753. [Google Scholar] [CrossRef]
- Koopman, K.E.; Caan, M.W.; Nederveen, A.J.; Pels, A.; Ackermans, M.T.; Fliers, E.; la Fleur, S.E.; Serlie, M.J. Hypercaloric diets with increased meal frequency, but not meal size, increase intrahepatic triglycerides: A randomized controlled trial. Hepatology 2014, 60, 545–553. [Google Scholar] [CrossRef] [PubMed]
- De Ruyter, J.C.; Olthof, M.R.; Seidell, J.C.; Katan, M.B. A Trial of Sugar-free or Sugar-Sweetened Beverages and Body Weight in Children. N. Engl. J. Med. 2012, 367, 1397–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebbeling, C.B.; Feldman, H.A.; Chomitz, V.R.; Antonelli, T.A.; Gortmaker, S.L.; Osganian, S.K.; Ludwig, D.S. A Randomized Trial of Sugar-Sweetened Beverages and Adolescent Body Weight. N. Engl. J. Med. 2012, 367, 1407–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlton, M.R.; Burns, J.M.; Pedersen, R.A.; Watt, K.D.; Heimbach, J.K.; Dierkhising, R.A. Frequency and Outcomes of Liver Transplantation for Nonalcoholic Steatohepatitis in the United States. Gastroenterology 2011, 141, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.T.; Chacko, S.K.; Sunehag, A.L.; Haymond, M.W. Measurements of Gluconeogenesis and Glycogenolysis: A Methodological Review. Diabetes 2015, 64, 3996–4010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.-M.; Krüger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. 2015, 90, 927–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahill, G.F., Jr. Starvation in man. N. Engl. J. Med. 1970, 282, 668–675. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [Green Version]
- Bentourkia, M.; Tremblay, S.; Pifferi, F.; Rousseau, J.; Lecomte, R.; Cunnane, S. PET study of 11C-acetoacetate kinetics in rat brain during dietary treatments affecting ketosis. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E796–E801. [Google Scholar] [CrossRef] [Green Version]
- Reichard, G.A., Jr.; Owen, O.E.; Haff, A.C.; Paul, P.; Bortz, W.M. Ketone-body production and oxidation in fasting obese humans. J. Clin. Investig. 1974, 53, 508–515. [Google Scholar] [CrossRef]
- Barrows, B.R.; Parks, E.J. Contributions of different fatty acid sources to very low-density lipoprotein-triacylglycerol in the fasted and fed states. J. Clin. Endocrinol. Metab. 2006, 91, 1446–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashek, D.G. Hepatic Fatty Acid Trafficking: Multiple Forks in the Road. Adv. Nutr. 2013, 4, 697–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.; Newsholme, E.A. Glycerol kinase activities in rat heart and adipose tissue. Biochem. J. 1967, 104, 2C–4C. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newsholme, E.A.; Taylor, K. Glycerol kinase activities in muscles from vertebrates and invertebrates. Biochem. J. 1969, 112, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Neese, R.A.; Turner, S.; Dare, D.; Hellerstein, M.K. Short-term alterations in carbohydrate energy intake in humans. Striking effects on hepatic glucose production, de novo lipogenesis, lipolysis, and whole-body fuel selection. J. Clin. Investig. 1995, 96, 2735–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chascione, C.; Elwyn, D.H.; Davila, M.; Gil, K.M.; Askanazi, J.; Kinney, J.M. Effect of carbohydrate intake on de novo lipogenesis in human adipose tissue. Am. J. Physiol. Endocrinol. Metab. 1987, 253, E664–E669. [Google Scholar] [CrossRef] [PubMed]
- Acheson, K.J.; Schutz, Y.; Bessard, T.; Anantharaman, K.; Flatt, J.P.; Jequier, E. Glycogen storage capacity and de novo lipogenesis during massive carbohydrate overfeeding in man. Am. J. Clin. Nutr. 1988, 48, 240–247. [Google Scholar] [CrossRef] [Green Version]
- Parks, E.J.; Krauss, R.M.; Christiansen, M.P.; Neese, R.A.; Hellerstein, M.K. Effects of a low-fat, high-carbohydrate diet on VLDL-triglyceride assembly, production, and clearance. J. Clin. Investig. 1999, 104, 1087–1096. [Google Scholar] [CrossRef] [Green Version]
- Ten Have, G.A.; Engelen, M.P.; Luiking, Y.C.; Deutz, N.E. Absorption kinetics of amino acids, peptides, and intact proteins. Int. J. Sport Nutr. Exerc. Metab. 2007, 17, S23–S36. [Google Scholar] [CrossRef]
- Wu, G. Intestinal mucosal amino acid catabolism. J. Nutr. 1998, 128, 1249–1252. [Google Scholar] [CrossRef] [Green Version]
- Felier, D.D.; Feist, E. Conversion of aromatic amino acids to fatty acids by adipose tissue. Biochim. Biophys. Acta 1963, 70, 85. [Google Scholar] [CrossRef]
- Gannon, M.C.; Nuttall, F.Q. Amino acid ingestion and glucose metabolism—A review. IUBMB Life 2010, 62, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Yudkoff, M.; Daikhin, Y.; Melø, T.M.; Nissim, I.; Sonnewald, U.; Nissim, I. The ketogenic diet and brain metabolism of amino acids: Relationship to the anticonvulsant effect. Annu. Rev. Nutr. 2007, 27, 415–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodney, S.; Boneh, A. Amino Acid Profiles in Patients with Urea Cycle Disorders at Admission to Hospital due to Metabolic Decompensation. JIMD Rep. 2013, 9, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G. Amino acids: Metabolism, functions, and nutrition. Amino Acids 2009, 37, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Floyd, J.C., Jr.; Fajans, S.S.; Conn, J.W.; Knopf, R.F.; Rull, J. Stimulation of insulin secretion by amino acids. J. Clin. Investig. 1966, 45, 1487–1502. [Google Scholar] [CrossRef]
- Nishitani, S.; Takehana, K.; Fujitani, S.; Sonaka, I. Branched-chain amino acids improve glucose metabolism in rats with liver cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1292–G1300. [Google Scholar] [CrossRef]
- Fischer, J.E.; Funovics, J.M.; Aguirre, A.; James, J.H.; Keane, J.M.; Wesdorp, R.I.; Yoshimura, N.; Westman, T. The role of plasma amino acids in hepatic encephalopathy. Surgery 1975, 78, 276–290. [Google Scholar]
- Yamakado, M.; Tanaka, T.; Nagao, K.; Imaizumi, A.; Komatsu, M.; Daimon, T.; Miyano, H.; Tani, M.; Toda, A.; Yamamoto, H.; et al. Plasma amino acid profile associated with fatty liver disease and co-occurrence of metabolic risk factors. Sci. Rep. 2017, 7, 14485. [Google Scholar] [CrossRef] [Green Version]
- Rao, M.; Afshin, A.; Singh, G.; Mozaffarian, D. Do healthier foods and diet patterns cost more than less healthy options? A systematic review and meta-analysis. BMJ Open 2013, 3, e004277. [Google Scholar] [CrossRef]
- Wehmeyer, M.H.; Zyriax, B.C.; Jagemann, B.; Roth, E.; Windler, E.; Schulze Zur Wiesch, J.; Lohse, A.W.; Kluwe, J. Nonalcoholic fatty liver disease is associated with excessive calorie intake rather than a distinctive dietary pattern. Medicine 2016, 95, e3887. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.L.; Howe, L.D.; Fraser, A.; Macdonald-Wallis, C.; Callaway, M.P.; Sattar, N.; Day, C.; Tilling, K.; Lawlor, D.A. Childhood energy intake is associated with nonalcoholic fatty liver disease in adolescents. J. Nutr. 2015, 145, 983–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen-Duy, T.-B.; Nichaman, M.Z.; Church, T.S.; Blair, S.N.; Ross, R. Visceral fat and liver fat are independent predictors of metabolic risk factors in men. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E1065–E1071. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Fontana, L.; Young, V.L.; Coggan, A.R.; Kilo, C.; Patterson, B.W.; Mohammed, B.S. Absence of an effect of liposuction on insulin action and risk factors for coronary heart disease. N. Engl. J. Med. 2004, 350, 2549–2557. [Google Scholar] [CrossRef]
- Biddinger, S.B.; Hernandez-Ono, A.; Rask-Madsen, C.; Haas, J.T.; Aleman, J.O.; Suzuki, R.; Scapa, E.F.; Agarwal, C.; Carey, M.C.; Stephanopoulos, G.; et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. 2008, 7, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Vatner, D.F.; Majumdar, S.K.; Kumashiro, N.; Petersen, M.C.; Rahimi, Y.; Gattu, A.K.; Bears, M.; Camporez, J.P.; Cline, G.W.; Jurczak, M.J.; et al. Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids. Proc. Natl. Acad. Sci. USA 2015, 112, 1143–1148. [Google Scholar] [CrossRef] [Green Version]
- Jin, E.S.; Browning, J.D.; Murphy, R.E.; Malloy, C.R. Fatty liver disrupts glycerol metabolism in gluconeogenic and lipogenic pathways in humans. J. Lipid Res. 2018, 59, 1685–1694. [Google Scholar] [CrossRef] [Green Version]
- Korenblat, K.M.; Fabbrini, E.; Mohammed, B.S.; Klein, S. Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology 2008, 134, 1369–1375. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, M.; Vandenborne, K.; Goldsmith, R.; Simoneau, J.-A.; Heymsfield, S.; Joanisse, D.R.; Hirsch, J.; Murphy, E.; Matthews, D.; Segal, K.R.; et al. Effects of experimental weight perturbation on skeletal muscle work efficiency in human subjects. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R183–R192. [Google Scholar] [CrossRef] [Green Version]
- Jin, E.S.; Lee, M.H.; Murphy, R.E.; Malloy, C.R. Pentose phosphate pathway activity parallels lipogenesis but not antioxidant processes in rat liver. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E543–E551. [Google Scholar] [CrossRef] [Green Version]
- Mannisto, V.T.; Simonen, M.; Hyysalo, J.; Soininen, P.; Kangas, A.J.; Kaminska, D.; Matte, A.K.; Venesmaa, S.; Kakela, P.; Karja, V.; et al. Ketone body production is differentially altered in steatosis and non-alcoholic steatohepatitis in obese humans. Liver Int. Off. J. Int. Assoc. Study Liver 2015, 35, 1853–1861. [Google Scholar] [CrossRef]
- Inokuchi, T.; Orita, M.; Imamura, K.; Takao, T.; Isogai, S. Resistance to ketosis in moderately obese patients: Influence of fatty liver. Intern. Med. 1992, 31, 978–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satapati, S.; Sunny, N.E.; Kucejova, B.; Fu, X.; He, T.T.; Méndez-Lucas, A.; Shelton, J.M.; Perales, J.C.; Browning, J.D.; Burgess, S.C. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012, 53, 1080–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, G.F.; Carpentier, A.; Adeli, K.; Giacca, A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr. Rev. 2002, 23, 201–229. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Linfoot, P.; Dare, D.; Aghajanian, K. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am. J. Clin. Nutr. 2003, 77, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011, 14, 804–810. [Google Scholar] [CrossRef] [Green Version]
- Gaggini, M.; Carli, F.; Rosso, C.; Buzzigoli, E.; Marietti, M.; Della Latta, V.; Ciociaro, D.; Abate, M.L.; Gambino, R.; Cassader, M.; et al. Altered amino acid concentrations in NAFLD: Impact of obesity and insulin resistance. Hepatology 2018, 67, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Wiklund, P.; Autio, R.; Borra, R.; Ojanen, X.; Xu, L.; Törmäkangas, T.; Alen, M. Adipose Tissue Dysfunction and Altered Systemic Amino Acid Metabolism Are Associated with Non-Alcoholic Fatty Liver Disease. PLoS ONE 2015, 10, e0138889. [Google Scholar] [CrossRef]
- De Chiara, F.; Heebøll, S.; Marrone, G.; Montoliu, C.; Hamilton-Dutoit, S.; Ferrandez, A.; Andreola, F.; Rombouts, K.; Grønbæk, H.; Felipo, V.; et al. Urea cycle dysregulation in non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 905–915. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holt, H.B.; Wild, S.H.; Wood, P.J.; Zhang, J.; Darekar, A.A.; Dewbury, K.; Poole, R.B.; Holt, R.I.; Phillips, D.I.; Byrne, C.D. Non-esterified fatty acid concentrations are independently associated with hepatic steatosis in obese subjects. Diabetologia 2006, 49, 141–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittendorfer, B.; Magkos, F.; Fabbrini, E.; Mohammed, B.S.; Klein, S. Relationship between body fat mass and free fatty acid kinetics in men and women. Obesity 2009, 17, 1872–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claessens, M.; van Baak, M.A.; Monsheimer, S.; Saris, W.H.M. The effect of a low-fat, high-protein or high-carbohydrate ad libitum diet on weight loss maintenance and metabolic risk factors. Int. J. Obes. 2009, 33, 296. [Google Scholar] [CrossRef] [Green Version]
- Tentolouris, N.; Tsigos, C.; Perea, D.; Koukou, E.; Kyriaki, D.; Kitsou, E.; Daskas, S.; Daifotis, Z.; Makrilakis, K.; Raptis, S.A.; et al. Differential effects of high-fat and high-carbohydrate isoenergetic meals on cardiac autonomic nervous system activity in lean and obese women. Metab. Clin. Exp. 2003, 52, 1426–1432. [Google Scholar] [CrossRef]
- Marques-Lopes, I.; Forga, L.; Martinez, J.A. Thermogenesis induced by a high-carbohydrate meal in fasted lean and overweight young men: Insulin, body fat, and sympathetic nervous system involvement. Nutrition 2003, 19, 25–29. [Google Scholar] [CrossRef]
- Browning, J.D.; Baker, J.A.; Rogers, T.; Davis, J.; Satapati, S.; Burgess, S.C. Short-term weight loss and hepatic triglyceride reduction: Evidence of a metabolic advantage with dietary carbohydrate restriction. Am. J. Clin. Nutr. 2011, 93, 1048–1052. [Google Scholar] [CrossRef]
- Jang, E.C.; Jun, D.W.; Lee, S.M.; Cho, Y.K.; Ahn, S.B. Comparison of efficacy of low-carbohydrate and low-fat diet education programs in non-alcoholic fatty liver disease: A randomized controlled study. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2018, 48, E22–E29. [Google Scholar] [CrossRef] [Green Version]
- Tendler, D.; Lin, S.; Yancy, W.S., Jr.; Mavropoulos, J.; Sylvestre, P.; Rockey, D.C.; Westman, E.C. The effect of a low-carbohydrate, ketogenic diet on nonalcoholic fatty liver disease: A pilot study. Dig. Dis. Sci. 2007, 52, 589–593. [Google Scholar] [CrossRef]
- Hartweg, J.; Perera, R.; Montori, V.; Dinneen, S.; Neil, H.A.; Farmer, A. Omega-3 polyunsaturated fatty acids (PUFA) for type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2008. [Google Scholar] [CrossRef]
- Liu, K.; Wang, B.; Zhou, R.; Lang, H.D.; Ran, L.; Wang, J.; Li, L.; Kang, C.; Zhu, X.H.; Zhang, Q.Y.; et al. Effect of combined use of a low-carbohydrate, high-protein diet with omega-3 polyunsaturated fatty acid supplementation on glycemic control in newly diagnosed type 2 diabetes: A randomized, double-blind, parallel-controlled trial. Am. J. Clin. Nutr. 2018, 108, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Greenson, J.K.; Omo, J.T.; Chao, C.; Peterman, D.; Anderson, L.; Foess-Wood, L.; Sherbondy, M.A.; Conjeevaram, H.S. Metabolic syndrome is associated with greater histologic severity, higher carbohydrate, and lower fat diet in patients with NAFLD. Am. J. Gastroenterol. 2006, 101, 2247–2253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feingold, K.R.; Grunfeld, C. Lipids: A key player in the battle between the host and microorganisms. J. Lipid Res. 2012, 53, 2487–2489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, J.; Das, S.; Guha, R.; Ghosh, D.; Naskar, K.; Das, A.; Roy, S. Hyperlipidemia offers protection against Leishmania donovani infection: Role of membrane cholesterol. J. Lipid Res. 2012, 53, 2560–2572. [Google Scholar] [CrossRef] [Green Version]
- Veech, R.L. The therapeutic implications of ketone bodies: The effects of ketone bodies in pathological conditions: Ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 309–319. [Google Scholar] [CrossRef]
- Keene, D.L. A Systematic Review of the Use of the Ketogenic Diet in Childhood Epilepsy. Pediatric Neurol. 2006, 35, 1–5. [Google Scholar] [CrossRef]
- Trumbo, P.; Schlicker, S.; Yates, A.A.; Poos, M. Dietary reference intakes for energy, carbohydrate, fiber, fat, fatty acids, cholesterol, protein and amino acids. J. Am. Diet. Assoc. 2002, 102, 1621–1630. [Google Scholar] [CrossRef]
- Moghaddam, E.; Vogt, J.A.; Wolever, T.M. The effects of fat and protein on glycemic responses in nondiabetic humans vary with waist circumference, fasting plasma insulin, and dietary fiber intake. J. Nutr. 2006, 136, 2506–2511. [Google Scholar] [CrossRef]
- Atkins, R.C. Dr. Atkins’ New Diet Revolution; M. Evans: New York, NY, USA, 1995. [Google Scholar]
- Agatston, A. The South Beach Diet; Random House: New York, NY, USA, 2003. [Google Scholar]
- Sears, B. The Zone Diet: 150 Fast and Simple Healthy Recipes from the Bestselling Author of “The Zone” and “Mastering the Zone”; Regan Books: New York, NY, USA, 1999. [Google Scholar]
- Stillman, I.M.; Baker, S.S. The Doctor’s Quick Weight Loss Diet, by Irwin Maxwell Stillman with Samm Sinclair Baker; Prentice-Hall: Englewood Cliffs, NJ, USA, 1967. [Google Scholar]
- Rickman, F.; Mitchell, N.; Dingman, J.; Dalen, J.E. Changes in serum cholesterol during the Stillman Diet. Nutr. Rev. 1974, 32, 24–26. [Google Scholar] [CrossRef]
- Agatston, A. The South Beach Diet: The Delicious, Doctor-Designed, Foolproof Plan for Fast and Healthy Weight Loss; Macmillan: London, UK, 2005. [Google Scholar]
- Hu, F.B. Protein, body weight, and cardiovascular health. Am. J. Clin. Nutr. 2005, 82, 242S–247S. [Google Scholar] [CrossRef]
- Pasiakos, S.M.; Lieberman, H.R.; Fulgoni, V.L., 3rd. Higher-protein diets are associated with higher HDL cholesterol and lower BMI and waist circumference in US adults. J. Nutr. 2015, 145, 605–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowen, J.; Noakes, M.; Clifton, P.M. Appetite Regulatory Hormone Responses to Various Dietary Proteins Differ by Body Mass Index Status Despite Similar Reductions in ad Libitum Energy Intake. J. Clin. Endocrinol. Metab. 2006, 91, 2913–2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerterp-Plantenga, M.S.; Rolland, V.; Wilson, S.A.; Westerterp, K.R. Satiety related to 24 h diet-induced thermogenesis during high protein/carbohydrate vs high fat diets measured in a respiration chamber. Eur. J. Clin. Nutr. 1999, 53, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latner, J.D.; Schwartz, M. The effects of a high-carbohydrate, high-protein or balanced lunch upon later food intake and hunger ratings. Appetite 1999, 33, 119–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkelsen, P.B.; Toubro, S.; Astrup, A. Effect of fat-reduced diets on 24-h energy expenditure: Comparisons between animal protein, vegetable protein, and carbohydrate. Am. J. Clin. Nutr. 2000, 72, 1135–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clifton, P.M.; Bastiaans, K.; Keogh, J.B. High protein diets decrease total and abdominal fat and improve CVD risk profile in overweight and obese men and women with elevated triacylglycerol. Nutr. Metab. Cardiovasc. Dis. NMCD 2009, 19, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Tharrey, M.; Mariotti, F.; Mashchak, A.; Barbillon, P.; Delattre, M.; Fraser, G.E. Patterns of plant and animal protein intake are strongly associated with cardiovascular mortality: The Adventist Health Study-2 cohort. Int. J. Epidemiol. 2018, 47, 1603–1612. [Google Scholar] [CrossRef]
- Huang, T.; Yang, B.; Zheng, J.; Li, G.; Wahlqvist, M.L.; Li, D. Cardiovascular disease mortality and cancer incidence in vegetarians: A meta-analysis and systematic review. Ann. Nutr. Metab. 2012, 60, 233–240. [Google Scholar] [CrossRef]
- Shang, X.; Scott, D.; Hodge, A.M.; English, D.R.; Giles, G.G.; Ebeling, P.R.; Sanders, K.M. Dietary protein intake and risk of type 2 diabetes: Results from the Melbourne Collaborative Cohort Study and a meta-analysis of prospective studies. Am. J. Clin. Nutr. 2016, 104, 1352–1365. [Google Scholar] [CrossRef]
- Navas-Carretero, S.; San-Cristobal, R.; Livingstone, K.M.; Celis-Morales, C.; Marsaux, C.F.; Macready, A.L.; Fallaize, R.; O’Donovan, C.B.; Forster, H.; Woolhead, C.; et al. Higher vegetable protein consumption, assessed by an isoenergetic macronutrient exchange model, is associated with a lower presence of overweight and obesity in the web-based Food4me European study. Int. J. Food Sci. Nutr. 2019, 70, 240–253. [Google Scholar] [CrossRef] [Green Version]
- Sinha, R.; Cross, A.J.; Graubard, B.I.; Leitzmann, M.F.; Schatzkin, A. Meat intake and mortality: A prospective study of over half a million people. Arch. Intern. Med. 2009, 169, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, N.; Chiu, S.; Williams, P.T.; M King, S.; Krauss, R.M. Effects of red meat, white meat, and nonmeat protein sources on atherogenic lipoprotein measures in the context of low compared with high saturated fat intake: A randomized controlled trial. Am. J. Clin. Nutr. 2019, 110, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Zelber-Sagi, S.; Nitzan-Kaluski, D.; Goldsmith, R.; Webb, M.; Blendis, L.; Halpern, Z.; Oren, R. Long term nutritional intake and the risk for non-alcoholic fatty liver disease (NAFLD): A population based study. J. Hepatol. 2007, 47, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Zhang, Y.; Wang, W.; Zhuang, P.; Wu, F.; Jiao, J. Plant-sourced and animal-sourced monounsaturated fatty acid intakes in relation to mortality: A prospective nationwide cohort study. Eur. J. Nutr. 2019. [Google Scholar] [CrossRef] [PubMed]
- Guasch-Ferré, C.M.; Zong, L.G.; Willett, J.W.; Zock, B.P.; Wanders, B.A.; Hu, B.F.; Sun, B.Q. Associations of Monounsaturated Fatty Acids From Plant and Animal Sources With Total and Cause-Specific Mortality in Two US Prospective Cohort Studies. Circ. Res. 2019, 124, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Zhu, J.; Meng, D.; Yu, C.; Li, Y. Association of homocysteine level with biopsy-proven non-alcoholic fatty liver disease: A meta-analysis. J. Clin. Biochem. Nutr. 2016, 58, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Tan, S.; Xu, Y.; Chandra, A.; Shi, C.; Song, B.; Qin, J.; Gao, Y. Vitamin B supplementation, homocysteine levels, and the risk of cerebrovascular disease: A meta-analysis. Neurology 2013, 81, 1298–1307. [Google Scholar] [CrossRef]
- Yang, J.; Hu, X.; Zhang, Q.; Cao, H.; Wang, J.; Liu, B. Homocysteine level and risk of fracture: A meta-analysis and systematic review. Bone 2012, 51, 376–382. [Google Scholar] [CrossRef]
- Adeva, M.M.; Souto, G. Diet-induced metabolic acidosis. Clin. Nutr. 2011, 30, 416–421. [Google Scholar] [CrossRef]
- Altorf-van der Kuil, W.; Brink, E.J.; Boetje, M.; Siebelink, E.; Bijlsma, S.; Engberink, M.F.; van’t Veer, P.; Tome, D.; Bakker, S.J.; van Baak, M.A.; et al. Identification of biomarkers for intake of protein from meat, dairy products and grains: A controlled dietary intervention study. Br. J. Nutr. 2013, 110, 810–822. [Google Scholar] [CrossRef] [Green Version]
- Remer, T. Influence of nutrition on acid-base balance–metabolic aspects. Eur. J. Nutr. 2001, 40, 214–220. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, F.; Thomsen, K.L.; Habtesion, A.; Jones, H.; Davies, N.; Gracia-Sancho, J.; Manicardi, N.; Hall, A.; Andreola, F.; Paish, H.L.; et al. Ammonia Scavenging Prevents Progression of Fibrosis in Experimental Nonalcoholic Fatty Liver Disease. Hepatology 2019. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.P.; Marchesini, G.; Fabbri, A.; Rondelli, A.; Bugianesi, E.; Zoli, M.; Pisi, E. Vegetable versus animal protein diet in cirrhotic patients with chronic encephalopathy. A randomized cross-over comparison. J. Intern. Med. 1993, 233, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Greenberger, N.J.; Carley, J.; Schenker, S.; Bettinger, I.; Stamnes, C.; Beyer, P. Effect of vegetable and animal protein diets in chronic hepatic encephalopathy. Am. J. Dig. Dis. 1977, 22, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.L., Jr.; Minco, D.; Fresard, K.M.; Banwell, J.G. Effects of vegetable diets on nitrogen metabolism in cirrhotic subjects. Gastroenterology 1985, 89, 538–544. [Google Scholar] [CrossRef]
- Kani, A.H.; Alavian, S.M.; Esmaillzadeh, A.; Adibi, P.; Azadbakht, L. Effects of a novel therapeutic diet on liver enzymes and coagulating factors in patients with non-alcoholic fatty liver disease: A parallel randomized trial. Nutrition 2014, 30, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Kontessis, P.; Jones, S.; Dodds, R.; Trevisan, R.; Nosadini, R.; Fioretto, P.; Borsato, M.; Sacerdoti, D.; Viberti, G. Renal, metabolic and hormonal responses to ingestion of animal and vegetable proteins. Kidney Int. 1990, 38, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Bosch, J.P.; Saccaggi, A.; Lauer, A.; Ronco, C.; Belledonne, M.; Glabman, S. Renal functional reserve in humans. Effect of protein intake on glomerular filtration rate. Am. J. Med. 1983, 75, 943–950. [Google Scholar] [CrossRef]
- Uribe, M.; Márquez, M.A.; Ramos, G.G.; Ramos-Uribe, M.H.; Vargas, F.; Villalobos, A.; Ramos, C. Treatment of chronic portal—Systemic encephalopathy with vegetable and animal protein diets. Dig. Dis. Sci. 1982, 27, 1109–1116. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Nagao, Y.; Matsuoka, H.; Ide, T.; Sata, M. Branched-chain amino acid-enriched supplementation improves insulin resistance in patients with chronic liver disease. Int. J. Mol. Med. 2008, 22, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, Y.; Hiroshima, Y.; Matsuo, K.; Kawaguchi, D.; Murakami, T.; Yabushita, Y.; Endo, I.; Taguri, M.; Koda, K.; Tanaka, K. A Randomized Clinical Trial of Preoperative Administration of Branched-Chain Amino Acids to Prevent Postoperative Ascites in Patients with Liver Resection for Hepatocellular Carcinoma. Ann. Surg. Oncol. 2016, 23, 3727–3735. [Google Scholar] [CrossRef] [PubMed]
- Hayaishi, S.; Chung, H.; Kudo, M.; Ishikawa, E.; Takita, M.; Ueda, T.; Kitai, S.; Inoue, T.; Yada, N.; Hagiwara, S.; et al. Oral branched-chain amino acid granules reduce the incidence of hepatocellular carcinoma and improve event-free survival in patients with liver cirrhosis. Dig. Dis. 2011, 29, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Imanaka, K.; Ohkawa, K.; Tatsumi, T.; Katayama, K.; Inoue, A.; Imai, Y.; Oshita, M.; Iio, S.; Mita, E.; Fukui, H.; et al. Impact of branched-chain amino acid supplementation on survival in patients with advanced hepatocellular carcinoma treated with sorafenib: A multicenter retrospective cohort study. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2016, 46, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Marliss, E.B.; Chevalier, S.; Gougeon, R.; Morais, J.A.; Lamarche, M.; Adegoke, O.A.J.; Wu, G. Elevations of plasma methylarginines in obesity and ageing are related to insulin sensitivity and rates of protein turnover. Diabetologia 2006, 49, 351–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Collins, J.K.; Perkins-Veazie, P.; Siddiq, M.; Dolan, K.D.; Kelly, K.A.; Heaps, C.L.; Meininger, C.J. Dietary supplementation with watermelon pomace juice enhances arginine availability and ameliorates the metabolic syndrome in Zucker diabetic fatty rats. J. Nutr. 2007, 137, 2680–2685. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Flores, M.; Cruz, M.; Duran-Reyes, G.; Munguia-Miranda, C.; Loza-Rodriguez, H.; Pulido-Casas, E.; Torres-Ramirez, N.; Gaja-Rodriguez, O.; Kumate, J.; Baiza-Gutman, L.A.; et al. Oral supplementation with glycine reduces oxidative stress in patients with metabolic syndrome, improving their systolic blood pressure. Can. J. Physiol. Pharmacol. 2013, 91, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Yan-Do, R.; Duong, E.; Manning Fox, J.E.; Dai, X.; Suzuki, K.; Khan, S.; Bautista, A.; Ferdaoussi, M.; Lyon, J.; Wu, X.; et al. A Glycine-Insulin Autocrine Feedback Loop Enhances Insulin Secretion From Human beta-Cells and Is Impaired in Type 2 Diabetes. Diabetes 2016, 65, 2311–2321. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Ortiz, M.; Medina-Santillan, R.; Martinez-Abundis, E.; von Drateln, C.R. Effect of glycine on insulin secretion and action in healthy first-degree relatives of type 2 diabetes mellitus patients. Horm. Metab. Res. 2001, 33, 358–360. [Google Scholar] [CrossRef]
- Okekunle, A.P.; Li, Y.; Liu, L.; Du, S.; Wu, X.; Chen, Y.; Qi, J.; Sun, C.; Feng, R. Abnormal circulating amino acid profiles in multiple metabolic disorders. Diabetes Res. Clin. Pract. 2017, 132, 45–58. [Google Scholar] [CrossRef]
- Liu, Z.; Jeppesen, P.B.; Gregersen, S.; Bach Larsen, L.; Hermansen, K. Chronic Exposure to Proline Causes Aminoacidotoxicity and Impaired Beta-Cell Function: Studies In Vitro. Rev. Diabet. Stud. 2016, 13, 66–78. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Jinzu, H.; Nagao, K.; Noguchi, Y.; Shimba, N.; Miyano, H.; Watanabe, T.; Iseki, K. Plasma amino acid profiles are associated with insulin, C-peptide and adiponectin levels in type 2 diabetic patients. Nutr. Diabetes 2014, 4, e133. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.J. High Dietary Protein Intake and Protein-Related Acid Load on Bone Health. Curr. Osteoporos. Rep. 2017, 15, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Barzel, U.S.; Massey, L.K. Excess dietary protein can adversely affect bone. J. Nutr. 1998, 128, 1051–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, R.; Cross, A.J.; Pollock, J.R.; Bingham, S. Dose-dependent effect of dietary meat on endogenous colonic N-nitrosation. Carcinogenesis 2001, 22, 199–202. [Google Scholar] [CrossRef]
- Norat, T.; Riboli, E. Meat consumption and colorectal cancer: A review of epidemiologic evidence. Nutr. Rev. 2001, 59, 37–47. [Google Scholar] [CrossRef]
- Meyer, T.W.; Anderson, S.; Brenner, B.M. Dietary protein intake and progressive glomerular sclerosis: The role of capillary hypertension and hyperperfusion in the progression of renal disease. Ann. Intern. Med. 1983, 98, 832–838. [Google Scholar] [CrossRef] [Green Version]
- Klahr, S.; Levey, A.S.; Beck, G.J.; Caggiula, A.W.; Hunsicker, L.; Kusek, J.W.; Striker, G. The effects of dietary protein restriction and blood-pressure control on the progression of chronic renal disease. Modification of Diet in Renal Disease Study Group. N. Engl. J. Med. 1994, 330, 877–884. [Google Scholar] [CrossRef]
- Frank, H.; Graf, J.; Amann-Gassner, U.; Bratke, R.; Daniel, H.; Heemann, U.; Hauner, H. Effect of short-term high-protein compared with normal-protein diets on renal hemodynamics and associated variables in healthy young men. Am. J. Clin. Nutr. 2009, 90, 1509–1516. [Google Scholar] [CrossRef] [Green Version]
- Newgard, C.B. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 2012, 15, 606–614. [Google Scholar] [CrossRef] [Green Version]
- Jensen, M.D.; Ryan, D.H.; Apovian, C.M.; Ard, J.D.; Comuzzie, A.G.; Donato, K.A.; Hu, F.B.; Hubbard, V.S.; Jakicic, J.M.; Kushner, R.F.; et al. 2013 AHA/ACC/TOS Guideline for the Management of Overweight and Obesity in Adults. Circulation 2014, 129, S102–S138. [Google Scholar] [CrossRef] [Green Version]
- Haufe, S.; Haas, V.; Utz, W.; Birkenfeld, A.L.; Jeran, S.; Bohnke, J.; Mahler, A.; Luft, F.C.; Schulz-Menger, J.; Boschmann, M.; et al. Long-lasting improvements in liver fat and metabolism despite body weight regain after dietary weight loss. Diabetes Care 2013, 36, 3786–3792. [Google Scholar] [CrossRef] [Green Version]
- White, P.J.; Lapworth, A.L.; An, J.; Wang, L.; McGarrah, R.W.; Stevens, R.D.; Ilkayeva, O.; George, T.; Muehlbauer, M.J.; Bain, J.R.; et al. Branched-chain amino acid restriction in Zucker-fatty rats improves muscle insulin sensitivity by enhancing efficiency of fatty acid oxidation and acyl-glycine export. Mol. Metab. 2016, 5, 538–551. [Google Scholar] [CrossRef] [PubMed]
- Um, S.H.; D’Alessio, D.; Thomas, G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 2006, 3, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metter, E.J.; Conwit, R.; Tobin, J.; Fozard, J.L. Age-associated loss of power and strength in the upper extremities in women and men. J. Gerontol. A Biol. Sci. Med. Sci. 1997, 52, B267–B276. [Google Scholar] [CrossRef]
- Boirie, Y. Physiopathological mechanism of sarcopenia. J. Nutr. Health Aging 2009, 13, 717–723. [Google Scholar] [CrossRef]
- Fearon, K.; Evans, W.J.; Anker, S.D. Myopenia-a new universal term for muscle wasting. J. Cachexia Sarcopenia Muscle 2011, 2, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Manini, T.M.; Clark, B.C. Dynapenia and aging: An update. J. Gerontol. A Biol. Sci. Med. Sci. 2012, 67, 28–40. [Google Scholar] [CrossRef] [Green Version]
- Srikanthan, P.; Hevener, A.L.; Karlamangla, A.S. Sarcopenia exacerbates obesity-associated insulin resistance and dysglycemia: Findings from the National Health and Nutrition Examination Survey III. PLoS ONE 2010, 5, e10805. [Google Scholar] [CrossRef]
- Goodpaster, B.H.; Carlson, C.L.; Visser, M.; Kelley, D.E.; Scherzinger, A.; Harris, T.B.; Stamm, E.; Newman, A.B. Attenuation of skeletal muscle and strength in the elderly: The Health ABC Study. J. Appl. Physiol. 2001, 90, 2157–2165. [Google Scholar] [CrossRef]
- Nye, C.K.; Hanson, R.W.; Kalhan, S.C. Glyceroneogenesis is the dominant pathway for triglyceride glycerol synthesis in vivo in the rat. J. Biol. Chem. 2008, 283, 27565–27574. [Google Scholar] [CrossRef] [Green Version]
- Montano-Loza, A.J.; Angulo, P.; Meza-Junco, J.; Prado, C.M.M.; Sawyer, M.B.; Beaumont, C.; Esfandiari, N.; Ma, M.; Baracos, V.E. Sarcopenic obesity and myosteatosis are associated with higher mortality in patients with cirrhosis. J. Cachexia Sarcopenia Muscle 2016, 7, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Montano–Loza, A.J.; Meza–Junco, J.; Prado, C.M.M.; Lieffers, J.R.; Baracos, V.E.; Bain, V.G.; Sawyer, M.B. Muscle Wasting Is Associated With Mortality in Patients With Cirrhosis. Clin. Gastroenterol. Hepatol. 2012, 10, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Hussaini, S.H.; Oldroyd, B.; Stewart, S.P.; Soo, S.; Roman, F.; Smith, M.A.; Pollard, S.; Lodge, P.; O’Grady, J.G.; Losowsky, M.S. Effects of orthotopic liver transplantation on body composition. Liver 1998, 18, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Plank, L.D.; Metzger, D.J.; McCall, J.L.; Barclay, K.L.; Gane, E.J.; Streat, S.J.; Munn, S.R.; Hill, G.L. Sequential changes in the metabolic response to orthotopic liver transplantation during the first year after surgery. Ann. Surg. 2001, 234, 245–255. [Google Scholar] [CrossRef]
- Kalafateli, M.; Mantzoukis, K.; Choi Yau, Y.; Mohammad, A.O.; Arora, S.; Rodrigues, S.; de Vos, M.; Papadimitriou, K.; Thorburn, D.; O’Beirne, J.; et al. Malnutrition and sarcopenia predict post-liver transplantation outcomes independently of the Model for End-stage Liver Disease score. J. Cachexia Sarcopenia Muscle 2017, 8, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Bhanji, R.A.; Takahashi, N.; Moynagh, M.R.; Narayanan, P.; Angirekula, M.; Mara, K.C.; Dierkhising, R.A.; Watt, K.D. The evolution and impact of sarcopenia pre- and post-liver transplantation. Aliment. Pharmacol. Ther. 2019, 49, 807–813. [Google Scholar] [CrossRef]
- Hanai, T.; Shiraki, M.; Nishimura, K.; Ohnishi, S.; Imai, K.; Suetsugu, A.; Takai, K.; Shimizu, M.; Moriwaki, H. Sarcopenia impairs prognosis of patients with liver cirrhosis. Nutrition 2015, 31, 193–199. [Google Scholar] [CrossRef]
- Cheung, K.; Lee, S.S.; Raman, M. Prevalence and mechanisms of malnutrition in patients with advanced liver disease, and nutrition management strategies. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2012, 10, 117–125. [Google Scholar] [CrossRef]
- Román, E.; Torrades, M.T.; Nadal, M.J.; Cárdenas, G.; Nieto, J.C.; Vidal, S.; Bascuñana, H.; Juárez, C.; Guarner, C.; Córdoba, J.; et al. Randomized Pilot Study: Effects of an Exercise Programme and Leucine Supplementation in Patients with Cirrhosis. Dig. Dis. Sci. 2014, 59, 1966–1975. [Google Scholar] [CrossRef]
- Frassetto, L.; Morris, R.C., Jr.; Sebastian, A. Potassium bicarbonate reduces urinary nitrogen excretion in postmenopausal women. J. Clin. Endocrinol. Metab. 1997, 82, 254–259. [Google Scholar] [CrossRef]
- Dawson-Hughes, B.; Castaneda-Sceppa, C.; Harris, S.S.; Palermo, N.J.; Cloutier, G.; Ceglia, L.; Dallal, G.E. Impact of supplementation with bicarbonate on lower-extremity muscle performance in older men and women. Osteoporos. Int. 2010, 21, 1171–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, T.; Abe, M.; Furukawa, S.; Tokumoto, Y.; Toshimitsu, K.; Ueda, T.; Yamamoto, S.; Hirooka, M.; Kumagi, T.; Hiasa, Y. Long-term branched-chain amino acid supplementation improves glucose tolerance in patients with nonalcoholic steatohepatitis-related cirrhosis. Intern. Med. 2012, 51, 2151–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellmann, C.; Degen, C.; Jin, C.J.; Nier, A.; Engstler, A.J.; Hasan Alkhatib, D.; De Bandt, J.-P.; Bergheim, I. Oral arginine supplementation protects female mice from the onset of non-alcoholic steatohepatitis. Amino Acids 2017, 49, 1215–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jobgen, W.; Meininger, C.J.; Jobgen, S.C.; Li, P.; Lee, M.J.; Smith, S.B.; Spencer, T.E.; Fried, S.K.; Wu, G. Dietary L-arginine supplementation reduces white fat gain and enhances skeletal muscle and brown fat masses in diet-induced obese rats. J. Nutr. 2009, 139, 230–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, P.A.; Simsolo, R.B.; Fournier, M. Effect of weight loss on muscle fiber type, fiber size, capillarity, and succinate dehydrogenase activity in humans. J. Clin. Endocrinol. Metab. 1999, 84, 4185–4190. [Google Scholar] [CrossRef] [PubMed]
- Bak, A.M.; Moller, A.B.; Vendelbo, M.H.; Nielsen, T.S.; Viggers, R.; Rungby, J.; Pedersen, S.B.; Jorgensen, J.O.; Jessen, N.; Moller, N. Differential regulation of lipid and protein metabolism in obese vs. lean subjects before and after a 72-h fast. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E224–E235. [Google Scholar] [CrossRef] [Green Version]
- Larson-Meyer, D.E.; Newcomer, B.R.; Hunter, G.R.; McLean, J.E.; Hetherington, H.P.; Weinsier, R.L. Effect of weight reduction, obesity predisposition, and aerobic fitness on skeletal muscle mitochondrial function. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E153–E161. [Google Scholar] [CrossRef]
- Poole, D.C.; Henson, L.C. Effect of acute caloric restriction on work efficiency. Am. J. Clin. Nutr. 1988, 47, 15–18. [Google Scholar] [CrossRef]
- Hambrecht, R.; Gielen, S. Essay: Hunter-gatherer to sedentary lifestyle. Lancet 2005, 366, S60–S61. [Google Scholar] [CrossRef]
- Laaksonen, D.E.; Lakka, H.M.; Salonen, J.T.; Niskanen, L.K.; Rauramaa, R.; Lakka, T.A. Low levels of leisure-time physical activity and cardiorespiratory fitness predict development of the metabolic syndrome. Diabetes Care 2002, 25, 1612–1618. [Google Scholar] [CrossRef] [Green Version]
- Eckard, C.; Cole, R.; Lockwood, J.; Torres, D.M.; Williams, C.D.; Shaw, J.C.; Harrison, S.A. Prospective histopathologic evaluation of lifestyle modification in nonalcoholic fatty liver disease: A randomized trial. Ther. Adv. Gastroenterol. 2013, 6, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Promrat, K.; Kleiner, D.E.; Niemeier, H.M.; Jackvony, E.; Kearns, M.; Wands, J.R.; Fava, J.L.; Wing, R.R. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 2010, 51, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.E.; Hackett, D.A.; Parker, H.M.; O’Connor, H.T.; Gerofi, J.A.; Sainsbury, A.; Baker, M.K.; Chuter, V.H.; Caterson, I.D.; George, J.; et al. Effect of aerobic exercise training dose on liver fat and visceral adiposity. J. Hepatol. 2015, 63, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Seppala-Lindroos, A.; Vehkavaara, S.; Hakkinen, A.M.; Goto, T.; Westerbacka, J.; Sovijarvi, A.; Halavaara, J.; Yki-Jarvinen, H. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J. Clin. Endocrinol. Metab. 2002, 87, 3023–3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Church, T.S.; Kuk, J.L.; Ross, R.; Priest, E.L.; Biltoft, E.; Blair, S.N. Association of cardiorespiratory fitness, body mass index, and waist circumference to nonalcoholic fatty liver disease. Gastroenterology 2006, 130, 2023–2030. [Google Scholar] [CrossRef]
- Sui, X.; LaMonte, M.J.; Laditka, J.N.; Hardin, J.W.; Chase, N.; Hooker, S.P.; Blair, S.N. Cardiorespiratory fitness and adiposity as mortality predictors in older adults. JAMA 2007, 298, 2507–2516. [Google Scholar] [CrossRef] [Green Version]
- Stefan, N.; Kantartzis, K.; Haring, H.U. Cardiorespiratory fitness, adiposity, and mortality. JAMA 2008, 299, 1013–1014. [Google Scholar] [CrossRef]
- Kantartzis, K.; Thamer, C.; Peter, A.; Machann, J.; Schick, F.; Schraml, C.; Königsrainer, A.; Königsrainer, I.; Kröber, S.; Niess, A.; et al. High cardiorespiratory fitness is an independent predictor of the reduction in liver fat during a lifestyle intervention in non-alcoholic fatty liver disease. Gut 2009, 58, 1281. [Google Scholar] [CrossRef]
- Roepstorff, C.; Steffensen, C.H.; Madsen, M.; Stallknecht, B.; Kanstrup, I.L.; Richter, E.A.; Kiens, B. Gender differences in substrate utilization during submaximal exercise in endurance-trained subjects. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E435–E447. [Google Scholar] [CrossRef] [Green Version]
- Friedlander, A.L.; Casazza, G.A.; Horning, M.A.; Huie, M.J.; Piacentini, M.F.; Trimmer, J.K.; Brooks, G.A. Training-induced alterations of carbohydrate metabolism in women: Women respond differently from men. J. Appl. Physiol. 1998, 85, 1175–1186. [Google Scholar] [CrossRef]
- Sondergaard, E.; Rahbek, I.; Sørensen, L.P.; Christiansen, J.S.; Gormsen, L.C.; Jensen, M.D.; Nielsen, S. Effects of exercise on VLDL-triglyceride oxidation and turnover. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E939–E944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watt, M.J.; Holmes, A.G.; Steinberg, G.R.; Mesa, J.L.; Kemp, B.E.; Febbraio, M.A. Reduced plasma FFA availability increases net triacylglycerol degradation, but not GPAT or HSL activity, in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E120–E127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Loon, L.J.; Thomason-Hughes, M.; Constantin-Teodosiu, D.; Koopman, R.; Greenhaff, P.L.; Hardie, D.G.; Keizer, H.A.; Saris, W.H.; Wagenmakers, A.J. Inhibition of adipose tissue lipolysis increases intramuscular lipid and glycogen use in vivo in humans. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E482–E493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magkos, F.; Wright, D.C.; Patterson, B.W.; Mohammed, B.S.; Mittendorfer, B. Lipid metabolism response to a single, prolonged bout of endurance exercise in healthy young men. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E355–E362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsekouras, Y.E.; Magkos, F.; Prentzas, K.I.; Basioukas, K.N.; Matsama, S.G.; Yanni, A.E.; Kavouras, S.A.; Sidossis, L.S. A single bout of whole-body resistance exercise augments basal VLDL-triacylglycerol removal from plasma in healthy untrained men. Clin. Sci. 2009, 116, 147–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Loon, L.J.; Koopman, R.; Stegen, J.H.; Wagenmakers, A.J.; Keizer, H.A.; Saris, W.H. Intramyocellular lipids form an important substrate source during moderate intensity exercise in endurance-trained males in a fasted state. J. Physiol. 2003, 553, 611–625. [Google Scholar] [CrossRef] [Green Version]
- Skrypnik, D.; Ratajczak, M.; Karolkiewicz, J.; Mądry, E.; Pupek-Musialik, D.; Hansdorfer-Korzon, R.; Walkowiak, J.; Jakubowski, H.; Bogdański, P. Effects of endurance and endurance–strength exercise on biochemical parameters of liver function in women with abdominal obesity. Biomed. Pharmacother. 2016, 80, 1–7. [Google Scholar] [CrossRef]
- VanDusseldorp, T.A.; Escobar, K.A.; Johnson, K.E.; Stratton, M.T.; Moriarty, T.; Cole, N.; McCormick, J.J.; Kerksick, C.M.; Vaughan, R.A.; Dokladny, K.; et al. Effect of Branched-Chain Amino Acid Supplementation on Recovery Following Acute Eccentric Exercise. Nutrients 2018, 10, 1389. [Google Scholar] [CrossRef] [Green Version]
- Lucotti, P.; Setola, E.; Monti, L.D.; Galluccio, E.; Costa, S.; Sandoli, E.P.; Fermo, I.; Rabaiotti, G.; Gatti, R.; Piatti, P. Beneficial effects of a long-term oral L-arginine treatment added to a hypocaloric diet and exercise training program in obese, insulin-resistant type 2 diabetic patients. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E906–E912. [Google Scholar] [CrossRef]
- Arentson-Lantz, E.J.; Galvan, E.; Ellison, J.; Wacher, A.; Paddon-Jones, D. Improving Dietary Protein Quality Reduces the Negative Effects of Physical Inactivity on Body Composition and Muscle Function. J. Gerontol. Ser. A 2019. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compartment/Organ | Health | NAFLD | |
|---|---|---|---|
| Glycolysis /gluconeogenesis | Cytosol-all organs | Removal of Excess of glucose in the blood after meals trough glucose oxidation and glycogen storage in the liver and muscle. The liver is also able to release glucose in the blood during fasting trough glycogenolysis and gluconeogenesis to avoid hypoglycemic events. | Liver and muscle cells become insulin resistant. In the liver, hepatocytes increase the production rate of glycogenolysis and gluconeogenesis as well as cholesterol and triglyceride synthesis [65,66,67]. The skeletal muscle cells decrease blood glucose uptake and their work efficiency [68,69]. |
| Pentose phosphate pathway (PPP) | Cytosol-Liver, mammary gland and adrenal cortex. | The PPP generates either the ribose 5-phosphate, one of the precursors for the synthesis of nucleotides and erythrose-4-phosphate used in the synthesis of aromatic amino acids. | Hepatic PPP increases in parallel with lipogenesis [70]. |
| Ketogenesis (Kt) | Mitochondria-Liver | The Ketogenesis breakdown ketogenic amino acids and fatty acids under fasting or caloric restriction conditions. | Obesogenic diets diminish the free fatty acid-induced ketogenesis according to the stage of the disease [71,72,73]. |
| Fatty acid synthesis (FAs)/β oxidation (β-Ox) | Cytoplasm/Mitochondria-Liver and adipose tissue | The FAs uses the end product of glucose metabolism, the acetyl-CoA, and convert it to fatty acids for the synthesis of cellular membranes, energy storage, and intracellular signaling pathways. Acetyl-CoA can be also esterified with glycerol to form triacylglycerol, packed in VLDL and secreted from the liver. With β-Ox, fatty acids molecules are used to generate acetyl-CoA. | IR increases lipolysis from peripheral adipose tissue as well as adipose-derived NEFA influx to the liver [74]. In addition, β-Ox is impaired due to mitochondrial dysfunction [24]. |
| De Novo Lipogenesis (DNL) | Cytosol-Liver | DNL synthetizes FA from acetyl-CoA produced when glycolysis is increased. DNL is suppressed by fasting [48]. | IR induces an increase in DNL which contribute to synthesis and accumulation of TG in the liver [75,76]. |
| Citric Acid Cycle (TCA) | Mitochondria-all organs | The TCA oxidize amino acids, fatty acids, and carbohydrates to provide most of the energy used by cells in presence of oxygen. | Lipids overload induce increase in hepatic mitochondrial oxidative and anaplerotic TCA cycle activity [73,77]. |
| Amino acid degradation and Urea Cycle | Cytosol/mitochondria-Small intestine, liver, kidney and skeletal muscle. | Amino acids are precursors for the synthesis of a variety of molecules vital to the health, growth, development, reproduction, and homeostasis of the organism. | Intrahepatic fat accumulation induces increase of amino acids in plasma, especially for the branched ones which correlates with more liver damage [78,79]. In addition, progressive deactivation of urea cycle take place with subsequent ammonia accumulation and progression of liver disease [80] as well as loss of muscle mass. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Chiara, F.; Ureta Checcllo, C.; Ramón Azcón, J. High Protein Diet and Metabolic Plasticity in Non-Alcoholic Fatty Liver Disease: Myths and Truths. Nutrients 2019, 11, 2985. https://doi.org/10.3390/nu11122985
De Chiara F, Ureta Checcllo C, Ramón Azcón J. High Protein Diet and Metabolic Plasticity in Non-Alcoholic Fatty Liver Disease: Myths and Truths. Nutrients. 2019; 11(12):2985. https://doi.org/10.3390/nu11122985
Chicago/Turabian StyleDe Chiara, Francesco, Cynthia Ureta Checcllo, and Javier Ramón Azcón. 2019. "High Protein Diet and Metabolic Plasticity in Non-Alcoholic Fatty Liver Disease: Myths and Truths" Nutrients 11, no. 12: 2985. https://doi.org/10.3390/nu11122985
APA StyleDe Chiara, F., Ureta Checcllo, C., & Ramón Azcón, J. (2019). High Protein Diet and Metabolic Plasticity in Non-Alcoholic Fatty Liver Disease: Myths and Truths. Nutrients, 11(12), 2985. https://doi.org/10.3390/nu11122985