Effects of Pharmacological Thermogenic Adipocyte Activation on Metabolism and Atherosclerotic Plaque Regression

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals, Housing Conditions, Diets, and Animal Experiments
2.2. Plasma Analysis
2.3. Gene Expression Analysis
2.4. Histology
2.5. Liver Analysis
2.6. Analysis of Atherosclerosis
2.7. Statistical Analysis
3. Results
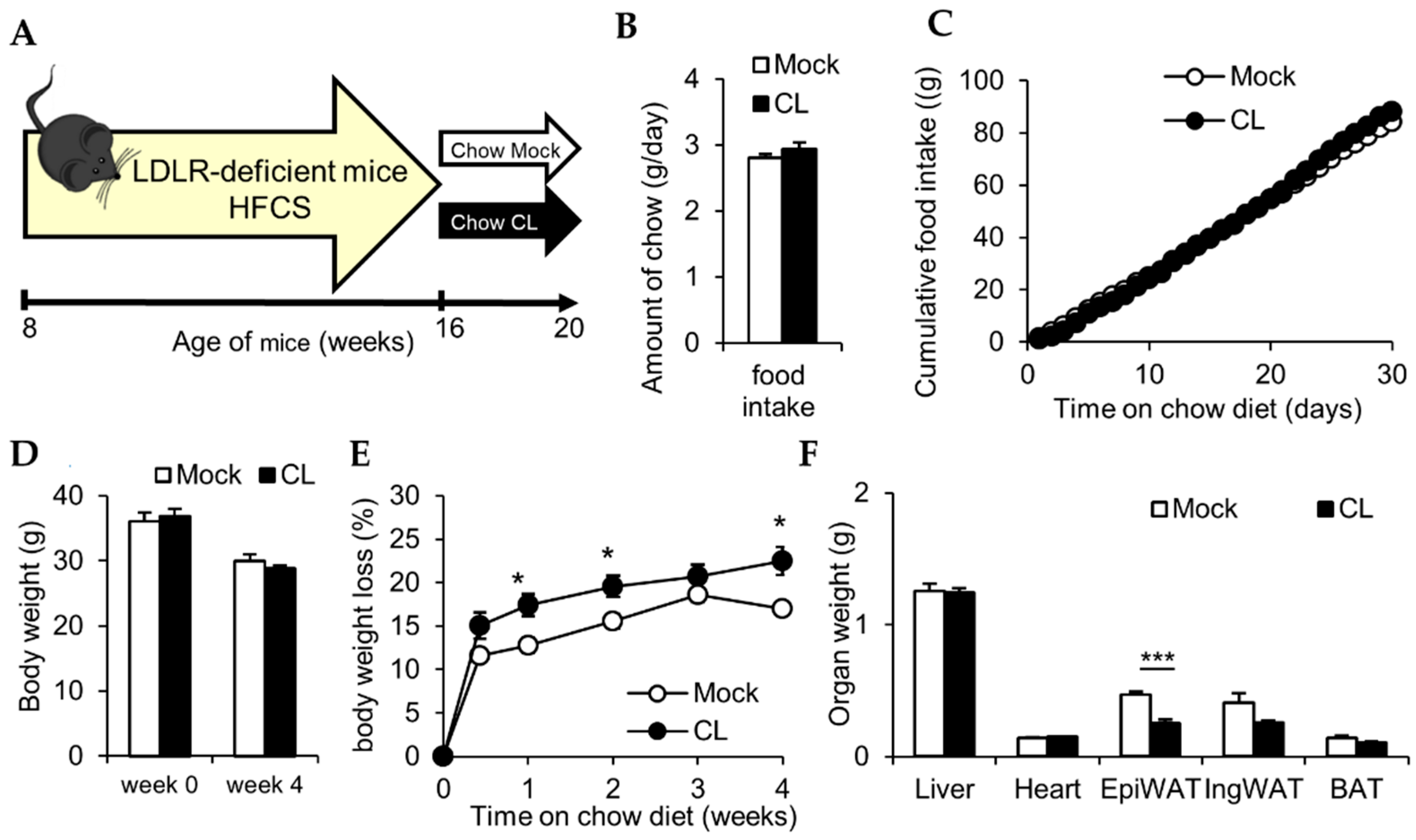
3.1. CL Treatment Reduces Adiposity Irrespective of Food Intake
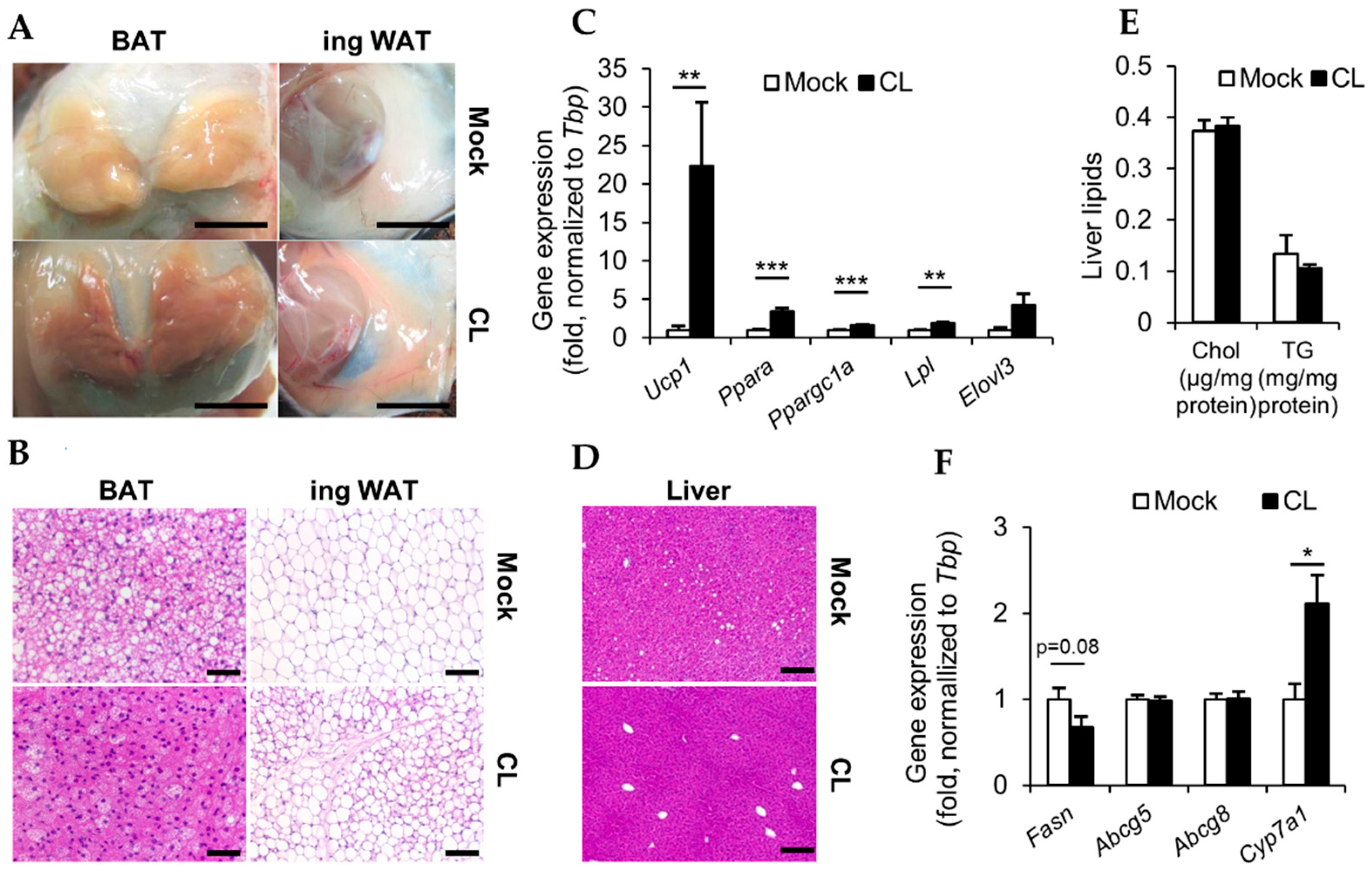
3.2. CL Treatment Activates Thermogenic Fat Depots
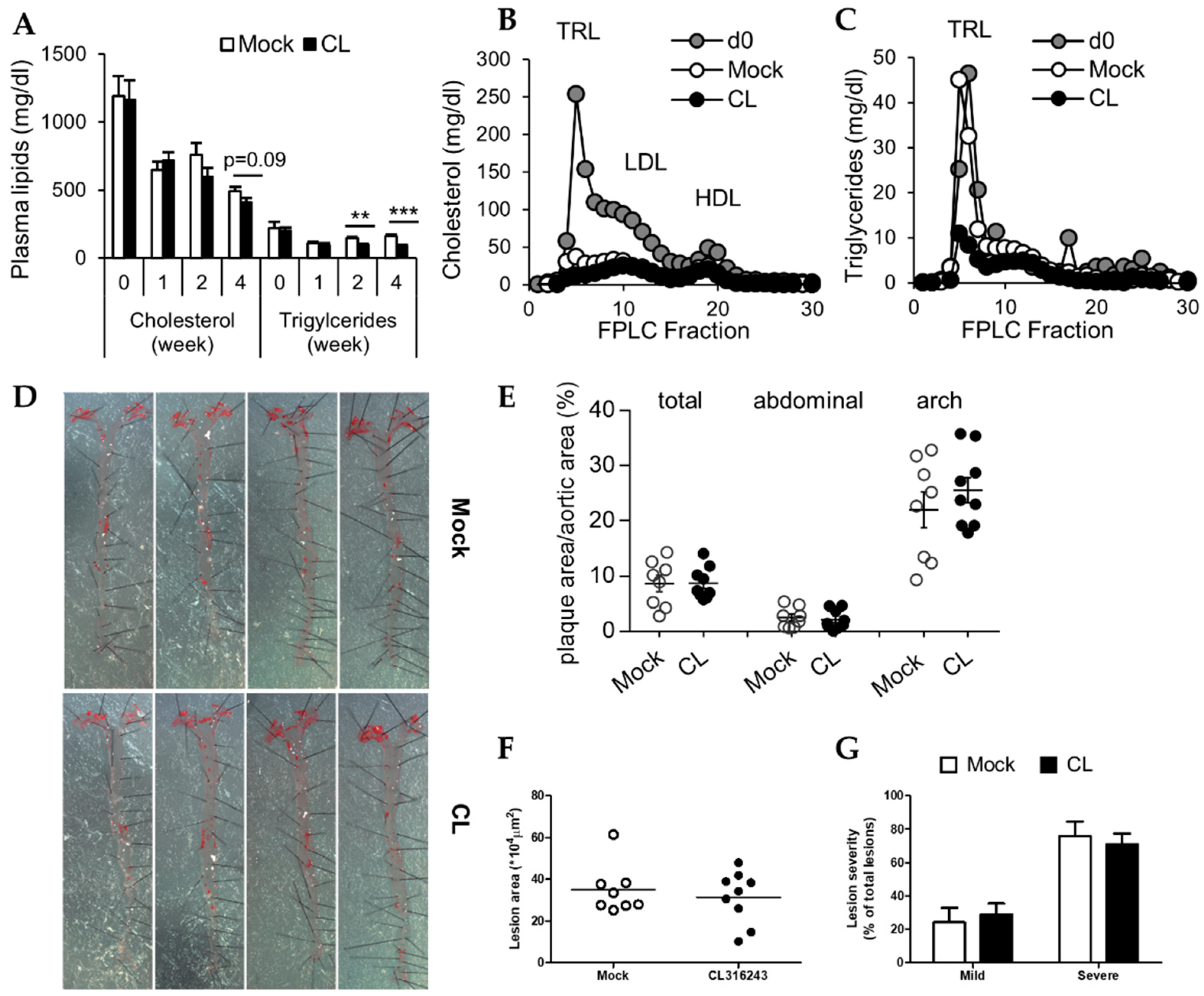
3.3. Decreased Plasma Triglycerides after CL Treatment Do Not Translate into Reduced Atherosclerosis
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McAloon, C.J.; Boylan, L.M. The changing face of cardiovascular disease 2000-2012: An analysis of the world health organisation global health estimates data. Int. J. Cardiol. 2016, 224, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Bruns, O.T. Brown adipose tissue activity controls triglyceride clearance. Nat. Med. 2011, 17, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Berbee, J.F.P.; Boon, M.R. Brown fat activation reduces hypercholesterolaemia and protects from atherosclerosis development. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Sakane, N. Anti-obesity and anti-diabetic effects of CL 316,243, a highly specific beta 3-adrenoceptor agonist, in yellow KK mice. Life Sci. 1994, 54, 491–498. [Google Scholar] [CrossRef]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Heeren, J. Adipose tissue browning and metabolic health. Nat. Rev. Endocrinol. 2014, 10, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; John, C. Thermogenic adipocytes promote HDL turnover and reverse cholesterol transport. Nat. Commun. 2017, 8, 15010. [Google Scholar] [CrossRef] [PubMed]
- Worthmann, A.; John, C. Cold-induced conversion of cholesterol to bile acids in mice shapes the gut microbiome and promotes adaptive thermogenesis. Nat. Med. 2017. [Google Scholar] [CrossRef]
- Saito, M.; Okamatsu-Ogura, Y. High incidence of metabolically active brown adipose tissue in healthy adult humans: Effects of cold exposure and adiposity. Diabetes 2009, 58, 1526–1531. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; Lehman, S. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef]
- Virtanen, K.A.; Lidell, M.E. Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 2009, 360, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Van Marken Lichtenbelt, W.D.; Vanhommerig, J.W. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, J.; Bengtsson, T. Unexpected evidence for active brown adipose tissue in adult humans. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E444–E452. [Google Scholar] [CrossRef] [PubMed]
- Zingaretti, M.C.; Crosta, F. The presence of UCP1 demonstrates that metabolically active adipose tissue in the neck of adult humans truly represents brown adipose tissue. FASEB J. 2009, 23, 3113–3120. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, V.; Routhier-Labadie, A. Outdoor temperature, age, sex, body mass index, and diabetic status determine the prevalence, mass, and glucose-uptake activity of 18F-FDG-detected BAT in humans. J. Clin. Endocrinol. Metab. 2011, 96, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Vijgen, G.H.; Bouvy, N.D. Brown adipose tissue in morbidly obese subjects. PLoS ONE 2011, 6, e17247. [Google Scholar] [CrossRef] [PubMed]
- Van der Lans, A.A.; Hoeks, J. Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J. Clin. Investig. 2013, 123, 3395–3403. [Google Scholar] [CrossRef] [PubMed]
- Yoneshiro, T.; Aita, S. Recruited brown adipose tissue as an antiobesity agent in humans. J. Clin. Investig. 2013, 123, 3404–3408. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, V.; Labbe, S.M. Brown adipose tissue oxidative metabolism contributes to energy expenditure during acute cold exposure in humans. J. Clin. Investig. 2012, 122, 545–552. [Google Scholar] [CrossRef]
- Dong, M.; Yang, X. Cold exposure promotes atherosclerotic plaque growth and instability via UCP1-dependent lipolysis. Cell Metab. 2013, 18, 118–129. [Google Scholar] [CrossRef]
- Williams, J.W.; Elvington, A. Thermoneutrality but Not UCP1 Deficiency Suppresses Monocyte Mobilization Into Blood. Circ. Res. 2017, 121, 662–676. [Google Scholar] [CrossRef] [PubMed]
- Feig, J.E. Regression of atherosclerosis: Insights from animal and clinical studies. Ann. Glob. Health 2014, 80, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Wissler, R.W.; Vesselinovitch, D. Studies of regression of advanced atherosclerosis in experimental animals and man. Ann. N. Y. Acad. Sci. 1976, 275, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Friedman, M.; Byers, S.O. Resolution of aortic atherosclerotic infiltration in the rabbit by phosphatide infusion. Proc. Soc. Exp. Biol. Med. 1957, 95, 586–588. [Google Scholar] [CrossRef] [PubMed]
- Maruffo, C.A.; Portman, O.W. Nutritional control of coronary artery atherosclerosis in the squirrel monkey. J. Atheroscler. Res. 1968, 8, 237–247. [Google Scholar] [CrossRef]
- Tsukamoto, K.; Tangirala, R. Rapid regression of atherosclerosis induced by liver-directed gene transfer of ApoE in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2162–2170. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Oka, K. Reversal of hypercholesterolemia in low density lipoprotein receptor knockout mice by adenovirus-mediated gene transfer of the very low density lipoprotein receptor. J. Biol. Chem. 1996, 271, 6852–6860. [Google Scholar] [CrossRef]
- Marais, A.D. Familial hypercholesterolaemia. Clin. Biochem. Rev. 2004, 25, 49–68. [Google Scholar]
- Getz, G.S.; Reardon, C.A. Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J. Lipid Res. 2009, 50, S156–S161. [Google Scholar] [CrossRef]
- Emini Veseli, B.; Perrotta, P. Animal models of atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef]
- Nagareddy, P.R.; Murphy, A.J. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013, 17, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, S.; Brown, M.S. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J. Clin. Investig. 1993, 92, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Widenmaier, S.B. Brown adipose tissue thermogenic adaptation requires Nrf1-mediated proteasomal activity. Nat. Med. 2018, 24, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Schlein, C.; Talukdar, S. FGF21 Lowers Plasma Triglycerides by Accelerating Lipoprotein Catabolism in White and Brown Adipose Tissues. Cell Metab. 2016, 23, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Orlando, P. Altered endocannabinoid signalling after a high-fat diet in Apoe(-/-) mice: Relevance to adipose tissue inflammation, hepatic steatosis and insulin resistance. Diabetologia 2011, 54, 2900–2910. [Google Scholar] [CrossRef] [PubMed]
- Himms-Hagen, J.; Melnyk, A. Multilocular fat cells in WAT of CL-316243-treated rats derive directly from white adipocytes. Am. J. Physiol. Cell Physiol. 2000, 279, C670–C681. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, R.; Kooijman, S. A Diurnal Rhythm in Brown Adipose Tissue Causes Rapid Clearance and Combustion of Plasma Lipids at Wakening. Cell Rep. 2018, 22, 3521–3533. [Google Scholar] [CrossRef]
- Bartelt, A.; Weigelt, C. Effects of adipocyte lipoprotein lipase on de novo lipogenesis and white adipose tissue browning. Biochim. Biophys. Acta 2013, 1831, 934–942. [Google Scholar] [CrossRef]
- Eissing, L.; Scherer, T. De novo lipogenesis in human fat and liver is linked to ChREBP-beta and metabolic health. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Guerra, C.; Koza, R.A. Emergence of brown adipocytes in white fat in mice is under genetic control. Effects on body weight and adiposity. J. Clin. Investig. 1998, 102, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Llodra, J.; Angeli, V. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc. Natl. Acad. Sci. USA 2004, 101, 11779–11784. [Google Scholar] [CrossRef] [PubMed]
- Trogan, E.; Fayad, Z.A. Serial studies of mouse atherosclerosis by in vivo magnetic resonance imaging detect lesion regression after correction of dyslipidemia. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1714–1719. [Google Scholar] [CrossRef] [PubMed]
- Trogan, E.; Feig, J.E. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc. Natl. Acad. Sci. USA 2006, 103, 3781–3786. [Google Scholar] [CrossRef] [PubMed]
- Willecke, F.; Yuan, C. Effects of High Fat Feeding and Diabetes on Regression of Atherosclerosis Induced by Low-Density Lipoprotein Receptor Gene Therapy in LDL Receptor-Deficient Mice. PLoS ONE 2015, 10, e0128996. [Google Scholar] [CrossRef] [PubMed]
- Hewing, B.; Parathath, S. Rapid regression of atherosclerosis with MTP inhibitor treatment. Atherosclerosis 2013, 227, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Rayner, K.J.; Sheedy, F.J. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J. Clin. Investig. 2011, 121, 2921–2931. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.H.; Reddick, R.L. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 1992, 258, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Plump, A.S.; Smith, J.D. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell 1992, 71, 343–353. [Google Scholar] [CrossRef]
- Van Vlijmen, B.J.; Van den Maagdenberg, A.M. Diet-induced hyperlipoproteinemia and atherosclerosis in apolipoprotein E3-Leiden transgenic mice. J. Clin. Investig. 1994, 93, 1403–1410. [Google Scholar] [CrossRef]
- Hoeke, G.; Nahon, K.J. Short-term cooling increases serum triglycerides and small high-density lipoprotein levels in humans. J. Clin. Lipidol. 2017, 11, 920–928. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Worthmann, A.; Schlein, C.; Berbée, J.F.P.; Rensen, P.C.N.; Heeren, J.; Bartelt, A. Effects of Pharmacological Thermogenic Adipocyte Activation on Metabolism and Atherosclerotic Plaque Regression. Nutrients 2019, 11, 463. https://doi.org/10.3390/nu11020463
Worthmann A, Schlein C, Berbée JFP, Rensen PCN, Heeren J, Bartelt A. Effects of Pharmacological Thermogenic Adipocyte Activation on Metabolism and Atherosclerotic Plaque Regression. Nutrients. 2019; 11(2):463. https://doi.org/10.3390/nu11020463
Chicago/Turabian StyleWorthmann, Anna, Christian Schlein, Jimmy F. P. Berbée, Patrick C. N. Rensen, Joerg Heeren, and Alexander Bartelt. 2019. "Effects of Pharmacological Thermogenic Adipocyte Activation on Metabolism and Atherosclerotic Plaque Regression" Nutrients 11, no. 2: 463. https://doi.org/10.3390/nu11020463
APA StyleWorthmann, A., Schlein, C., Berbée, J. F. P., Rensen, P. C. N., Heeren, J., & Bartelt, A. (2019). Effects of Pharmacological Thermogenic Adipocyte Activation on Metabolism and Atherosclerotic Plaque Regression. Nutrients, 11(2), 463. https://doi.org/10.3390/nu11020463