A Mitochondrial Specific Antioxidant Reverses Metabolic Dysfunction and Fatty Liver Induced by Maternal Cigarette Smoke in Mice


 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Bioassays
2.3. Real Time-PCR
2.4. Western Blotting
2.5. Immunohistochemistry
2.6. Second Harmonic Generation
2.7. Mitochondrial Density and ROS
2.8. Lipidomics
2.9. Statistical Analysis
3. Results
3.1. Body and Liver Weights
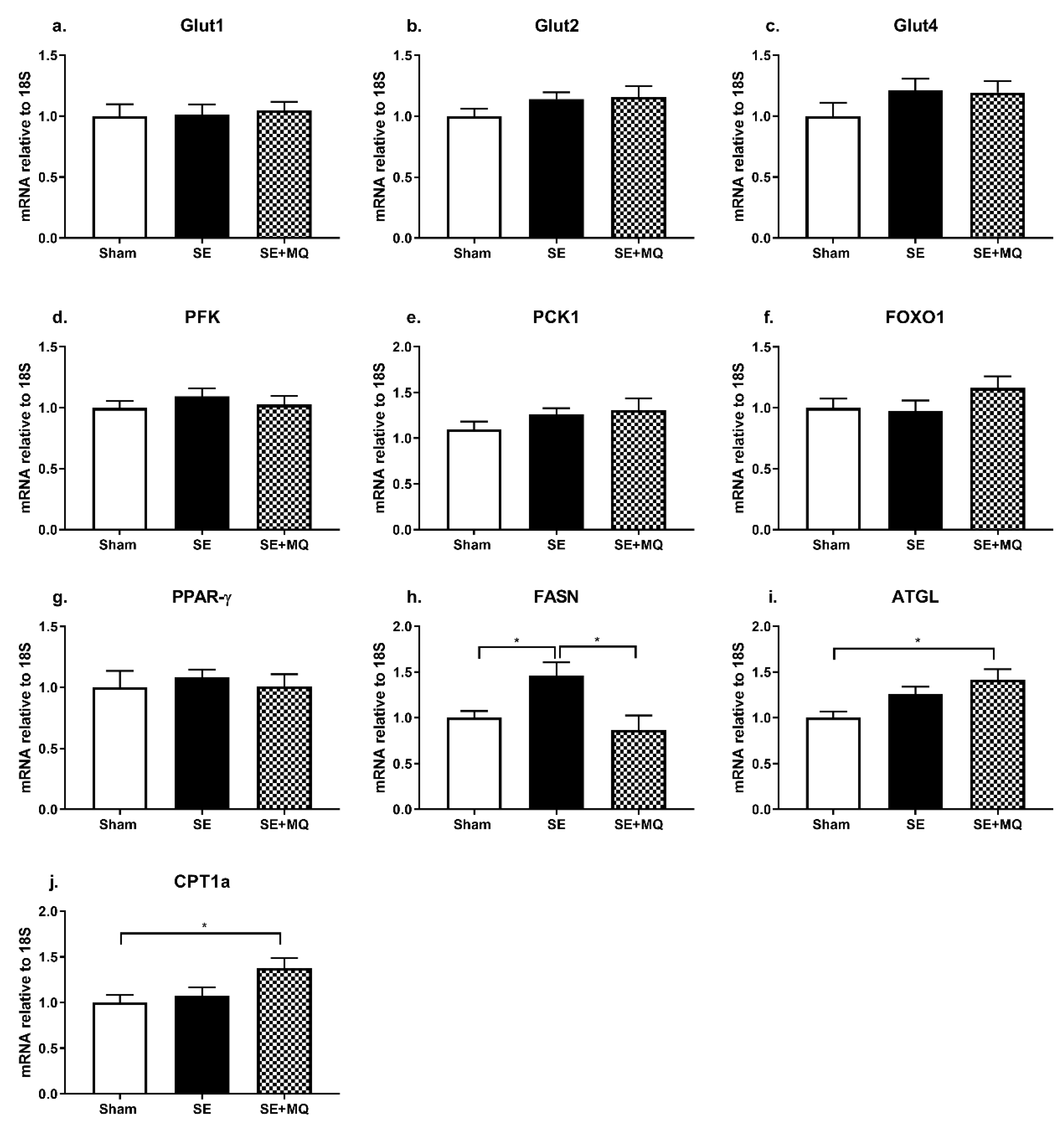
3.2. Glucose Metabolic Markers
3.3. Lipid Metabolic Markers
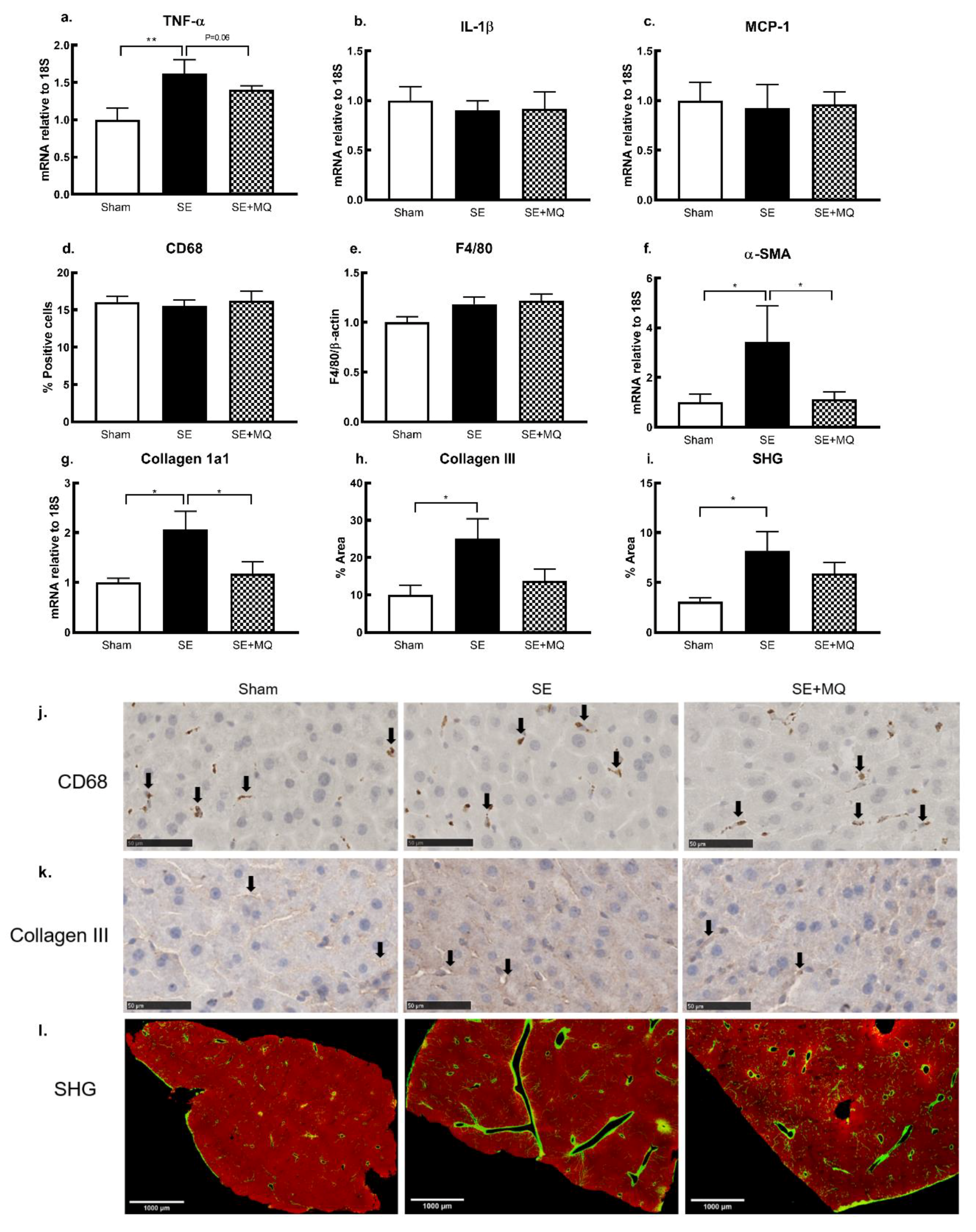
3.4. Liver Injury Markers
3.5. Oxidative Stress and Mitochondrial Integrity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jaddoe, V.W.; De Jonge, L.L.; Van Dam, R.M.; Willett, W.C.; Harris, H.; Stampfer, M.J.; Hu, F.B.; Michels, K.B. Fetal exposure to parental smoking and the risk of type 2 diabetes in adult women. Diabetes Care 2014, 37, 2966–2973. [Google Scholar] [CrossRef] [PubMed]
- Izzotti, A.; Balansky, R.M.; Cartiglia, C.; Camoirano, A.; Longobardi, M.; De Flora, S. Genomic and transcriptional alterations in mouse fetus liver after transplacental exposure to cigarette smoke. FASEB J. 2003, 17, 1127–1129. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Liu, F. Mitochondrial stress: A bridge between mitochondrial dysfunction and metabolic diseases? Cell. Signal. 2011, 23, 1528–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Harrison, C.M.; Chuang, G.C.; Ballinger, S.W. The role of tobacco smoke induced mitochondrial damage in vascular dysfunction and atherosclerosis. Mutat. Res. 2007, 621, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, Y.L.; Saad, S.; Al-Odat, I.; Oliver, B.G.; Pollock, C.; Jones, N.M.; Chen, H. Maternal l-carnitine supplementation improves brain health in offspring from cigarette smoke exposed mothers. Front. Mol. Neurosci. 2017, 10, 33. [Google Scholar] [CrossRef]
- Chan, Y.L.; Saad, S.; Pollock, C.; Oliver, B.; Al-Odat, I.; Zaky, A.A.; Jones, N.; Chen, H. Impact of maternal cigarette smoke exposure on brain inflammation and oxidative stress in male mice offspring. Sci. Rep. 2016, 6, 25881. [Google Scholar] [CrossRef] [Green Version]
- Stangenberg, S.; Nguyen, L.T.; Chen, H.; Al-Odat, I.; Killingsworth, M.C.; Gosnell, M.E.; Anwer, A.G.; Goldys, E.M.; Pollock, C.A.; Saad, S. Oxidative stress, mitochondrial perturbations and fetal programming of renal disease induced by maternal smoking. Int. J. Biochem. Cell Biol. 2015, 64, 81–90. [Google Scholar] [CrossRef]
- Smith, R.A.; Murphy, M.P. Animal and human studies with the mitochondria-targeted antioxidant mitoq. Ann. N. Y. Acad. Sci. 2010, 1201, 96–103. [Google Scholar] [CrossRef]
- Al-Bazi, M.M.; Elshal, M.F.; Khoja, S.M. Reduced coenzyme q(10) in female smokers and its association with lipid profile in a young healthy adult population. Arch. Med Sci. 2011, 7, 948–954. [Google Scholar] [CrossRef]
- Lim, S.C.; Tan, H.H.; Goh, S.K.; Subramaniam, T.; Sum, C.F.; Tan, I.K.; Lee, B.L.; Ong, C.N. Oxidative burden in prediabetic and diabetic individuals: Evidence from plasma coenzyme q(10). Diabet. Med. A J. Br. Diabet. Assoc. 2006, 23, 1344–1349. [Google Scholar] [CrossRef]
- Sukjamnong, S.; Chan, Y.L.; Zakarya, R.; Saad, S.; Sharma, P.; Santiyanont, R.; Chen, H.; Oliver, B.G. Effect of long-term maternal smoking on the offspring’s lung health. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L416–L423. [Google Scholar] [CrossRef]
- Sukjamnong, S.; Chan, Y.L.; Zakarya, R.; Nguyen, L.T.; Anwer, A.G.; Zaky, A.A.; Santiyanont, R.; Oliver, B.G.; Goldys, E.; Pollock, C.A.; et al. Mitoq supplementation prevent long-term impact of maternal smoking on renal development, oxidative stress and mitochondrial density in male mice offspring. Sci. Rep. 2018, 8, 6631. [Google Scholar] [CrossRef]
- Al-Odat, I.; Chen, H.; Chan, Y.L.; Amgad, S.; Wong, M.G.; Gill, A.; Pollock, C.; Saad, S. The impact of maternal cigarette smoke exposure in a rodent model on renal development in the offspring. PLoS ONE 2014, 9, e103443. [Google Scholar] [CrossRef]
- Vivekanandarajah, A.; Chan, Y.L.; Chen, H.; Machaalani, R. Prenatal cigarette smoke exposure effects on apoptotic and nicotinic acetylcholine receptor expression in the infant mouse brainstem. Neurotoxicology 2016, 53, 53–63. [Google Scholar] [CrossRef]
- Smith, R.A.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar] [CrossRef]
- Chen, H.; Simar, D.; Pegg, K.; Saad, S.; Palmer, C.; Morris, M.J. Exendin-4 is effective against metabolic disorders induced by intrauterine and postnatal overnutrition in rodents. Diabetologia 2014, 57, 614–622. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Hutchins, P.D.; Russell, J.D.; Coon, J.J. Lipidex: An integrated software package for high-confidence lipid identification. Cell Syst. 2018, 6, 621–625. [Google Scholar] [CrossRef]
- Owens, L.; Laing, I.A.; Murdzoska, J.; Zhang, G.; Turner, S.W.; Le Souef, P.N. Glutathione s-transferase genotype protects against in utero tobacco linked lung function deficits. Am. J. Respir. Crit. Care Med. 2019. [Google Scholar] [CrossRef]
- Horta, B.L.; Victora, C.G.; Menezes, A.M.; Halpern, R.; Barros, F.C. Low birthweight, preterm births and intrauterine growth retardation in relation to maternal smoking. Paediatr. Perinat. Epidemiol. 1997, 11, 140–151. [Google Scholar] [CrossRef]
- Thiering, E.; Brüske, I.; Kratzsch, J.; Thiery, J.; Sausenthaler, S.; Meisinger, C.; Koletzko, S.; Bauer, C.-P.; Schaaf, B.; Von Berg, A.; et al. Prenatal and postnatal tobacco smoke exposure and development of insulin resistance in 10 year old children. Int. J. Hyg. Environ. Health 2011, 214, 361–368. [Google Scholar] [CrossRef]
- Ayonrinde, O.T.; Adams, L.A.; Mori, T.A.; Beilin, L.J.; De Klerk, N.; Pennell, C.E.; White, S.; Olynyk, J.K. Sex differences between parental pregnancy characteristics and nonalcoholic fatty liver disease in adolescents. Hepatology 2018, 67, 108–122. [Google Scholar] [CrossRef]
- Chen, H.; Morris, M.J. Maternal smoking-a contributor to the obesity epidemic? Obes. Res. Clin. Pract. 2007, 1, 155–163. [Google Scholar] [CrossRef]
- Saad, S.; Al-Odat, I.; Chan, Y.L.; McGrath, K.C.; Pollock, C.A.; Oliver, B.G.; Chen, H. Maternal l-carnitine supplementation improves glucose and lipid profiles in female offspring of dams exposed to cigarette smoke. Clin. Exp. Pharmacol. Physiol. 2018, 45, 694–703. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Stangenberg, S.; Chen, H.; Al-Odat, I.; Chan, Y.L.; Gosnell, M.E.; Anwer, A.G.; Goldys, E.M.; Pollock, C.A.; Saad, S. L-carnitine reverses maternal cigarette smoke exposure-induced renal oxidative stress and mitochondrial dysfunction in mouse offspring. Am. J. Physiol. Ren. Physiol. 2015, 308, F689–F696. [Google Scholar] [CrossRef]
- Montgomery, S.M.; Ekbom, A. Smoking during pregnancy and diabetes mellitus in a british longitudinal birth cohort. BMJ 2002, 324, 26–27. [Google Scholar] [CrossRef]
- Song, M.; Schuschke, D.A.; Zhou, Z.; Zhong, W.; Zhang, J.; Zhang, X.; Wang, Y.; McClain, C.J. Kupffer cell depletion protects against the steatosis, but not the liver damage, induced by marginal-copper, high-fructose diet in male rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G934–G945. [Google Scholar] [CrossRef] [Green Version]
- Filis, P.; Nagrath, N.; Fraser, M.; Hay, D.C.; Iredale, J.P.; O’Shaughnessy, P.; Fowler, P.A. Maternal smoking dysregulates protein expression in second trimester human fetal livers in a sex-specific manner. J. Clin. Endocrinol. Metab. 2015, 100, E861–E870. [Google Scholar] [CrossRef]
- Ma, D.W.; Arendt, B.M.; Hillyer, L.M.; Fung, S.K.; McGilvray, I.; Guindi, M.; Allard, J.P. Plasma phospholipids and fatty acid composition differ between liver biopsy-proven nonalcoholic fatty liver disease and healthy subjects. Nutr. Diabetes 2016, 6, e220. [Google Scholar] [CrossRef]
- Rossman, M.J.; Santos-Parker, J.R.; Steward, C.A.C.; Bispham, N.Z.; Cuevas, L.M.; Rosenberg, H.L.; Woodward, K.A.; Chonchol, M.; Gioscia-Ryan, R.A.; Murphy, M.P.; et al. Chronic supplementation with a mitochondrial antioxidant (mitoq) improves vascular function in healthy older adults. Hypertension 2018, 71, 1056–1063. [Google Scholar] [CrossRef]
- Gioscia-Ryan, R.A.; LaRocca, T.J.; Sindler, A.L.; Zigler, M.C.; Murphy, M.P.; Seals, D.R. Mitochondria-targeted antioxidant (mitoq) ameliorates age-related arterial endothelial dysfunction in mice. J. Physiol. 2014, 592, 2549–2561. [Google Scholar] [CrossRef]
- Toda, N.; Okamura, T. Cigarette smoking impairs nitric oxide-mediated cerebral blood flow increase: Implications for alzheimer’s disease. J. Pharmacol. Sci. 2016, 131, 223–232. [Google Scholar] [CrossRef]
- Xu, Z.; Huo, J.; Ding, X.; Yang, M.; Li, L.; Dai, J.; Hosoe, K.; Kubo, H.; Mori, M.; Higuchi, K.; et al. Coenzyme q10 improves lipid metabolism and ameliorates obesity by regulating camkii-mediated pde4 inhibition. Sci. Rep. 2017, 7, 8253. [Google Scholar] [CrossRef]
- Feillet-Coudray, C.; Fouret, G.; Ebabe Elle, R.; Rieusset, J.; Bonafos, B.; Chabi, B.; Crouzier, D.; Zarkovic, K.; Zarkovic, N.; Ramos, J.; et al. The mitochondrial-targeted antioxidant mitoq ameliorates metabolic syndrome features in obesogenic diet-fed rats better than apocynin or allopurinol. Free Radic. Res. 2014, 48, 1232–1246. [Google Scholar] [CrossRef]
- Mercer, J.R.; Yu, E.; Figg, N.; Cheng, K.K.; Prime, T.A.; Griffin, J.L.; Masoodi, M.; Vidal-Puig, A.; Murphy, M.P.; Bennett, M.R. The mitochondria-targeted antioxidant mitoq decreases features of the metabolic syndrome in atm+/-/apoe-/-mice. Free Radic. Biol. Med. 2012, 52, 841–849. [Google Scholar] [CrossRef]
- Caldwell, S.H.; Swerdlow, R.H.; Khan, E.M.; Iezzoni, J.C.; Hespenheide, E.E.; Parks, J.K.; Parker, W.D., Jr. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J. Hepatol. 1999, 31, 430–434. [Google Scholar] [CrossRef]
- Seki, S.; Kitada, T.; Yamada, T.; Sakaguchi, H.; Nakatani, K.; Wakasa, K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J. Hepatol. 2002, 37, 56–62. [Google Scholar] [CrossRef]
- Amir, M.; Czaja, M.J. Autophagy in nonalcoholic steatohepatitis. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Spencer, N.Y.; Zhou, W.; Li, Q.; Zhang, Y.; Luo, M.; Yan, Z.; Lynch, T.J.; Abbott, D.; Banfi, B.; Engelhardt, J.F. Hepatocytes produce tnf-alpha following hypoxia-reoxygenation and liver ischemia-reperfusion in a nadph oxidase-and c-src-dependent manner. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G84–G94. [Google Scholar] [CrossRef]
- Fetterman, J.L.; Pompilius, M.; Westbrook, D.G.; Uyeminami, D.; Brown, J.; Pinkerton, K.E.; Ballinger, S.W. Developmental exposure to second-hand smoke increases adult atherogenesis and alters mitochondrial DNA copy number and deletions in apoe(-/-) mice. PLoS ONE 2013, 8, e66835. [Google Scholar] [CrossRef]
- Westbrook, D.G.; Anderson, P.G.; Pinkerton, K.E.; Ballinger, S.W. Perinatal tobacco smoke exposure increases vascular oxidative stress and mitochondrial damage in non-human primates. Cardiovasc. Toxicol. 2010, 10, 216–226. [Google Scholar] [CrossRef]
- Sanchez-Valle, V.; Chavez-Tapia, N.C.; Uribe, M.; Mendez-Sanchez, N. Role of oxidative stress and molecular changes in liver fibrosis: A review. Curr. Med. Chem. 2012, 19, 4850–4860. [Google Scholar] [CrossRef]
- Seth, R.K.; Kumar, A.; Das, S.; Kadiiska, M.B.; Michelotti, G.; Diehl, A.M.; Chatterjee, S. Environmental toxin-linked nonalcoholic steatohepatitis and hepatic metabolic reprogramming in obese mice. Toxicol. Sci. 2013, 134, 291–303. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sham (n = 19) | SE (n = 20) | SE + MQ (n = 14) | |
|---|---|---|---|
| Birth weight (g) | 1.51 ± 0.03 | 1.30 ± 0.06 ** | 1.65 ± 0.05 ## |
| Body weight at 13 weeks (g) | 25.2 ± 0.2 | 24.2 ± 0.2 ** | 25.1 ± 0.2 ## |
| Liver weight (g) | 1.12 ± 0.019 | 1.05 ± 0.019 * | 1.15 ± 0.02 ## |
| Liver weight (% of body weight) | 4.42 ± 0.073 | 4.37 ± 0.079 | 4.55 ± 0.08 |
| Liver triglycerides (mg/g liver) | 4.08 ± 0.41 | 6.03 ± 0.49 * | 4.04 ± 0.61 # |
| Liver PE 34:1 intensity (cps) | 121,000 ± 8900 | 203,000 ± 29,000 * | 170,000 ± 19,000 |
| Liver PE 38:1 intensity (cps) | 13,100 ± 1600 | 21,700 ± 3000 * | 17,000 ± 1900 |
| Liver PE 38:2 intensity (cps) | 12,500 ± 2400 | 23,000 ± 4700 * | 12,700 ± 1200 # |
| Liver PE 40:5 intensity (cps) | 88,200 ± 7000 | 156,000 ± 25,000 * | 104,000 ± 15,000 |
| Plasma triglycerides (mg/mL) | 2.36 ± 0.19 | 2.31 ± 0.21 | 2.40 ± 0.29 |
| Plasma ALT (U/L) | 8.03 ± 1.1 | 11.2 ± 0.9 * | 7.75 ± 0.7 # |
| Plasma insulin (ng/mL) | 0.0702 ± 0.014 | 0.103 ± 0.083 | 0.111 ± 0.010 |
| AUC of IPGTT (mM·min) | 1150 ± 20 | 1280 ± 41 * | 1130 ± 38 # |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Chan, Y.L.; Sukjamnong, S.; Anwer, A.G.; Vindin, H.; Padula, M.; Zakarya, R.; George, J.; Oliver, B.G.; Saad, S.; et al. A Mitochondrial Specific Antioxidant Reverses Metabolic Dysfunction and Fatty Liver Induced by Maternal Cigarette Smoke in Mice. Nutrients 2019, 11, 1669. https://doi.org/10.3390/nu11071669
Li G, Chan YL, Sukjamnong S, Anwer AG, Vindin H, Padula M, Zakarya R, George J, Oliver BG, Saad S, et al. A Mitochondrial Specific Antioxidant Reverses Metabolic Dysfunction and Fatty Liver Induced by Maternal Cigarette Smoke in Mice. Nutrients. 2019; 11(7):1669. https://doi.org/10.3390/nu11071669
Chicago/Turabian StyleLi, Gerard, Yik Lung Chan, Suporn Sukjamnong, Ayad G. Anwer, Howard Vindin, Matthew Padula, Razia Zakarya, Jacob George, Brian G. Oliver, Sonia Saad, and et al. 2019. "A Mitochondrial Specific Antioxidant Reverses Metabolic Dysfunction and Fatty Liver Induced by Maternal Cigarette Smoke in Mice" Nutrients 11, no. 7: 1669. https://doi.org/10.3390/nu11071669
APA StyleLi, G., Chan, Y. L., Sukjamnong, S., Anwer, A. G., Vindin, H., Padula, M., Zakarya, R., George, J., Oliver, B. G., Saad, S., & Chen, H. (2019). A Mitochondrial Specific Antioxidant Reverses Metabolic Dysfunction and Fatty Liver Induced by Maternal Cigarette Smoke in Mice. Nutrients, 11(7), 1669. https://doi.org/10.3390/nu11071669