Metabolic Defects Caused by High-Fat Diet Modify Disease Risk through Inflammatory and Amyloidogenic Pathways in a Mouse Model of Alzheimer’s Disease

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Procedures
2.1. Experimental Animals
2.2. Dietary Treatment
2.3. Glucose Measurements
2.4. Serum Biochemistries
2.5. Indirect Calorimetry
2.6. Hippocampal mRNA Quantitation
2.7. 16S rRNA Library Preparation and Sequencing
2.8. Informatics Analysis of 16S rRNA Sequences
2.9. Statistics
2.10. Statistical Analysis of Annotated Sequences
3. Results
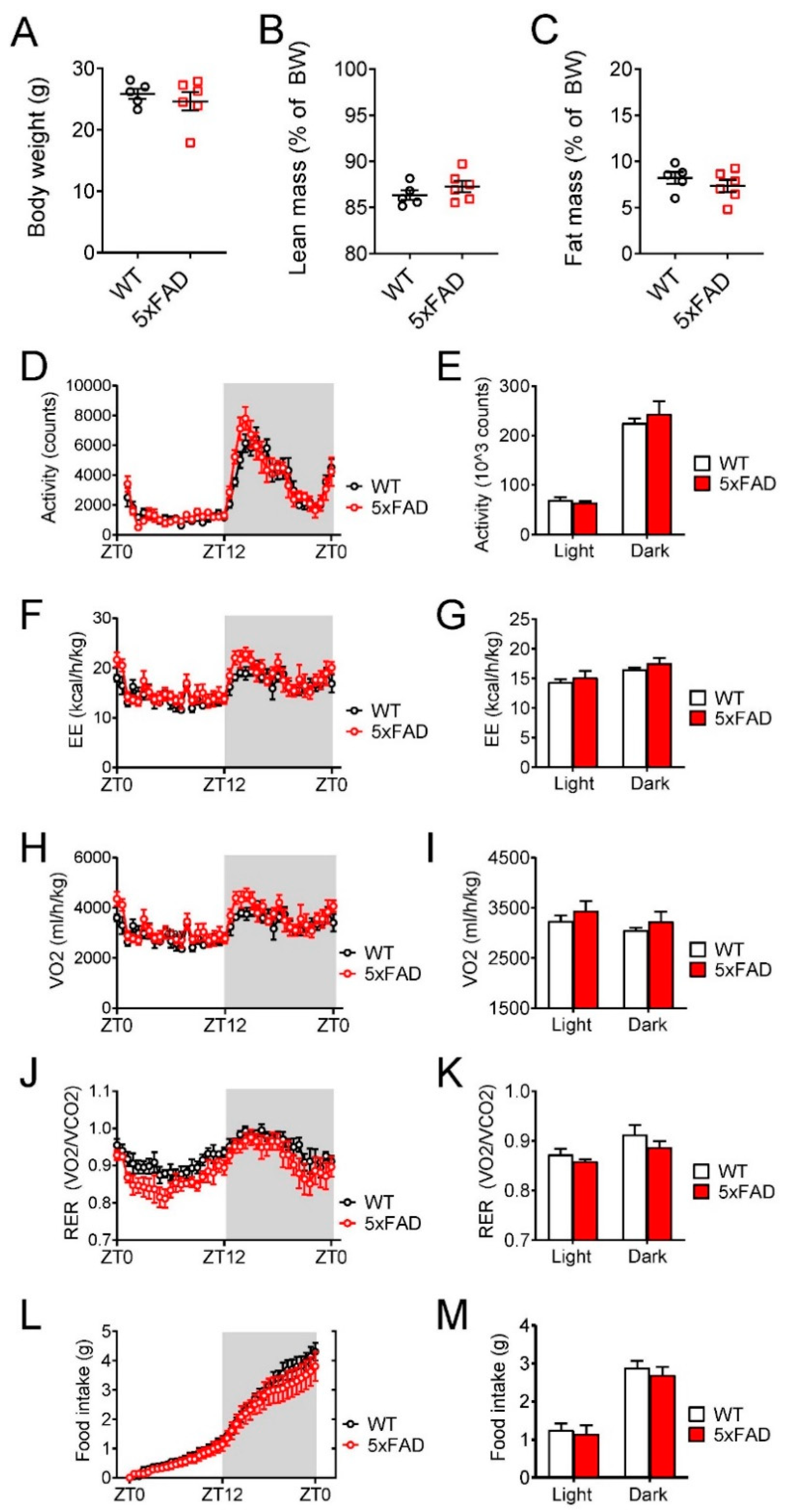
3.1. Young Male 5xFAD Mice Exhibited Normal Overall Energy Homeostasis When Fed Normal Chow Diet (NCD)
3.2. 5xFAD Mice Fed Normal Chow Diet Exhibited Age-Dependent Sexual Dimorphic Effects on Body Weight Maintenance
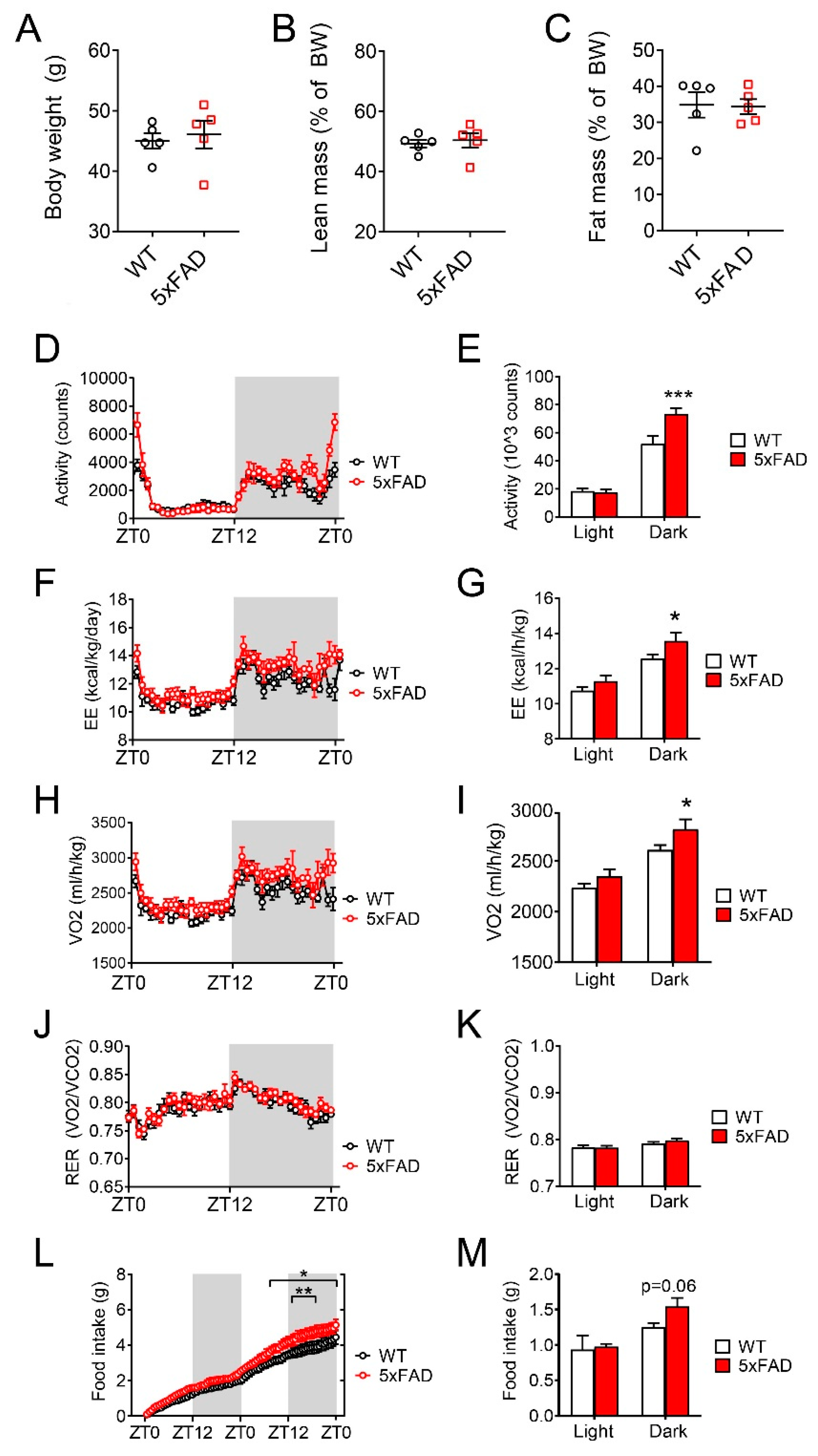
3.3. 5xFAD Mice Displayed Metabolic Defects after High-Fat Diet (HFD) Feeding
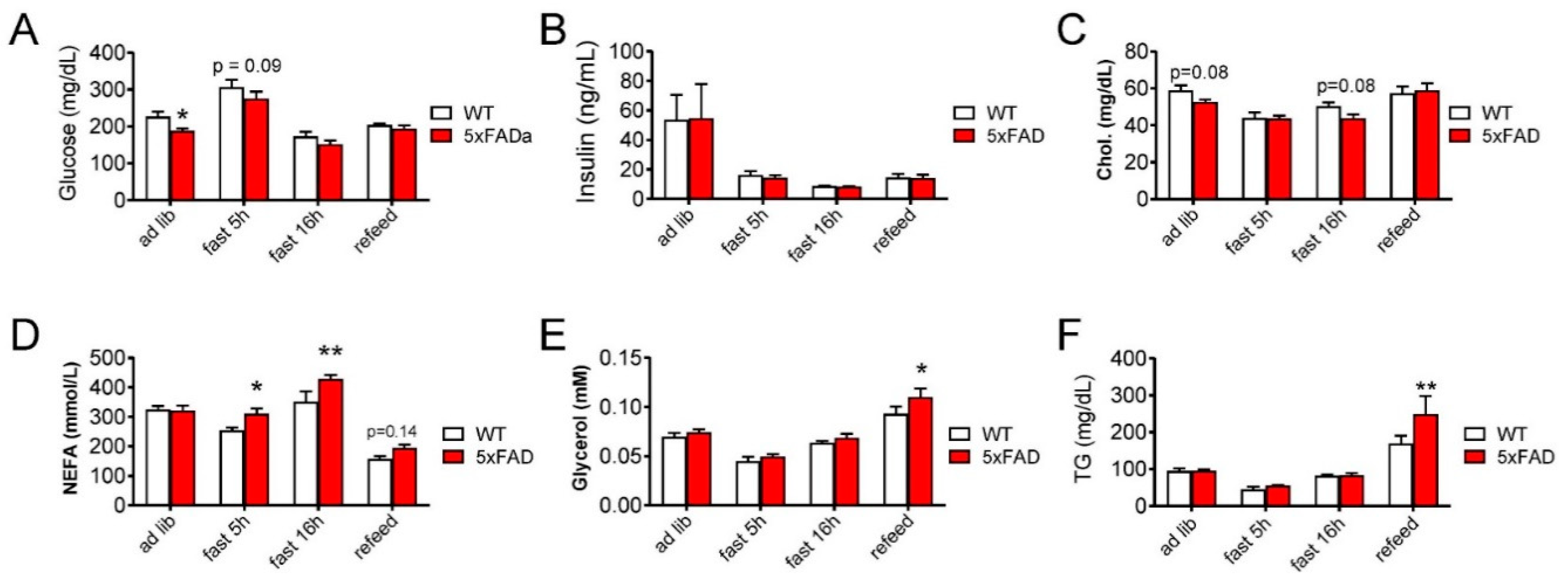
3.4. 5xFAD Mice on High-Fat Diet (HFD) Have Altered Glycemia and Blood Lipid Profile Compared with WT Mice
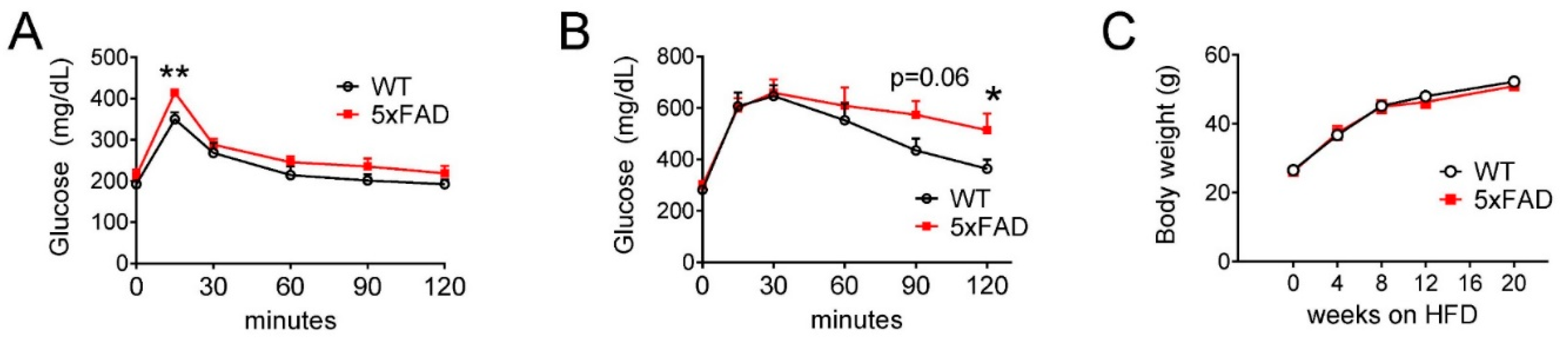
3.5. High-Fat Diet (HFD) Exacerbated the Glucose Intolerance Phenotype in Male 5xFAD Mice
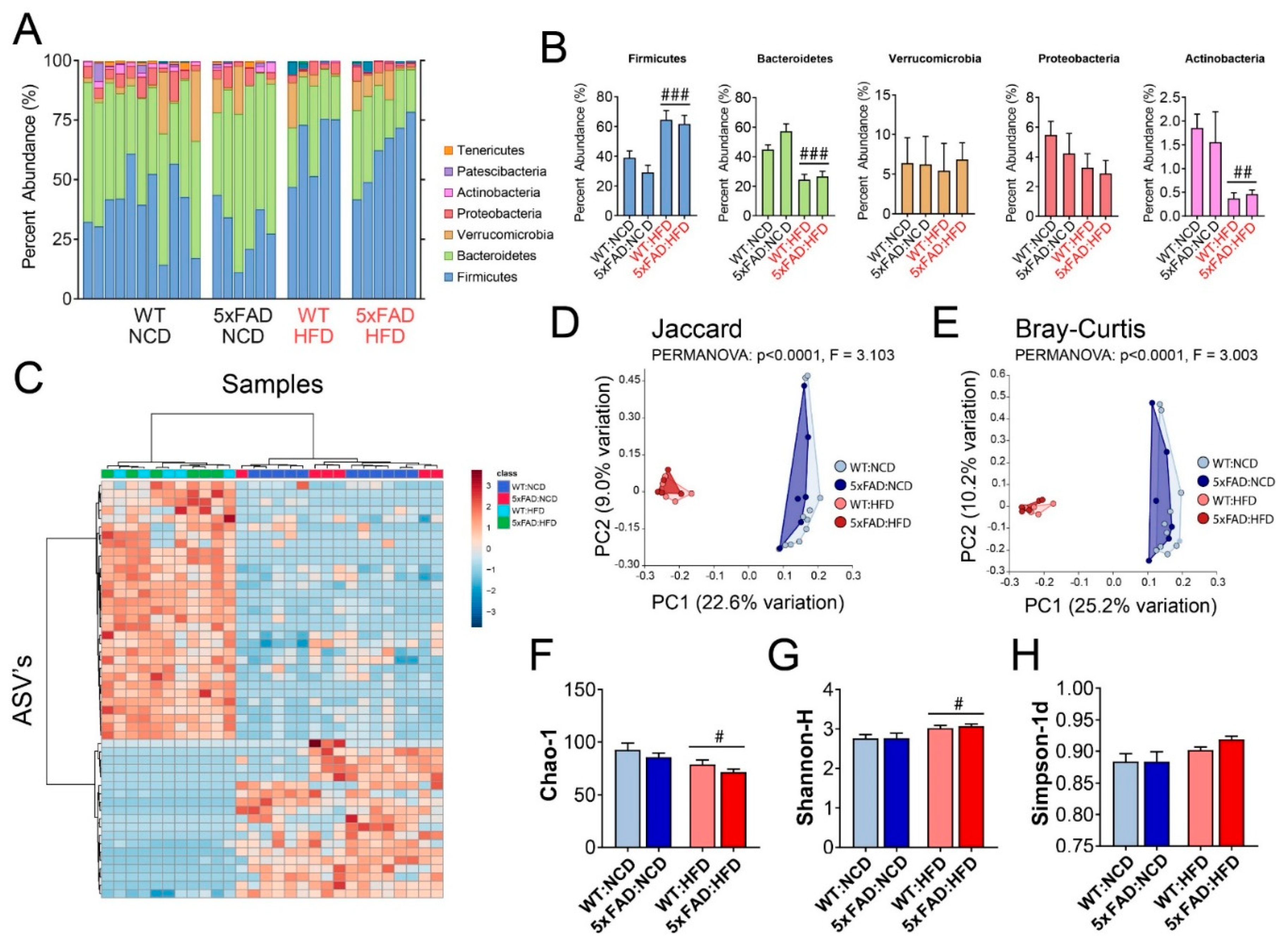
3.6. High-Fat Diet (HFD) Altered the Gut Microbiome Composition in both WT and 5xFAD Mice
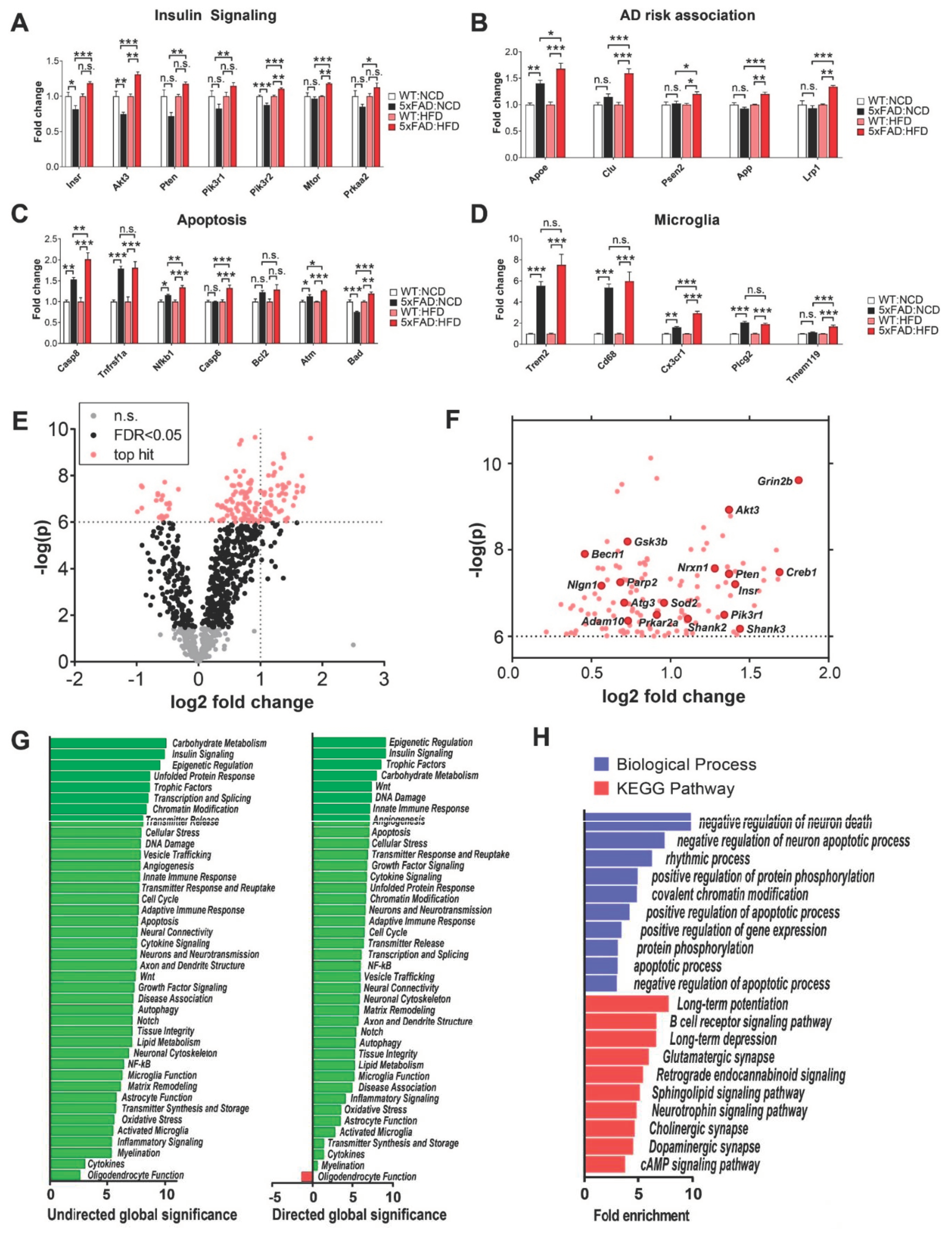
3.7. Amyloidogenic and Inflammatory Pathways in the Hippocampus of 5xFAD Mice Are Exacerbated by High-Fat Diet (HFD)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.M.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.-I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507.e6. [Google Scholar] [CrossRef]
- Ulland, T.K.; Song, W.M.; Huang, S.C.-C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e13. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement 2016, 12, 719–732. [Google Scholar] [CrossRef]
- McCrimmon, R.J.; Ryan, C.M.; Frier, B.M. Diabetes and cognitive dysfunction. Lancet 2012, 379, 2291–2299. [Google Scholar] [CrossRef]
- Strachan, M.W.J.; Reynolds, R.M.; Marioni, R.E.; Price, J.F. Cognitive function, dementia and type 2 diabetes mellitus in the elderly. Nat. Rev. Endocrinol. 2011, 7, 108–114. [Google Scholar] [CrossRef]
- Gudala, K.; Bansal, D.; Schifano, F.; Bhansali, A. Diabetes mellitus and risk of dementia: A meta-analysis of prospective observational studies. J. Diabetes Investig. 2013, 4, 640–650. [Google Scholar] [CrossRef] [Green Version]
- Enzinger, C.; Fazekas, F.; Matthews, P.M.; Ropele, S.; Schmidt, H.; Smith, S.; Schmidt, R. Risk factors for progression of brain atrophy in aging: Six-year follow-up of normal subjects. Neurology 2005, 64, 1704–1711. [Google Scholar] [CrossRef] [PubMed]
- Macauley, S.L.; Stanley, M.; Caesar, E.E.; Yamada, S.A.; Raichle, M.E.; Perez, R.; Mahan, T.E.; Sutphen, C.L.; Holtzman, D.M. Hyperglycemia modulates extracellular amyloid-β concentrations and neuronal activity in vivo. J. Clin. Investig. 2015, 125, 2463–2467. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jais, A.; Solas, M.; Backes, H.; Chaurasia, B.; Kleinridders, A.; Theurich, S.; Mauer, J.; Steculorum, S.M.; Hampel, B.; Goldau, J.; et al. Myeloid-Cell-Derived VEGF Maintains Brain Glucose Uptake and Limits Cognitive Impairment in Obesity. Cell 2016, 165, 882–895. [Google Scholar] [CrossRef] [Green Version]
- Cholerton, B.; Baker, L.D.; Craft, S. Insulin resistance and pathological brain ageing. Diabet. Med. 2011, 28, 1463–1475. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.-Y.; Kazi, H.; Han, L.-Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [Green Version]
- Neth, B.J.; Craft, S. Insulin Resistance and Alzheimer’s Disease: Bioenergetic Linkages. Front. Aging Neurosci. 2017, 9, 345. [Google Scholar] [CrossRef]
- Yusufov, M.; Weyandt, L.L.; Piryatinsky, I. Alzheimer’s disease and diet: A systematic review. Int. J. Neurosci. 2017, 127, 161–175. [Google Scholar] [CrossRef]
- Capurso, C.; Bellanti, F.; Lo Buglio, A.; Vendemiale, G. The Mediterranean Diet Slows Down the Progression of Aging and Helps to Prevent the Onset of Frailty: A Narrative Review. Nutrients 2019, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Feart, C.; Samieri, C.; Barberger-Gateau, P. Mediterranean diet and cognitive health: An update of available knowledge. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 51–62. [Google Scholar] [CrossRef]
- McGrattan, A.M.; McGuinness, B.; McKinley, M.C.; Kee, F.; Passmore, P.; Woodside, J.V.; McEvoy, C.T. Diet and Inflammation in Cognitive Ageing and Alzheimer’s Disease. Curr. Nutr. Rep. 2019, 8, 53–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Féart, C.; Samieri, C.; Rondeau, V.; Amieva, H.; Portet, F.; Dartigues, J.-F.; Scarmeas, N.; Barberger-Gateau, P. Adherence to a Mediterranean diet, cognitive decline, and risk of dementia. JAMA 2009, 302, 638–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef]
- Lin, B.; Hasegawa, Y.; Takane, K.; Koibuchi, N.; Cao, C.; Kim-Mitsuyama, S. High-Fat-Diet Intake Enhances Cerebral Amyloid Angiopathy and Cognitive Impairment in a Mouse Model of Alzheimer’s Disease, Independently of Metabolic Disorders. JAHA 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medrano-Jiménez, E.; Jiménez-Ferrer Carrillo, I.; Pedraza-Escalona, M.; Ramírez-Serrano, C.E.; Álvarez-Arellano, L.; Cortés-Mendoza, J.; Herrera-Ruiz, M.; Jiménez-Ferrer, E.; Zamilpa, A.; Tortoriello, J.; et al. Malva parviflora extract ameliorates the deleterious effects of a high fat diet on the cognitive deficit in a mouse model of Alzheimer’s disease by restoring microglial function via a PPAR-γ-dependent mechanism. J. Neuroinflamm. 2019, 16, 143. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Reilly, A.M.; Zhou, S.; Panigrahi, S.K.; Yan, S.; Conley, J.M.; Sheets, P.L.; Wardlaw, S.L.; Ren, H. Gpr17 deficiency in POMC neurons ameliorates the metabolic derangements caused by long-term high-fat diet feeding. Nutr. Diabetes 2019, 9, 29. [Google Scholar] [CrossRef]
- Walters, W.A.; Caporaso, J.G.; Lauber, C.L.; Berg-Lyons, D.; Fierer, N.; Knight, R. PrimerProspector: De novo design and taxonomic analysis of barcoded polymerase chain reaction primers. Bioinformatics 2011, 27, 1159–1161. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kuczynski, J.; Stombaugh, J.; Walters, W.A.; González, A.; Caporaso, J.G.; Knight, R. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Bioinform. 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernath, M.M.; Bhattacharyya, S.; Nho, K.; Barupal, D.K.; Fiehn, O.; Baillie, R.; Risacher, S.; Arnold, M.; Jacobson, T.; Trojanowski, J.Q.; et al. Serum triglycerides in Alzheimer’s disease: Relation to neuroimaging and CSF biomarkers. Neuroscience 2018, 94, e2088–e2098. [Google Scholar]
- Choi, H.J.; Byun, M.S.; Yi, D.; Choe, Y.M.; Sohn, B.K.; Baek, H.W.; Lee, J.H.; Kim, H.J.; Han, J.Y.; Yoon, E.J.; et al. Association Between Serum Triglycerides and Cerebral Amyloidosis in Cognitively Normal Elderly. Am. J. Geriatr. Psychiatry 2016, 24, 604–612. [Google Scholar] [CrossRef]
- Raffaitin, C.; Gin, H.; Empana, J.-P.; Helmer, C.; Berr, C.; Tzourio, C.; Portet, F.; Dartigues, J.-F.; Alperovitch, A.; Barberger-Gateau, P. Metabolic Syndrome and Risk for Incident Alzheimer’s Disease or Vascular Dementia: The Three-City Study. Diabetes Care 2009, 32, 169–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jais, A.; Brüning, J.C. Hypothalamic inflammation in obesity and metabolic disease. J. Clin. Investig. 2017, 127, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Valdearcos, M.; Douglass, J.D.; Robblee, M.M.; Dorfman, M.D.; Stifler, D.R.; Bennett, M.L.; Gerritse, I.; Fasnacht, R.; Barres, B.A.; Thaler, J.P.; et al. Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell Metab. 2017, 26, 185–197.e3. [Google Scholar] [CrossRef] [Green Version]
- Scearce-Levie, K.; Sanchez, P.E.; Lewcock, J.W. Leveraging preclinical models for the development of Alzheimer disease therapeutics. Nat. Rev. Drug Discov. 2020, 19, 447–462. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Seeley, R.J.; Tschöp, M.H.; Woods, S.C.; Morton, G.J.; Myers, M.G.; D’Alessio, D. Cooperation between brain and islet in glucose homeostasis and diabetes. Nature 2013, 503, 59–66. [Google Scholar] [CrossRef]
- Bentsen, M.A.; Mirzadeh, Z.; Schwartz, M.W. Revisiting How the Brain Senses Glucose-And Why. Cell Metab. 2019, 29, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A.; Farr, S.A.; Salameh, T.S.; Niehoff, M.L.; Rhea, E.M.; Morley, J.E.; Hanson, A.J.; Hansen, K.M.; Craft, S. Triglycerides cross the blood–brain barrier and induce central leptin and insulin receptor resistance. Int. J. Obes. 2018, 42, 391–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaler, J.P.; Yi, C.-X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitz, N.F.; Cronican, A.; Pham, T.; Fogg, A.; Fauq, A.H.; Chapman, R.; Lefterov, I.; Koldamova, R. Liver X Receptor Agonist Treatment Ameliorates Amyloid Pathology and Memory Deficits Caused by High-Fat Diet in APP23 Mice. J. Neurosci. 2010, 30, 6862–6872. [Google Scholar] [CrossRef] [Green Version]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.; Figueroa-Romero, C.; Pacut, C.; Backus, C.; Feldman, E.L. Insulin Resistance Prevents AMPK-induced Tau Dephosphorylation through Akt-mediated Increase in AMPK Ser-485 Phosphorylation. J. Biol. Chem. 2015, 290, 19146–19157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vingtdeux, V.; Davies, P.; Dickson, D.W.; Marambaud, P. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol. 2011, 121, 337–349. [Google Scholar] [CrossRef] [Green Version]
- Arnold, S.E.; Lucki, I.; Brookshire, B.R.; Carlson, G.C.; Browne, C.A.; Kazi, H.; Bang, S.; Choi, B.-R.; Chen, Y.; McMullen, M.F.; et al. High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol. Dis. 2014, 67, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Park, H.-S.; Park, S.-S.; Kim, C.-J.; Shin, M.-S.; Kim, T.-W. Exercise Alleviates Cognitive Functions by Enhancing Hippocampal Insulin Signaling and Neuroplasticity in High-Fat Diet-Induced Obesity. Nutrients 2019, 11, 1603. [Google Scholar] [CrossRef] [Green Version]
- McNay, E.C.; Ong, C.T.; McCrimmon, R.J.; Cresswell, J.; Bogan, J.S.; Sherwin, R.S. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol. Learn. Mem. 2010, 93, 546–553. [Google Scholar] [CrossRef] [Green Version]
- Stranahan, A.M.; Norman, E.D.; Lee, K.; Cutler, R.G.; Telljohann, R.S.; Egan, J.M.; Mattson, M.P. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus 2008, 18, 1085–1088. [Google Scholar] [CrossRef] [Green Version]
- Hao, S.; Dey, A.; Yu, X.; Stranahan, A.M. Dietary obesity reversibly induces synaptic stripping by microglia and impairs hippocampal plasticity. Brain Behav. Immun. 2016, 51, 230–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucsek, Z.; Toth, P.; Sosnowska, D.; Gautam, T.; Mitschelen, M.; Koller, A.; Szalai, G.; Sonntag, W.E.; Ungvari, Z.; Csiszar, A. Obesity in Aging Exacerbates Blood-Brain Barrier Disruption, Neuroinflammation, and Oxidative Stress in the Mouse Hippocampus: Effects on Expression of Genes Involved in Beta-Amyloid Generation and Alzheimer’s Disease. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, 1212–1226. [Google Scholar] [CrossRef] [PubMed]
- Vinuesa, A.; Bentivegna, M.; Calfa, G.; Filipello, F.; Pomilio, C.; Bonaventura, M.M.; Lux-Lantos, V.; Matzkin, M.E.; Gregosa, A.; Presa, J.; et al. Early Exposure to a High-Fat Diet Impacts on Hippocampal Plasticity: Implication of Microglia-Derived Exosome-like Extracellular Vesicles. Mol. Neurobiol. 2019, 56, 5075–5094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beli, E.; Prabakaran, S.; Krishnan, P.; Evans-Molina, C.; Grant, M.B. Loss of Diurnal Oscillatory Rhythms in Gut Microbiota Correlates with Changes in Circulating Metabolites in Type 2 Diabetic db/db Mice. Nutrients 2019, 11, 2310. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, K.; Mulak, A. Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J. Neurogastroenterol. Motil. 2019, 25, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.; Neher, J.J.; Fåk, F.; Jucker, M.; Lasser, T.; et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 41802. [Google Scholar] [CrossRef]
- Bruce-Keller, A.J.; Salbaum, J.M.; Luo, M.; Blanchard, E.; Taylor, C.M.; Welsh, D.A.; Berthoud, H.-R. Obese-type Gut Microbiota Induce Neurobehavioral Changes in the Absence of Obesity. Biol. Psychiatry 2015, 77, 607–615. [Google Scholar] [CrossRef] [Green Version]
- De Morais Fabrício, D.; Chagas, M.H.N.; Diniz, B.S. Frailty and cognitive decline. Transl. Res. 2020, 221, 58–64. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reilly, A.M.; Tsai, A.P.; Lin, P.B.; Ericsson, A.C.; Oblak, A.L.; Ren, H. Metabolic Defects Caused by High-Fat Diet Modify Disease Risk through Inflammatory and Amyloidogenic Pathways in a Mouse Model of Alzheimer’s Disease. Nutrients 2020, 12, 2977. https://doi.org/10.3390/nu12102977
Reilly AM, Tsai AP, Lin PB, Ericsson AC, Oblak AL, Ren H. Metabolic Defects Caused by High-Fat Diet Modify Disease Risk through Inflammatory and Amyloidogenic Pathways in a Mouse Model of Alzheimer’s Disease. Nutrients. 2020; 12(10):2977. https://doi.org/10.3390/nu12102977
Chicago/Turabian StyleReilly, Austin M., Andy P. Tsai, Peter B. Lin, Aaron C. Ericsson, Adrian L. Oblak, and Hongxia Ren. 2020. "Metabolic Defects Caused by High-Fat Diet Modify Disease Risk through Inflammatory and Amyloidogenic Pathways in a Mouse Model of Alzheimer’s Disease" Nutrients 12, no. 10: 2977. https://doi.org/10.3390/nu12102977
APA StyleReilly, A. M., Tsai, A. P., Lin, P. B., Ericsson, A. C., Oblak, A. L., & Ren, H. (2020). Metabolic Defects Caused by High-Fat Diet Modify Disease Risk through Inflammatory and Amyloidogenic Pathways in a Mouse Model of Alzheimer’s Disease. Nutrients, 12(10), 2977. https://doi.org/10.3390/nu12102977