Impact of Cholesterol Metabolism in Immune Cell Function and Atherosclerosis

 , and
, and 
{kind=link}
{kind=link}
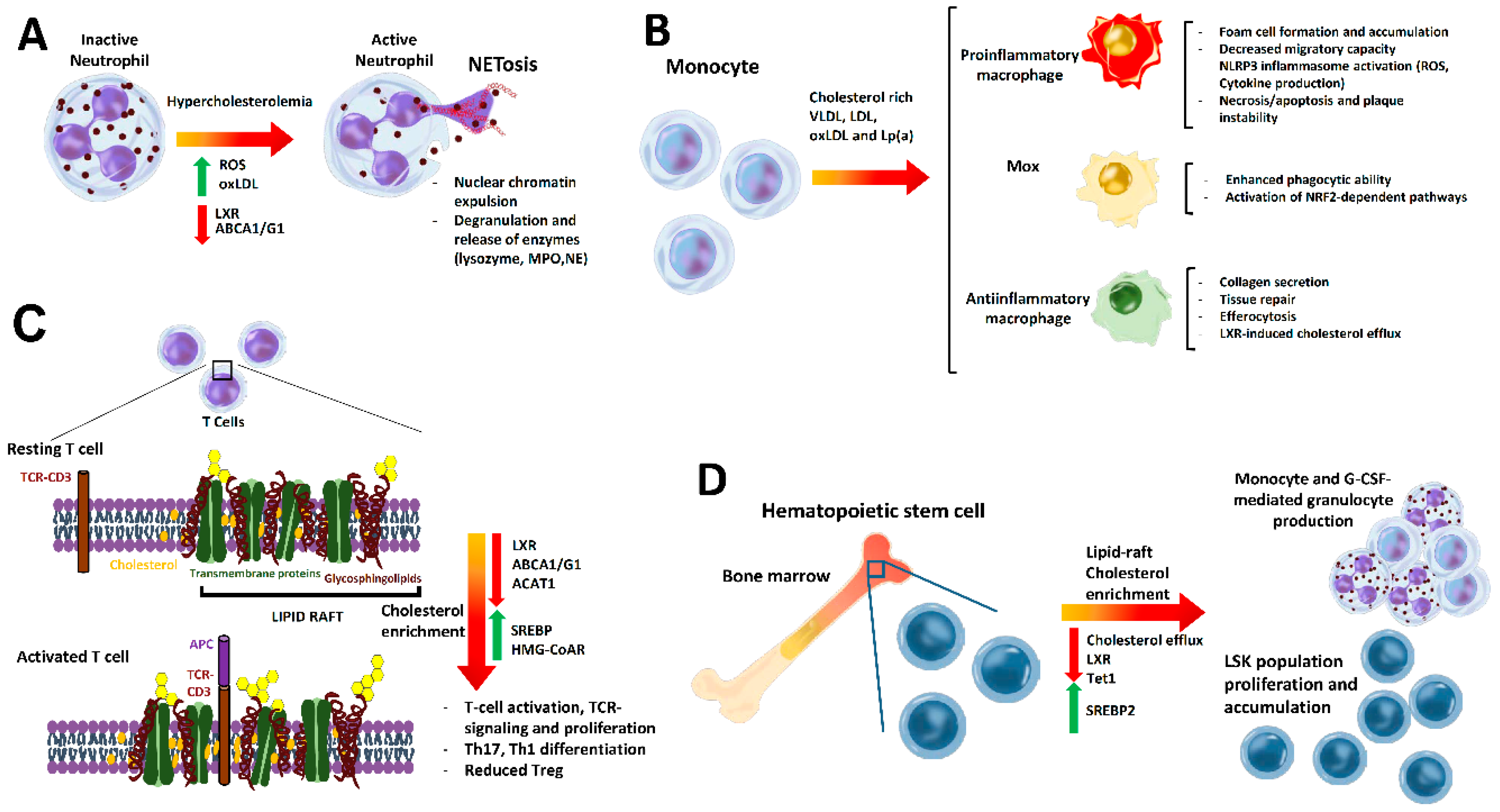{kind=link}
Abstract
:1. Introduction
2. Cholesterol Metabolism
2.1. Cholesterol Absorption and Metabolism

2.2. Endogenous Synthesis of Cholesterol
2.3. Cholesterol Transport and Lipoprotein Metabolism
2.4. Cellular Cholesterol Efflux and Storage
3. Cholesterol in Atherosclerosis
4. Cholesterol Metabolism in Immune Cells
4.1. Cholesterol Effect on Neutrophil Biology in Atherosclerosis
4.2. Cholesterol Impact on Monocytes and Macrophages
4.3. Cholesterol in T Cell Function and Phenotype
4.4. Effect of Cholesterol on Hematopoiesis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABCA1 | adenosine triphosphate-binding cassette transporter A1 |
| ABCB11 | adenosine triphosphate-binding cassette subfamily B member 11 |
| ABCG1 | adenosine triphosphate-binding cassette transporter G1 |
| ABCG8 | adenosine triphosphate-binding cassette transporter G8 |
| ABCG5 | adenosine triphosphate-binding cassette transporter G5 |
| Acetyl-CoA | acetyl coenzyme A |
| ACAT | acetyl-CoA acetyltransferase |
| ApoE | apolipoprotein E |
| BM | bone marrow |
| CETP | cholesteryl ester transfer protein |
| CRP | c-reactive protein |
| CV | cardiovascular |
| CVD | cardiovascular disease |
| CYP7A1 | cytochrome P450 family 7 subfamily A member 1 |
| FXR | farnesoid X receptor |
| G-CSF | granulocyte-colony stimulating factor |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| HDL | high-density lipoprotein |
| HFD | high-fat diet |
| HL | hepatic lipase |
| HMG-CoAR | 3-hydroxy-3-methyl-glutaryl coenzyme A reductase |
| HNF | hepatocyte nuclear factor |
| HSPC | hematopoietic stem and progenitor cell |
| IDL | intermediate-density lipoprotein |
| INSIG1 | insulin-induced gene 1 |
| LDL | low-density lipoprotein |
| LDL-C | low-density lipoprotein cholesterol |
| LDLr | low-density lipoprotein receptor |
| LPL | lipoprotein lipase |
| LPS | lipopolysaccharide |
| LSK+ | Lin-Sca+cKit+ |
| LXR | liver X receptor |
| MPO | myeloperoxidase |
| NCP1L1 | Niemann–Pick C1-Like 1 |
| NE | neutrophil elastase |
| NETs | neutrophil extracellular traps |
| NF-κb | nuclear factor kappa B |
| NF-Y | nuclear transcription factor Y subunit alpha |
| PCSK9 | proprotein convertase subtilisin kexin 9 |
| PPAR | peroxisome proliferator-activated receptor |
| PXR | pregnane X receptor |
| ROS | reactive oxidant species |
| RXR | retinoid X receptor |
| SCAP | SREBP cleavage activating protein |
| SP1 | site 1 protease |
| SQLE | squalene monooxygenase |
| SRA | scavenger receptor type A |
| SRBI | scavenger receptor class B type 1 |
| SRE | sterol regulatory element |
| SREBP2 | sterol regulatory element-binding protein 2 |
| SSD | sterol-sensing domains |
| TCR | T cell receptor |
| TET1 | ten-eleven translocation-1 |
| Th | T helper |
| TLR | toll-like receptor |
| Treg | regulatory T cells |
| VLDL | very low density lipoprotein |
| VSMCs | vascular smooth muscle cells |
References
- De Boer, J.F.; Kuipers, F.; Groen, A.K. Cholesterol Transport Revisited: A New Turbo Mechanism to Drive Cholesterol Excretion. Trends Endocrinol. Metab. 2018, 29, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.W.; Qu, J.; Black, D.D.; Tso, P. Regulation of intestinal lipid metabolism: Current concepts and relevance to disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef] [PubMed]
- Silva Afonso, M.; Marcondes Machado, R.; Ferrari Lavrador, M.S.; Rocha Quintao, E.C.; Moore, K.J.; Lottenberg, A.M. Molecular pathways underlying cholesterol homeostasis. Nutrients 2018, 10, 760. [Google Scholar] [CrossRef] [Green Version]
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. A century of cholesterol and coronaries: From plaques to genes to statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, A.; Goldstein, J.L.; McDonald, J.G.; Brown, M.S. Switch-like control of srebp-2 transport triggered by small changes in ER cholesterol: A delicate balance. Cell Metab. 2008, 8, 512–521. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, L.J.; Cook, E.C.L.; Zelcer, N.; Brown, A.J. The UPS and downs of cholesterol homeostasis. Trends Biochem. Sci. 2014, 39, 527–535. [Google Scholar] [CrossRef]
- Goedeke, L.; Fernández-Hernando, C. Regulation of cholesterol homeostasis. Cell. Mol. Life Sci. 2012, 69, 915–930. [Google Scholar] [CrossRef]
- Calkin, A.C.; Tontonoz, P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat. Rev. Mol. Cell Biol. 2012, 13, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Mazuy, C.; Helleboid, A.; Staels, B.; Lefebvre, P. Nuclear bile acid signaling through the farnesoid X receptor. Cell. Mol. Life Sci. 2015, 72, 1631–1650. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M.; Yurdagul, A.; Tabas, I.; Öörni, K.; Kovanen, P.T.; Lichtman, A.H.H. Monocyte-Macrophages and T Cells in Atherosclerosis. Immunity 2019, 16, 389–406. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Kashirskikh, D.A.; Khotina, V.A.; Grechko, A.V.; Orekhov, A.N. Immune-inflammatory responses in atherosclerosis: The role of myeloid cells. J. Clin. Med. 2019, 8, 1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäck, M.; Yurdagul, A.; Tabas, I.; Öörni, K.; Kovanen, P.T. Inflammation and its Resolution in Atherosclerosis: Mediators and Therapeutic Opportunities; Nature Publishing Group: Berlin, Germany, 2019; Volume 16, pp. 389–406. [Google Scholar]
- Weber, C.; Noels, H. Atherosclerosis: Current Pathogenesis and Therapeutic Options; Nature Publishing Group: Berlin, Germany, 2011; Volume 17, pp. 1410–1422. [Google Scholar]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Hedrick, C.C. Lymphocytes in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 253–257. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yao, S.; Dann, S.M.; Qin, H.; Elson, C.O.; Cong, Y. ERK differentially regulates Th17-and Treg-cell development and contributes to the pathogenesis of colitis. Eur. J. Immunol. 2013, 43, 1716–1726. [Google Scholar] [CrossRef]
- Wigren, M.; Björkbacka, H.; Andersson, L.; Ljungcrantz, I.; Fredrikson, G.N.; Persson, M.; Bryngelsson, C.; Hedblad, B.; Nilsson, J. Low levels of circulating CD4+ FoxP3+ T cells are associated with an increased risk for development of myocardial infarction but not for stroke. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2000–2007. [Google Scholar] [CrossRef] [Green Version]
- Potekhina, A.V.; Pylaeva, E.; Provatorov, S.; Ruleva, N.; Masenko, V.; Noeva, E.; Krasnikova, T.; Arefieva, T. Treg/Th17 balance in stable CAD patients with different stages of coronary atherosclerosis. Atherosclerosis 2015, 238, 17–21. [Google Scholar] [CrossRef]
- Martínez-Hervás, S.; Vinué, Á.; Nú, L.; André s-Blasco, I.; Piqueras, L.; Tomás Real, J.; Francisco Ascaso, J.; Jane Burks, D.; Jesú Sanz, M.; González-Navarro, H. Insulin resistance aggravates atherosclerosis by reducing vascular smooth muscle cell survival and increasing CX 3 CL1/CX 3 CR1 axis. Cardiovasc. Res. 2014, 103, 324–336. [Google Scholar] [CrossRef] [Green Version]
- Silvestre-Roig, C.; Braster, Q.; Ortega-Gomez, A.; Soehnlein, O. Neutrophils as regulators of cardiovascular inflammation. Nat. Rev. Cardiol. 2020, 17, 327–340. [Google Scholar] [CrossRef]
- Döring, Y.; Soehnlein, O.; Weber, C. Neutrophil extracellular traps in atherosclerosis and atherothrombosis. Circ. Res. 2017, 120, 736–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillipson, M.; Kubes, P. The neutrophil in vascular inflammation. Nat. Med. 2011, 17, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Drechsler, M.; Megens, R.T.A.; Van Zandvoort, M.; Weber, C.; Soehnlein, O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation 2010, 122, 1837–1845. [Google Scholar] [CrossRef]
- Westerterp, M.; Fotakis, P.; Ouimet, M.; Bochem, A.E.; Zhang, H.; Molusky, M.M.; Wang, W.; Abramowicz, S.; La Bastide-Van Gemert, S.; Wang, N.; et al. Cholesterol efflux pathways suppress inflammasome activation, NETosis, and atherogenesis. Circulation 2018, 138, 898–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yvan-Charvet, L.; Welch, C.; Pagler, T.A.; Ranalletta, M.; Lamkanfi, M.; Han, S.; Ishibashi, M.; Li, R.; Wang, N.; Tall, A.R. Increased inflammatory gene expression in ABC transporter-deficient macrophages: Free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation 2008, 118, 1837–1847. [Google Scholar] [CrossRef]
- Hong, C.; Kidani, Y.; Noelia, A.; Phung, T.; Ito, A.; Rong, X.; Ericson, K.; Mikkola, H.; Beaven, S.W.; Miller, L.S.; et al. Coordinate regulation of neutrophil homeostasis by liver X receptors in mice. J. Clin. Investig. 2012, 122, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.G.; Doran, A.C.; Fotakis, P.; Westerterp, M.; Antonson, P.; Jiang, H.; Jiang, X.C.; Gustafsson, J.Å.; Tabas, I.; Tall, A.R. LXR suppresses inflammatory gene expression and neutrophil migration through cis-repression and cholesterol efflux. Cell Rep. 2018, 25, 3774–3785. [Google Scholar] [CrossRef] [Green Version]
- Alba, G.; Reyes-Quiróz, M.E.; Sáenz, J.; Geniz, I.; Jiménez, J.; Martín-Nieto, J.; Pintado, E.; Sobrino, F.; Santa-María, C. 7-Keto-cholesterol and 25-hydroxy-1 cholesterol rapidly enhance ROS production in human neutrophils. Eur. J. Nutr. 2016, 55, 2485–2492. [Google Scholar] [CrossRef]
- Awasthi, D.; Nagarkoti, S.; Kumar, A.; Dubey, M.; Singh, A.K.; Pathak, P.; Chandra, T.; Barthwal, M.K.; Dikshit, M. Oxidized LDL induced extracellular trap formation in human neutrophils via TLR-PKC-IRAK-MAPK and NADPH-oxidase activation. Free Radic. Biol. Med. 2016, 93, 190–203. [Google Scholar] [CrossRef]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [Green Version]
- Wen, G.; An, W.; Chen, J.; Maguire, E.M.; Chen, Q.; Yang, F.; Pearce, S.W.A.; Kyriakides, M.; Zhang, L.; Ye, S.; et al. Genetic and pharmacologic inhibition of the neutrophil elastase inhibits experimental atherosclerosis. J. Am. Heart Assoc. 2018, 7, 1–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Sjöberg, S.; Tang, T.T.; Öörni, K.; Wu, W.; Liu, C.; Secco, B.; Tia, V.; Sukhova, G.K.; Fernandes, C.; et al. Cathepsin G activity lowers plasma LDL and reduces atherosclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 2174–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazor, R.; Shurtz-Swirski, R.; Farah, R.; Kristal, B.; Shapiro, G.; Dorlechter, F.; Cohen-Mazor, M.; Meilin, E.; Tamara, S.; Sela, S. Primed polymorphonuclear leukocytes constitute a possible link between inflammation and oxidative stress in hyperlipidemic patients. Atherosclerosis 2008, 197, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Puntoni, M.; Sbrana, F.; Bigazzi, F.; Minichilli, F.; Ferdeghini, E.; Sampietro, T. Myeloperoxidase modulation by LDL apheresis in Familial Hypercholesterolemia. Lipids Health Dis. 2011, 10, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balta, S.; Celik, T.; Mikhailidis, D.P.; Ozturk, C.; Demirkol, S.; Aparci, M.; Iyisoy, A. The Relation between Atherosclerosis and the Neutrophil-Lymphocyte Ratio. Clin. Appl. Thromb. 2014, 22, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Kanter, J.E.; Hsu, C.-C.; Bornfeldt, K.E. Monocytes and Macrophages as Protagonists in Vascular Complications of Diabetes. Front. Cardiovasc. Med. 2020, 7, 1–16. [Google Scholar] [CrossRef]
- Orsó, E.; Schmitz, G. Lipoprotein(a) and its role in inflammation, atherosclerosis and malignancies. Clin. Res. Cardiol. Suppl. 2017, 12, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Bensinger, S.J.; Bradley, M.N.; Joseph, S.B.; Zelcer, N.; Janssen, E.M.; Hausner, M.A.; Shih, R.; Parks, J.S.; Edwards, P.A.; Jamieson, B.D.; et al. LXR Signaling Couples Sterol Metabolism to Proliferation in the Acquired Immune Response. Cell 2008, 134, 97–111. [Google Scholar] [CrossRef] [Green Version]
- Koelwyn, G.J.; Corr, E.M.; Erbay, E.; Moore, K.J. Regulation of macrophage immunometabolism in atherosclerosis. Nat. Immunol. 2018, 19, 526–537. [Google Scholar] [CrossRef] [Green Version]
- Aluganti Narasimhulu, C.; Fernandez-Ruiz, I.; Selvarajan, K.; Jiang, X.; Sengupta, B.; Riad, A.; Parthasarathy, S. Atherosclerosis—Do we know enough already to prevent it? Curr. Opin. Pharmacol. 2016, 27, 92–102. [Google Scholar] [CrossRef]
- Groh, L.; Keating, S.T.; Joosten, L.A.B.; Netea, M.G.; Riksen, N.P. Monocyte and macrophage immunometabolism in atherosclerosis. Semin. Immunopathol. 2018, 40, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Reboldi, A.; Dang, E. Cholesterol metabolism in innate and adaptive response [version 1; peer review: 2 approved]. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swirski, F.K.; Libby, P.; Aikawa, E.; Alcaide, P.; Luscinskas, F.W.; Weissleder, R.; Pittet, M.J. Ly-6C hi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J. Clin. Investig. 2007, 117. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Gower, R.M.; Wang, H.; Perrard, X.-Y.D.; Ma, R.; Bullard, D.C.; Burns, A.R.; Paul, A.; Smith, C.W.; Simon, S.I.; et al. Functional role of CD11c+ monocytes in atherogenesis associated with hypercholesterolemia. Circulation 2009, 119, 2708–2717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolani, S.; Pagler, T.A.; Murphy, A.J.; Bochem, A.E.; Abramowicz, S.; Welch, C.; Nagareddy, P.R.; Holleran, S.; Hovingh, G.K.; Kuivenhoven, J.A.; et al. Hypercholesterolemia and reduced HDL-C promote hematopoietic stem cell proliferation and monocytosis: Studies in mice and FH children. Atherosclerosis 2013, 229, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Shirai, T.; Nazarewicz, R.R.; Wallis, B.B.; Yanes, R.E.; Watanabe, R.; Hilhorst, M.; Tian, L.; Harrison, D.G.; Giacomini, J.C.; Assimes, T.L.; et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J. Exp. Med. 2016, 213, 337–354. [Google Scholar] [CrossRef] [Green Version]
- Fessler, M.B. Regulation of adaptive immunity in health and disease by cholesterol metabolism. Curr. Allergy Asthma Rep. 2015, 15, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Robinson, G.A.; Waddington, K.E.; Pineda-Torra, I.; Jury, E.C. Transcriptional regulation of T-cell lipid metabolism: Implications for plasma membrane lipid rafts and T-cell function. Front. Immunol. 2017, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.B.; Bishop-Bailey, D.; Estrada-Hernandez, T.; Hla, T.; Puddington, L.; Padula, S.J. The nuclear receptor PPARγ and immunoregulation: PPARγ mediates inhibition of helper T Cell responses. J. Immunol. 2000, 164, 1364–1371. [Google Scholar] [CrossRef] [Green Version]
- Kidani, Y.; Elsaesser, H.; Hock, M.B.; Vergnes, L.; Williams, K.J.; Argus, J.P.; Marbois, B.N.; Komisopoulou, E.; Wilson, E.B.; Osborne, T.F.; et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat. Immunol. 2013, 14, 489–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailer, R.K.W.; Gisterå, A.; Polyzos, K.A.; Ketelhuth, D.F.J.; Hansson, G.K. hypercholesterolemia induces differentiation of regulatory t cells in the liver. Circ. Res. 2017, 120, 1740–1753. [Google Scholar] [CrossRef] [PubMed]
- Proto, J.D.; Doran, A.C.; Subramanian, M.; Wang, H.; Zhang, M.; Sozen, E.; Rymond, C.C.; Kuriakose, G.; D’Agati, V.; Winchester, R.; et al. Hypercholesterolemia induces T cell expansion in humanized immune mice. J. Clin. Investig. 2018, 128, 2370–2375. [Google Scholar] [CrossRef] [Green Version]
- Surls, J.; Nazarov-Stoica, C.; Kehl, M.; Olsen, C.; Casares, S.; Brumeanu, T.-D. Increased membrane cholesterol in lymphocytes diverts T-Cells toward an inflammatory response. PLoS ONE 2012, 7, e38733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, A.J.; Gebre, A.K.; Parks, J.S.; Hedrick, C.C. ATP-binding cassette transporter G1 negatively regulates thymocyte and peripheral lymphocyte proliferation. J. Immunol. 2010, 184, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.Y.; Gaddis, D.E.; Wu, R.; McSkimming, C.; Haynes, L.D.; Taylor, A.M.; McNamara, C.A.; Sorci-Thomas, M.; Hedrick, C.C. Loss of ABCG1 influences regulatory T cell differentiation and atherosclerosis. J. Clin. Investig. 2016, 126, 3236–3246. [Google Scholar] [CrossRef] [Green Version]
- Chyu, K.Y.; Lio, W.M.; Dimayuga, P.C.; Zhou, J.; Zhao, X.; Yano, J.; Trinidad, P.; Honjo, T.; Cercek, B.; Shah, P.K. Cholesterol lowering modulates T cell function in vivo and in vitro. PLoS ONE 2014, 9, e92095. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, A.J.; Zabalawi, M.; Grayson, J.M.; Weant, A.E.; Major, A.S.; Owen, J.; Bharadwaj, M.; Walzem, R.; Chan, L.; Oka, K.; et al. Apolipoprotein A-I and its role in lymphocyte cholesterol homeostasis and autoimmunity. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 843–849. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Zhang, K.; Li, J.; Dong, M.; Yang, J.; An, G.; Qin, W.; Gao, F.; Zhang, C.; Zhang, Y. Statins induce the accumulation of regulatory T cells in atherosclerotic plaque. Mol. Med. 2012, 18, 598–605. [Google Scholar] [CrossRef]
- Josefowicz, S.Z.; Lu, L.-F.; Rudensky, A.Y. Regulatory T Cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Yan, C.; Li, H.; Huang, W.; Shi, X.; Huang, M.; Wang, Y.; Pan, W.; Cai, M.; Li, L.; et al. Lipid-dependent conformational dynamics underlie the functional versatility of T-cell receptor. Cell Res. 2017, 27, 505–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bietz, A.; Zhu, H.; Xue, M.; Xu, C. Cholesterol metabolism in T cells. Front. Immunol. 2017, 8, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swamy, M.; Beck-Garcia, K.; Beck-Garcia, E.; Hartl, F.A.; Morath, A.; Yousefi, O.S.; Dopfer, E.P.; Molnár, E.; Schulze, A.K.; Blanco, R.; et al. A cholesterol-based allostery model of t cell receptor phosphorylation. Immunity 2016, 44, 1091–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Beck-García, K.; Zorzin, C.; Schamel, W.W.A.; Davis, M.M. Inhibition of T cell receptor signaling by cholesterol sulfate, a naturally occurring derivative of membrane cholesterol. Nat. Immunol. 2016, 17, 844–850. [Google Scholar] [CrossRef]
- Zelcer, N.; Sharpe, L.J.; Loregger, A.; Kristiana, I.; Cook, E.C.L.; Phan, L.; Stevenson, J.; Brown, A.J. The E3 ubiquitin ligase MARCH6 degrades squalene monooxygenase and affects 3-hydroxy-3-methyl-glutaryl coenzyme A reductase and the cholesterol synthesis pathway. Mol. Cell. Biol. 2014, 34, 1262–1270. [Google Scholar] [CrossRef] [Green Version]
- Soroosh, P.; Wu, J.; Xue, X.; Song, J.; Sutton, S.W.; Sablad, M.; Yu, J.; Nelen, M.I.; Liu, X.; Castro, G.; et al. Oxysterols are agonist ligands of RORγt and drive Th17 cell differentiation. Proc. Natl. Acad. Sci. USA 2014, 111, 12163–12168. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Wang, Y.; Hao, L.Y.; Liu, X.; Lesch, C.A.; Sanchez, B.M.; Wendling, J.M.; Morgan, R.W.; Aicher, T.D.; Carter, L.L.; et al. Sterol metabolism controls TH17 differentiation by generating endogenous RORγ agonists. Nat. Chem. Biol. 2015, 11, 141–147. [Google Scholar] [CrossRef]
- Coller, B.S. Leukocytosis and ischemic vascular disease morbidity and mortality: Is it time to intervene? Arterioscler. Thromb. Vasc. Biol. 2005, 25, 658–670. [Google Scholar] [CrossRef] [Green Version]
- Soehnlein, O.; Swirski, F.K. Hypercholesterolemia links hematopoiesis with atherosclerosis. Trends Endocrinol. Metab. 2013, 24, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Westerterp, M.; Gourion-Arsiquaud, S.; Murphy, A.J.; Shih, A.; Cremers, S.; Levine, R.L.; Tall, A.R.; Yvan-Charvet, L. Regulation of hematopoietic stem and progenitor cell mobilization by cholesterol efflux pathways. Cell Stem Cell 2012, 11, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Murphy, A.J.; Akhtari, M.; Tolani, S.; Pagler, T.; Bijl, N.; Kuo, C.L.; Wang, M.; Sanson, M.; Abramowicz, S.; Welch, C.; et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J. Clin. Investig. 2011, 121, 4138–4149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yvan-Charvet, L.; Pagler, T.; Gautier, E.L.; Avagyan, S.; Siry, R.L.; Han, S.; Welch, C.L.; Wang, N.; Randolph, G.J.; Snoeck, H.W.; et al. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 2010, 328, 1689–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Schouteden, S.; Geenens, R.; van Duppen, V.; Herijgers, P.; Holvoet, P.; van Veldhoven, P.P.; Verfaillie, C.M. Hematopoietic stem/progenitor cell proliferation and differentiation is differentially regulated by high-density and low-density lipoproteins in mice. PLoS ONE 2012, 7, e47286. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Zhao, D.; Schouteden, S.; Sorci-Thomas, M.G.; Van Veldhoven, P.P.; Eggermont, K.; Liu, G.; Verfaillie, C.M.; Feng, Y. Regulation of high-density lipoprotein on hematopoietic stem/progenitor cells in atherosclerosis requires scavenger receptor type BI expression. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1900–1909. [Google Scholar] [CrossRef] [Green Version]
- Gu, Q.; Yang, X.; Lv, J.; Zhang, J.; Xia, B.; Kim, J.D.; Wang, R.; Xiong, F.; Meng, S.; Clements, T.P.; et al. AIBP-mediated cholesterol efflux instructs hematopoietic stem and progenitor cell fate. Science 2019, 363, 1085–1088. [Google Scholar] [CrossRef] [PubMed]
- Tie, G.; Yan, J.; Khair, L.; Tutto, A.; Messina, L.M. Hypercholesterolemia Accelerates the Aging Phenotypes of Hematopoietic Stem Cells by a Tet1-Dependent Pathway. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguilar-Ballester, M.; Herrero-Cervera, A.; Vinué, Á.; Martínez-Hervás, S.; González-Navarro, H. Impact of Cholesterol Metabolism in Immune Cell Function and Atherosclerosis. Nutrients 2020, 12, 2021. https://doi.org/10.3390/nu12072021
Aguilar-Ballester M, Herrero-Cervera A, Vinué Á, Martínez-Hervás S, González-Navarro H. Impact of Cholesterol Metabolism in Immune Cell Function and Atherosclerosis. Nutrients. 2020; 12(7):2021. https://doi.org/10.3390/nu12072021
Chicago/Turabian StyleAguilar-Ballester, María, Andrea Herrero-Cervera, Ángela Vinué, Sergio Martínez-Hervás, and Herminia González-Navarro. 2020. "Impact of Cholesterol Metabolism in Immune Cell Function and Atherosclerosis" Nutrients 12, no. 7: 2021. https://doi.org/10.3390/nu12072021
APA StyleAguilar-Ballester, M., Herrero-Cervera, A., Vinué, Á., Martínez-Hervás, S., & González-Navarro, H. (2020). Impact of Cholesterol Metabolism in Immune Cell Function and Atherosclerosis. Nutrients, 12(7), 2021. https://doi.org/10.3390/nu12072021