
Synthetic Microbiomes on the Rise—Application in Deciphering the Role of Microbes in Host Health and Disease


Abstract
:1. Introduction
2. Syncoms Representing Simplified Intestinal Microbiome
3. Utilizing Syncoms to Model Gut Disease Phenotypes
3.1. Infectious Diseases
3.2. Inflammatory Diseases
3.3. Metabolic Disorders
3.4. Colorectal Cancer
4. Differences between Human-Derived and Model-Specific Communities
5. Conclusions and Future Prospective
Author Contributions
Funding
Conflicts of Interest
References
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.J.; Ivanov, I.I.; Darce, J.; Hattori, K.; Shima, T.; Umesaki, Y.; Littman, D.R.; Benoist, C.; Mathis, D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 2010, 32, 815–827. [Google Scholar] [CrossRef] [Green Version]
- Wen, L.; Ley, R.E.; Volchkov, P.Y.; Stranges, P.B.; Avanesyan, L.; Stonebraker, A.C.; Hu, C.; Wong, F.S.; Szot, G.L.; Bluestone, J.A.; et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 2008, 455, 1109–1113. [Google Scholar] [CrossRef]
- Ussar, S.; Griffin, N.W.; Bezy, O.; Fujisaka, S.; Vienberg, S.; Softic, S.; Deng, L.; Bry, L.; Gordon, J.I.; Kahn, C.R. Interactions between Gut Microbiota, Host Genetics and Diet Modulate the Predisposition to Obesity and Metabolic Syndrome. Cell Metab. 2015, 22, 516–530. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.K.; Menezes, J.S.; Umesaki, Y.; Mazmanian, S.K. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4615–4622. [Google Scholar] [CrossRef] [Green Version]
- Liang, Q.; Chiu, J.; Chen, Y.; Huang, Y.; Higashimori, A.; Fang, J.; Brim, H.; Ashktorab, H.; Ng, S.C.; Ng, S.S.M.; et al. Fecal Bacteria Act as Novel Biomarkers for Noninvasive Diagnosis of Colorectal Cancer. Clin. Cancer Res. 2017, 23, 2061–2070. [Google Scholar] [CrossRef] [Green Version]
- Surana, N.K.; Kasper, D.L. Moving beyond microbiome-wide associations to causal microbe identification. Nature 2017, 552, 244–247. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Quinn, R.A.; Debelius, J.; Xu, Z.Z.; Morton, J.; Garg, N.; Jansson, J.K.; Dorrestein, P.C.; Knight, R. Microbiome-wide association studies link dynamic microbial consortia to disease. Nature 2016, 535, 94–103. [Google Scholar] [CrossRef]
- Basic, M.; Bleich, A. Gnotobiotics: Past, present and future. Lab. Anim. 2019, 53, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Tlaskalova-Hogenova, H.; Stepankova, R.; Kozakova, H.; Hudcovic, T.; Vannucci, L.; Tuckova, L.; Rossmann, P.; Hrncir, T.; Kverka, M.; Zakostelska, Z.; et al. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: Contribution of germ-free and gnotobiotic animal models of human diseases. Cell Mol. Immunol. 2011, 8, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Stecher, B.; Berry, D.; Loy, A. Colonization resistance and microbial ecophysiology: Using gnotobiotic mouse models and single-cell technology to explore the intestinal jungle. FEMS Microbiol. Rev. 2013, 37, 793–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavel, T.; Gomes-Neto, J.C.; Lagkouvardos, I.; Ramer-Tait, A.E. Deciphering interactions between the gut microbiota and the immune system via microbial cultivation and minimal microbiomes. Immunol. Rev. 2017, 279, 8–22. [Google Scholar] [CrossRef]
- Lagkouvardos, I.; Pukall, R.; Abt, B.; Foesel, B.U.; Meier-Kolthoff, J.P.; Kumar, N.; Bresciani, A.; Martinez, I.; Just, S.; Ziegler, C.; et al. The Mouse Intestinal Bacterial Collection (miBC) provides host-specific insight into cultured diversity and functional potential of the gut microbiota. Nat. Microbiol. 2016, 1, 16131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poyet, M.; Groussin, M.; Gibbons, S.M.; Avila-Pacheco, J.; Jiang, X.; Kearney, S.M.; Perrotta, A.R.; Berdy, B.; Zhao, S.; Lieberman, T.D.; et al. A library of human gut bacterial isolates paired with longitudinal multiomics data enables mechanistic microbiome research. Nat. Med. 2019, 25, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Wylensek, D.; Hitch, T.C.A.; Riedel, T.; Afrizal, A.; Kumar, N.; Wortmann, E.; Liu, T.; Devendran, S.; Lesker, T.R.; Hernandez, S.B.; et al. A collection of bacterial isolates from the pig intestine reveals functional and taxonomic diversity. Nat. Commun. 2020, 11, 6389. [Google Scholar] [CrossRef] [PubMed]
- Mabwi, H.A.; Kim, E.; Song, D.G.; Yoon, H.S.; Pan, C.H.; Komba, E.V.G.; Ko, G.; Cha, K.H. Synthetic gut microbiome: Advances and challenges. Comput. Struct. Biotechnol. J. 2021, 19, 363–371. [Google Scholar] [CrossRef]
- Schaedler, R.W.; Dubs, R.; Costello, R. Association of Germfree Mice with Bacteria Isolated from Normal Mice. J. Exp. Med. 1965, 122, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Schaedler, R.W.; Dubos, R.; Costello, R. The Development of the Bacterial Flora in the Gastrointestinal Tract of Mice. J. Exp. Med. 1965, 122, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Wymore Brand, M.; Wannemuehler, M.J.; Phillips, G.J.; Proctor, A.; Overstreet, A.M.; Jergens, A.E.; Orcutt, R.P.; Fox, J.G. The Altered Schaedler Flora: Continued Applications of a Defined Murine Microbial Community. ILAR J. 2015, 56, 169–178. [Google Scholar] [CrossRef] [Green Version]
- Orcutt, R.; Gianni, F.; Judge, R. Development of an “altered Schaedler flora” for NCI gnotobiotic rodents. Microecol. Ther. 1987, 17, 59. [Google Scholar]
- Dewhirst, F.E.; Chien, C.C.; Paster, B.J.; Ericson, R.L.; Orcutt, R.P.; Schauer, D.B.; Fox, J.G. Phylogeny of the defined murine microbiota: Altered Schaedler flora. Appl. Environ. Microbiol. 1999, 65, 3287–3292. [Google Scholar] [CrossRef] [Green Version]
- Bolsega, S.; Basic, M.; Smoczek, A.; Buettner, M.; Eberl, C.; Ahrens, D.; Odum, K.A.; Stecher, B.; Bleich, A. Composition of the Intestinal Microbiota Determines the Outcome of Virus-Triggered Colitis in Mice. Front. Immunol. 2019, 10, 1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stecher, B.; Chaffron, S.; Kappeli, R.; Hapfelmeier, S.; Freedrich, S.; Weber, T.C.; Kirundi, J.; Suar, M.; McCoy, K.D.; von Mering, C.; et al. Like will to like: Abundances of closely related species can predict susceptibility to intestinal colonization by pathogenic and commensal bacteria. PLoS Pathog. 2010, 6, e1000711. [Google Scholar] [CrossRef] [PubMed]
- Stehr, M.; Greweling, M.C.; Tischer, S.; Singh, M.; Blocker, H.; Monner, D.A.; Muller, W. Charles River altered Schaedler flora (CRASF) remained stable for four years in a mouse colony housed in individually ventilated cages. Lab. Anim. 2009, 43, 362–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basic, M.; Bolsega, S.; Smoczek, A.; Glasner, J.; Hiergeist, A.; Eberl, C.; Stecher, B.; Gessner, A.; Bleich, A. Monitoring and contamination incidence of gnotobiotic experiments performed in microisolator cages. Int. J. Med. Microbiol. 2021, 311, 151482. [Google Scholar] [CrossRef]
- Elie, C.; Mathieu, A.; Saliou, A.; Villain, A.; Darnaud, M.; Leulier, F.; Tamellini, A. Draft Genome Sequences of 15 Bacterial Species Constituting the Stable Defined Intestinal Microbiota of the GM15 Gnotobiotic Mouse Model. Microbiol. Resour. Announc. 2020, 9, e00686-20. [Google Scholar] [CrossRef]
- Li, H.; Limenitakis, J.P.; Fuhrer, T.; Geuking, M.B.; Lawson, M.A.; Wyss, M.; Brugiroux, S.; Keller, I.; Macpherson, J.A.; Rupp, S.; et al. The outer mucus layer hosts a distinct intestinal microbial niche. Nat. Commun. 2015, 6, 8292. [Google Scholar] [CrossRef]
- Brugiroux, S.; Beutler, M.; Pfann, C.; Garzetti, D.; Ruscheweyh, H.J.; Ring, D.; Diehl, M.; Herp, S.; Lotscher, Y.; Hussain, S.; et al. Genome-guided design of a defined mouse microbiota that confers colonization resistance against Salmonella enterica serovar Typhimurium. Nat. Microbiol. 2016, 2, 16215. [Google Scholar] [CrossRef]
- Eberl, C.; Ring, D.; Munch, P.C.; Beutler, M.; Basic, M.; Slack, E.C.; Schwarzer, M.; Srutkova, D.; Lange, A.; Frick, J.S.; et al. Reproducible Colonization of Germ-Free Mice with the Oligo-Mouse-Microbiota in Different Animal Facilities. Front. Microbiol. 2019, 10, 2999. [Google Scholar] [CrossRef] [PubMed]
- Streidl, T.; Karkossa, I.; Segura Munoz, R.R.; Eberl, C.; Zaufel, A.; Plagge, J.; Schmaltz, R.; Schubert, K.; Basic, M.; Schneider, K.M.; et al. The gut bacterium Extibacter muris produces secondary bile acids and influences liver physiology in gnotobiotic mice. Gut Microbes 2021, 13, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Eftychi, C.; Schwarzer, R.; Vlantis, K.; Wachsmuth, L.; Basic, M.; Wagle, P.; Neurath, M.F.; Becker, C.; Bleich, A.; Pasparakis, M. Temporally Distinct Functions of the Cytokines IL-12 and IL-23 Drive Chronic Colon Inflammation in Response to Intestinal Barrier Impairment. Immunity 2019, 51, 367–380.e364. [Google Scholar] [CrossRef] [PubMed]
- Kittana, H.; Gomes-Neto, J.C.; Heck, K.; Geis, A.L.; Segura Munoz, R.R.; Cody, L.A.; Schmaltz, R.J.; Bindels, L.B.; Sinha, R.; Hostetter, J.M.; et al. Commensal Escherichia coli Strains Can Promote Intestinal Inflammation via Differential Interleukin-6 Production. Front. Immunol. 2018, 9, 2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basic, M.; Keubler, L.M.; Buettner, M.; Achard, M.; Breves, G.; Schroder, B.; Smoczek, A.; Jorns, A.; Wedekind, D.; Zschemisch, N.H.; et al. Norovirus triggered microbiota-driven mucosal inflammation in interleukin 10-deficient mice. Inflamm. Bowel Dis. 2014, 20, 431–443. [Google Scholar] [CrossRef] [Green Version]
- Donohoe, D.R.; Holley, D.; Collins, L.B.; Montgomery, S.A.; Whitmore, A.C.; Hillhouse, A.; Curry, K.P.; Renner, S.W.; Greenwalt, A.; Ryan, E.P.; et al. A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov. 2014, 4, 1387–1397. [Google Scholar] [CrossRef] [Green Version]
- Studer, N.; Desharnais, L.; Beutler, M.; Brugiroux, S.; Terrazos, M.A.; Menin, L.; Schurch, C.M.; McCoy, K.D.; Kuehne, S.A.; Minton, N.P.; et al. Functional Intestinal Bile Acid 7alpha-Dehydroxylation by Clostridium scindens Associated with Protection from Clostridium difficile Infection in a Gnotobiotic Mouse Model. Front. Cell Infect. Microbiol. 2016, 6, 191. [Google Scholar] [CrossRef] [Green Version]
- Fischer, F.; Romero, R.; Hellhund, A.; Linne, U.; Bertrams, W.; Pinkenburg, O.; Eldin, H.S.; Binder, K.; Jacob, R.; Walker, A.; et al. Dietary cellulose induces anti-inflammatory immunity and transcriptional programs via maturation of the intestinal microbiota. Gut Microbes 2020, 12, 1–17. [Google Scholar] [CrossRef]
- Herp, S.; Brugiroux, S.; Garzetti, D.; Ring, D.; Jochum, L.M.; Beutler, M.; Eberl, C.; Hussain, S.; Walter, S.; Gerlach, R.G.; et al. Mucispirillum schaedleri Antagonizes Salmonella Virulence to Protect Mice against Colitis. Cell Host Microbe 2019, 25, 681–694.e688. [Google Scholar] [CrossRef]
- Goodman, A.L.; Kallstrom, G.; Faith, J.J.; Reyes, A.; Moore, A.; Dantas, G.; Gordon, J.I. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc. Natl. Acad. Sci. USA 2011, 108, 6252–6257. [Google Scholar] [CrossRef] [Green Version]
- Becker, N.; Kunath, J.; Loh, G.; Blaut, M. Human intestinal microbiota: Characterization of a simplified and stable gnotobiotic rat model. Gut Microbes 2011, 2, 25–33. [Google Scholar] [CrossRef]
- Kovatcheva-Datchary, P.; Shoaie, S.; Lee, S.; Wahlstrom, A.; Nookaew, I.; Hallen, A.; Perkins, R.; Nielsen, J.; Backhed, F. Simplified Intestinal Microbiota to Study Microbe-Diet-Host Interactions in a Mouse Model. Cell Rep. 2019, 26, 3772–3783.e3776. [Google Scholar] [CrossRef] [Green Version]
- Rezzonico, E.; Mestdagh, R.; Delley, M.; Combremont, S.; Dumas, M.E.; Holmes, E.; Nicholson, J.; Bibiloni, R. Bacterial adaptation to the gut environment favors successful colonization: Microbial and metabonomic characterization of a simplified microbiota mouse model. Gut Microbes 2011, 2, 307–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNulty, N.P.; Yatsunenko, T.; Hsiao, A.; Faith, J.J.; Muegge, B.D.; Goodman, A.L.; Henrissat, B.; Oozeer, R.; Cools-Portier, S.; Gobert, G.; et al. The impact of a consortium of fermented milk strains on the gut microbiome of gnotobiotic mice and monozygotic twins. Sci. Transl. Med. 2011, 3, 106ra106. [Google Scholar] [CrossRef] [Green Version]
- Mark Welch, J.L.; Hasegawa, Y.; McNulty, N.P.; Gordon, J.I.; Borisy, G.G. Spatial organization of a model 15-member human gut microbiota established in gnotobiotic mice. Proc. Natl. Acad. Sci. USA 2017, 114, E9105–E9114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denou, E.; Rezzonico, E.; Panoff, J.M.; Arigoni, F.; Brussow, H. A Mesocosm of Lactobacillus johnsonii, Bifidobacterium longum, and Escherichia coli in the mouse gut. DNA Cell Biol. 2009, 28, 413–422. [Google Scholar] [CrossRef]
- Rakoff-Nahoum, S.; Foster, K.R.; Comstock, L.E. The evolution of cooperation within the gut microbiota. Nature 2016, 533, 255–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, K.M.; Ferreyra, J.A.; Higginbottom, S.K.; Lynch, J.B.; Kashyap, P.C.; Gopinath, S.; Naidu, N.; Choudhury, B.; Weimer, B.C.; Monack, D.M.; et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 2013, 502, 96–99. [Google Scholar] [CrossRef] [Green Version]
- Woting, A.; Pfeiffer, N.; Loh, G.; Klaus, S.; Blaut, M. Clostridium ramosum promotes high-fat diet-induced obesity in gnotobiotic mouse models. mBio 2014, 5, e01530-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weitkunat, K.; Schumann, S.; Petzke, K.J.; Blaut, M.; Loh, G.; Klaus, S. Effects of dietary inulin on bacterial growth, short-chain fatty acid production and hepatic lipid metabolism in gnotobiotic mice. J. Nutr. Biochem. 2015, 26, 929–937. [Google Scholar] [CrossRef]
- Ring, C.; Klopfleisch, R.; Dahlke, K.; Basic, M.; Bleich, A.; Blaut, M. Akkermansia muciniphila strain ATCC BAA-835 does not promote short-term intestinal inflammation in gnotobiotic interleukin-10-deficient mice. Gut Microbes 2019, 10, 188–203. [Google Scholar] [CrossRef] [Green Version]
- Ganesh, B.P.; Klopfleisch, R.; Loh, G.; Blaut, M. Commensal Akkermansia muciniphila exacerbates gut inflammation in Salmonella Typhimurium-infected gnotobiotic mice. PLoS ONE 2013, 8, e74963. [Google Scholar] [CrossRef]
- Eun, C.S.; Mishima, Y.; Wohlgemuth, S.; Liu, B.; Bower, M.; Carroll, I.M.; Sartor, R.B. Induction of bacterial antigen-specific colitis by a simplified human microbiota consortium in gnotobiotic interleukin-10-/- mice. Infect. Immun. 2014, 82, 2239–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lengfelder, I.; Sava, I.G.; Hansen, J.J.; Kleigrewe, K.; Herzog, J.; Neuhaus, K.; Hofmann, T.; Sartor, R.B.; Haller, D. Complex Bacterial Consortia Reprogram the Colitogenic Activity of Enterococcus faecalis in a Gnotobiotic Mouse Model of Chronic, Immune-Mediated Colitis. Front. Immunol. 2019, 10, 1420. [Google Scholar] [CrossRef]
- Martz, S.L.; McDonald, J.A.; Sun, J.; Zhang, Y.G.; Gloor, G.B.; Noordhof, C.; He, S.M.; Gerbaba, T.K.; Blennerhassett, M.; Hurlbut, D.J.; et al. Administration of defined microbiota is protective in a murine Salmonella infection model. Sci. Rep. 2015, 5, 16094. [Google Scholar] [CrossRef]
- Luk, B.; Veeraragavan, S.; Engevik, M.; Balderas, M.; Major, A.; Runge, J.; Luna, R.A.; Versalovic, J. Postnatal colonization with human “infant-type” Bifidobacterium species alters behavior of adult gnotobiotic mice. PLoS ONE 2018, 13, e0196510. [Google Scholar] [CrossRef]
- Crost, E.H.; Pujol, A.; Ladire, M.; Dabard, J.; Raibaud, P.; Carlier, J.P.; Fons, M. Production of an antibacterial substance in the digestive tract involved in colonization-resistance against Clostridium perfringens. Anaerobe 2010, 16, 597–603. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNulty, N.P.; Wu, M.; Erickson, A.R.; Pan, C.; Erickson, B.K.; Martens, E.C.; Pudlo, N.A.; Muegge, B.D.; Henrissat, B.; Hettich, R.L.; et al. Effects of diet on resource utilization by a model human gut microbiota containing Bacteroides cellulosilyticus WH2, a symbiont with an extensive glycobiome. PLoS Biol. 2013, 11, e1001637. [Google Scholar] [CrossRef]
- Rey, F.E.; Gonzalez, M.D.; Cheng, J.; Wu, M.; Ahern, P.P.; Gordon, J.I. Metabolic niche of a prominent sulfate-reducing human gut bacterium. Proc. Natl. Acad. Sci. USA 2013, 110, 13582–13587. [Google Scholar] [CrossRef] [Green Version]
- Faith, J.J.; McNulty, N.P.; Rey, F.E.; Gordon, J.I. Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science 2011, 333, 101–104. [Google Scholar] [CrossRef] [Green Version]
- Narushima, S.; Itoha, K.; Miyamoto, Y.; Park, S.H.; Nagata, K.; Kuruma, K.; Uchida, K. Deoxycholic acid formation in gnotobiotic mice associated with human intestinal bacteria. Lipids 2006, 41, 835–843. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Devendran, S.; Alves, J.M.; Doden, H.; Wolf, P.G.; Pereira, G.V.; Ly, L.; Volland, A.; Takei, H.; Nittono, H.; et al. The ‘in vivo lifestyle’ of bile acid 7alpha-dehydroxylating bacteria: Comparative genomics, metatranscriptomic, and bile acid metabolomics analysis of a defined microbial community in gnotobiotic mice. Gut Microbes 2020, 11, 381–404. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.M.; Ma, J.; Prince, A.L.; Antony, K.M.; Seferovic, M.D.; Aagaard, K.M. Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat. Med. 2017, 23, 314–326. [Google Scholar] [CrossRef] [Green Version]
- Palmer, C.; Bik, E.M.; DiGiulio, D.B.; Relman, D.A.; Brown, P.O. Development of the human infant intestinal microbiota. PLoS Biol. 2007, 5, e177. [Google Scholar] [CrossRef] [Green Version]
- Arentsen, T.; Qian, Y.; Gkotzis, S.; Femenia, T.; Wang, T.; Udekwu, K.; Forssberg, H.; Diaz Heijtz, R. The bacterial peptidoglycan-sensing molecule Pglyrp2 modulates brain development and behavior. Mol. Psychiatry 2017, 22, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Lupp, C.; Robertson, M.L.; Wickham, M.E.; Sekirov, I.; Champion, O.L.; Gaynor, E.C.; Finlay, B.B. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2007, 2, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stecher, B.; Robbiani, R.; Walker, A.W.; Westendorf, A.M.; Barthel, M.; Kremer, M.; Chaffron, S.; Macpherson, A.J.; Buer, J.; Parkhill, J.; et al. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 2007, 5, 2177–2189. [Google Scholar] [CrossRef] [Green Version]
- Rohr, J.R.; Barrett, C.B.; Civitello, D.J.; Craft, M.E.; Delius, B.; DeLeo, G.A.; Hudson, P.J.; Jouanard, N.; Nguyen, K.H.; Ostfeld, R.S.; et al. Emerging human infectious diseases and the links to global food production. Nat. Sustain. 2019, 2, 445–456. [Google Scholar] [CrossRef]
- Dethlefsen, L.; Huse, S.; Sogin, M.L.; Relman, D.A. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008, 6, e280. [Google Scholar] [CrossRef]
- Chang, J.Y.; Antonopoulos, D.A.; Kalra, A.; Tonelli, A.; Khalife, W.T.; Schmidt, T.M.; Young, V.B. Decreased diversity of the fecal Microbiome in recurrent Clostridium difficile-associated diarrhea. J. Infect. Dis. 2008, 197, 435–438. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.D.; Myers, C.J.; Harris, S.C.; Kakiyama, G.; Lee, I.K.; Yun, B.S.; Matsuzaki, K.; Furukawa, M.; Min, H.K.; Bajaj, J.S.; et al. Bile Acid 7alpha-Dehydroxylating Gut Bacteria Secrete Antibiotics that Inhibit Clostridium difficile: Role of Secondary Bile Acids. Cell Chem. Biol. 2019, 26, 27–34.e24. [Google Scholar] [CrossRef] [Green Version]
- Narushima, S.; Itoh, K.; Takamine, F.; Uchida, K. Absence of cecal secondary bile acids in gnotobiotic mice associated with two human intestinal bacteria with the ability to dehydroxylate bile acids in vitro. Microbiol. Immunol. 1999, 43, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Derrien, M.; Belzer, C.; de Vos, W.M. Akkermansia muciniphila and its role in regulating host functions. Microb. Pathog. 2017, 106, 171–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keubler, L.M.; Buettner, M.; Hager, C.; Bleich, A. A Multihit Model: Colitis Lessons from the Interleukin-10-deficient Mouse. Inflamm. Bowel Dis. 2015, 21, 1967–1975. [Google Scholar] [CrossRef]
- Sellon, R.K.; Tonkonogy, S.; Schultz, M.; Dieleman, L.A.; Grenther, W.; Balish, E.; Rennick, D.M.; Sartor, R.B. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect. Immun. 1998, 66, 5224–5231. [Google Scholar] [CrossRef] [Green Version]
- Schaubeck, M.; Clavel, T.; Calasan, J.; Lagkouvardos, I.; Haange, S.B.; Jehmlich, N.; Basic, M.; Dupont, A.; Hornef, M.; von Bergen, M.; et al. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence. Gut 2016, 65, 225–237. [Google Scholar] [CrossRef] [Green Version]
- Stolzer, I.; Kaden-Volynets, V.; Ruder, B.; Letizia, M.; Bittel, M.; Rausch, P.; Basic, M.; Bleich, A.; Baines, J.F.; Neurath, M.F.; et al. Environmental Microbial Factors Determine the Pattern of Inflammatory Lesions in a Murine Model of Crohn’s Disease-Like Inflammation. Inflamm. Bowel Dis. 2020, 26, 66–79. [Google Scholar] [CrossRef]
- Kang, C.S.; Ban, M.; Choi, E.J.; Moon, H.G.; Jeon, J.S.; Kim, D.K.; Park, S.K.; Jeon, S.G.; Roh, T.Y.; Myung, S.J.; et al. Extracellular vesicles derived from gut microbiota, especially Akkermansia muciniphila, protect the progression of dextran sulfate sodium-induced colitis. PLoS ONE 2013, 8, e76520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Png, C.W.; Linden, S.K.; Gilshenan, K.S.; Zoetendal, E.G.; McSweeney, C.S.; Sly, L.I.; McGuckin, M.A.; Florin, T.H. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am. J. Gastroenterol. 2010, 105, 2420–2428. [Google Scholar] [CrossRef]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plovier, H.; Everard, A.; Druart, C.; Depommier, C.; Van Hul, M.; Geurts, L.; Chilloux, J.; Ottman, N.; Duparc, T.; Lichtenstein, L.; et al. A purified membrane protein from Akkermansia muciniphila or the pasteurized bacterium improves metabolism in obese and diabetic mice. Nat. Med. 2017, 23, 107–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depommier, C.; Van Hul, M.; Everard, A.; Delzenne, N.M.; De Vos, W.M.; Cani, P.D. Pasteurized Akkermansia muciniphila increases whole-body energy expenditure and fecal energy excretion in diet-induced obese mice. Gut Microbes 2020, 11, 1231–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadwell, K.; Patel, K.K.; Maloney, N.S.; Liu, T.C.; Ng, A.C.; Storer, C.E.; Head, R.D.; Xavier, R.; Stappenbeck, T.S.; Virgin, H.W. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 2010, 141, 1135–1145. [Google Scholar] [CrossRef] [Green Version]
- Khan, R.R.; Lawson, A.D.; Minnich, L.L.; Martin, K.; Nasir, A.; Emmett, M.K.; Welch, C.A.; Udall, J.N., Jr. Gastrointestinal norovirus infection associated with exacerbation of inflammatory bowel disease. J. Pediatr. Gastroenterol. Nutr. 2009, 48, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Lencioni, K.C.; Seamons, A.; Treuting, P.M.; Maggio-Price, L.; Brabb, T. Murine norovirus: An intercurrent variable in a mouse model of bacteria-induced inflammatory bowel disease. Comp. Med. 2008, 58, 522–533. [Google Scholar]
- Zeissig, S.; Bürgel, N.; Günzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, J.; Vogelsang, H.; Hubl, W.; Waldhoer, T.; Lochs, H. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet 1993, 341, 1437–1439. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef]
- Tedelind, S.; Westberg, F.; Kjerrulf, M.; Vidal, A. Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: A study with relevance to inflammatory bowel disease. World J. Gastroenterol. WJG 2007, 13, 2826–2832. [Google Scholar] [CrossRef]
- Hill, D.A.; Hoffmann, C.; Abt, M.C.; Du, Y.; Kobuley, D.; Kirn, T.J.; Bushman, F.D.; Artis, D. Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol. 2010, 3, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Dethlefsen, L.; Relman, D.A. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4554–4561. [Google Scholar] [CrossRef] [Green Version]
- Devkota, S.; Wang, Y.; Musch, M.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary fat-induced taurocholic acid production promotes pathobiont and colitis in IL-10(−/−) mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llewellyn, S.R.; Britton, G.J.; Contijoch, E.J.; Vennaro, O.H.; Mortha, A.; Colombel, J.F.; Grinspan, A.; Clemente, J.C.; Merad, M.; Faith, J.J. Interactions Between Diet and the Intestinal Microbiota Alter Intestinal Permeability and Colitis Severity in Mice. Gastroenterology 2018, 154, 1037–1046.e1032. [Google Scholar] [CrossRef]
- Daniel, H.; Gholami, A.M.; Berry, D.; Desmarchelier, C.; Hahne, H.; Loh, G.; Mondot, S.; Lepage, P.; Rothballer, M.; Walker, A.; et al. High-fat diet alters gut microbiota physiology in mice. ISME J. 2014, 8, 295–308. [Google Scholar] [CrossRef]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.W.; Ince, J.; Duncan, S.H.; Webster, L.M.; Holtrop, G.; Ze, X.; Brown, D.; Stares, M.D.; Scott, P.; Bergerat, A.; et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011, 5, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Knauf, C.; Iglesias, M.A.; Drucker, D.J.; Delzenne, N.M.; Burcelin, R. Improvement of glucose tolerance and hepatic insulin sensitivity by oligofructose requires a functional glucagon-like peptide 1 receptor. Diabetes 2006, 55, 1484–1490. [Google Scholar] [CrossRef]
- Cani, P.D.; Neyrinck, A.M.; Fava, F.; Knauf, C.; Burcelin, R.G.; Tuohy, K.M.; Gibson, G.R.; Delzenne, N.M. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 2007, 50, 2374–2383. [Google Scholar] [CrossRef] [Green Version]
- Woting, A.; Pfeiffer, N.; Hanske, L.; Loh, G.; Klaus, S.; Blaut, M. Alleviation of high fat diet-induced obesity by oligofructose in gnotobiotic mice is independent of presence of Bifidobacterium longum. Mol. Nutr. Food Res. 2015, 59, 2267–2278. [Google Scholar] [CrossRef] [Green Version]
- Rahat-Rozenbloom, S.; Fernandes, J.; Gloor, G.B.; Wolever, T.M. Evidence for greater production of colonic short-chain fatty acids in overweight than lean humans. Int. J. Obes. 2014, 38, 1525–1531. [Google Scholar] [CrossRef] [Green Version]
- Isken, F.; Klaus, S.; Osterhoff, M.; Pfeiffer, A.F.; Weickert, M.O. Effects of long-term soluble vs. insoluble dietary fiber intake on high-fat diet-induced obesity in C57BL/6J mice. J. Nutr. Biochem. 2010, 21, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Buffie, C.G.; Bucci, V.; Stein, R.R.; McKenney, P.T.; Ling, L.; Gobourne, A.; No, D.; Liu, H.; Kinnebrew, M.; Viale, A.; et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 2015, 517, 205–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2013, 62, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Keren, N.; Konikoff, F.M.; Paitan, Y.; Gabay, G.; Reshef, L.; Naftali, T.; Gophna, U. Interactions between the intestinal microbiota and bile acids in gallstones patients. Environ. Microbiol. Rep. 2015, 7, 874–880. [Google Scholar] [CrossRef] [PubMed]
- Baxter, N.T.; Zackular, J.P.; Chen, G.Y.; Schloss, P.D. Structure of the gut microbiome following colonization with human feces determines colonic tumor burden. Microbiome 2014, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.H.; Zhao, L.; Zhang, X.; Nakatsu, G.; Han, J.; Xu, W.; Xiao, X.; Kwong, T.N.Y.; Tsoi, H.; Wu, W.K.K.; et al. Gavage of Fecal Samples From Patients With Colorectal Cancer Promotes Intestinal Carcinogenesis in Germ-Free and Conventional Mice. Gastroenterology 2017, 153, 1621–1633.e1626. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Li, X.; Zhong, W.; Yang, M.; Xu, M.; Sun, Y.; Ma, J.; Liu, T.; Song, X.; Dong, W.; et al. Gut microbiota from colorectal cancer patients enhances the progression of intestinal adenoma in Apc(min/+) mice. EBioMedicine 2019, 48, 301–315. [Google Scholar] [CrossRef] [Green Version]
- Tomkovich, S.; Yang, Y.; Winglee, K.; Gauthier, J.; Muhlbauer, M.; Sun, X.; Mohamadzadeh, M.; Liu, X.; Martin, P.; Wang, G.P.; et al. Locoregional Effects of Microbiota in a Preclinical Model of Colon Carcinogenesis. Cancer Res. 2017, 77, 2620–2632. [Google Scholar] [CrossRef] [Green Version]
- Sui, H.; Zhang, L.; Gu, K.; Chai, N.; Ji, Q.; Zhou, L.; Wang, Y.; Ren, J.; Yang, L.; Zhang, B.; et al. YYFZBJS ameliorates colorectal cancer progression in Apc(Min/+) mice by remodeling gut microbiota and inhibiting regulatory T-cell generation. Cell Commun. Signal. 2020, 18, 113. [Google Scholar] [CrossRef] [PubMed]
- Tomkovich, S.; Gharaibeh, R.Z.; Dejea, C.M.; Pope, J.L.; Jiang, J.; Winglee, K.; Gauthier, J.; Newsome, R.C.; Yang, Y.; Fodor, A.A.; et al. Human Colon Mucosal Biofilms and Murine Host Communicate via Altered mRNA and microRNA Expression during Cancer. MSystems 2020, 5, e00451-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krych, L.; Hansen, C.H.; Hansen, A.K.; van den Berg, F.W.; Nielsen, D.S. Quantitatively different, yet qualitatively alike: A meta-analysis of the mouse core gut microbiome with a view towards the human gut microbiome. PLoS ONE 2013, 8, e62578. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, I.B.; O’Toole, P.W.; Ohman, L.; Claesson, M.J.; Deane, J.; Quigley, E.M.; Simren, M. An irritable bowel syndrome subtype defined by species-specific alterations in faecal microbiota. Gut 2012, 61, 997–1006. [Google Scholar] [CrossRef]
- Nagpal, R.; Wang, S.; Solberg Woods, L.C.; Seshie, O.; Chung, S.T.; Shively, C.A.; Register, T.C.; Craft, S.; McClain, D.A.; Yadav, H. Comparative Microbiome Signatures and Short-Chain Fatty Acids in Mouse, Rat, Non-human Primate, and Human Feces. Front. Microbiol. 2018, 9, 2897. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.; Pamp, S.J.; Hill, J.A.; Surana, N.K.; Edelman, S.M.; Troy, E.B.; Reading, N.C.; Villablanca, E.J.; Wang, S.; Mora, J.R.; et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell 2012, 149, 1578–1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wos-Oxley, M.; Bleich, A.; Oxley, A.P.; Kahl, S.; Janus, L.M.; Smoczek, A.; Nahrstedt, H.; Pils, M.C.; Taudien, S.; Platzer, M.; et al. Comparative evaluation of establishing a human gut microbial community within rodent models. Gut Microbes 2012, 3, 234–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayin, S.I.; Wahlstrom, A.; Felin, J.; Jantti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyotylainen, T.; Oresic, M.; Backhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Seedorf, H.; Griffin, N.W.; Ridaura, V.K.; Reyes, A.; Cheng, J.; Rey, F.E.; Smith, M.I.; Simon, G.M.; Scheffrahn, R.H.; Woebken, D.; et al. Bacteria from diverse habitats colonize and compete in the mouse gut. Cell 2014, 159, 253–266. [Google Scholar] [CrossRef] [Green Version]
- Marcobal, A.; Kashyap, P.C.; Nelson, T.A.; Aronov, P.A.; Donia, M.S.; Spormann, A.; Fischbach, M.A.; Sonnenburg, J.L. A metabolomic view of how the human gut microbiota impacts the host metabolome using humanized and gnotobiotic mice. ISME J. 2013, 7, 1933–1943. [Google Scholar] [CrossRef] [Green Version]
- Frese, S.A.; Benson, A.K.; Tannock, G.W.; Loach, D.M.; Kim, J.; Zhang, M.; Oh, P.L.; Heng, N.C.; Patil, P.B.; Juge, N.; et al. The evolution of host specialization in the vertebrate gut symbiont Lactobacillus reuteri. PLoS Genet. 2011, 7, e1001314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Kalyan, S.; Steck, N.; Turner, L.M.; Harr, B.; Kunzel, S.; Vallier, M.; Hasler, R.; Franke, A.; Oberg, H.H.; et al. Analysis of intestinal microbiota in hybrid house mice reveals evolutionary divergence in a vertebrate hologenome. Nat. Commun. 2015, 6, 6440. [Google Scholar] [CrossRef] [Green Version]
- Gulati, A.S.; Shanahan, M.T.; Arthur, J.C.; Grossniklaus, E.; von Furstenberg, R.J.; Kreuk, L.; Henning, S.J.; Jobin, C.; Sartor, R.B. Mouse background strain profoundly influences Paneth cell function and intestinal microbial composition. PLoS ONE 2012, 7, e32403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.L.; Vieira-Silva, S.; Liston, A.; Raes, J. How informative is the mouse for human gut microbiota research? Dis. Model. Mech. 2015, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Fisher, C.K.; Mehta, P. Identifying keystone species in the human gut microbiome from metagenomic timeseries using sparse linear regression. PLoS ONE 2014, 9, e102451. [Google Scholar] [CrossRef]
- Park, J.C.; Im, S.H. Of men in mice: The development and application of a humanized gnotobiotic mouse model for microbiome therapeutics. Exp. Mol. Med. 2020, 52, 1383–1396. [Google Scholar] [CrossRef]
- Lagkouvardos, I.; Overmann, J.; Clavel, T. Cultured microbes represent a substantial fraction of the human and mouse gut microbiota. Gut Microbes 2017, 8, 493–503. [Google Scholar] [CrossRef] [Green Version]
- Browne, H.P.; Forster, S.C.; Anonye, B.O.; Kumar, N.; Neville, B.A.; Stares, M.D.; Goulding, D.; Lawley, T.D. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature 2016, 533, 543–546. [Google Scholar] [CrossRef] [Green Version]
- Sohngen, C.; Podstawka, A.; Bunk, B.; Gleim, D.; Vetcininova, A.; Reimer, L.C.; Ebeling, C.; Pendarovski, C.; Overmann, J. BacDive--The Bacterial Diversity Metadatabase in 2016. Nucleic Acids Res. 2016, 44, D581–D585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, J.; Lee, J.H.; Jung, Y.; Kim, M.; Kim, S.; Kim, B.K.; Lim, Y.W. EzTaxon: A web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int. J. Syst. Evol. Microbiol. 2007, 57, 2259–2261. [Google Scholar] [CrossRef] [PubMed]



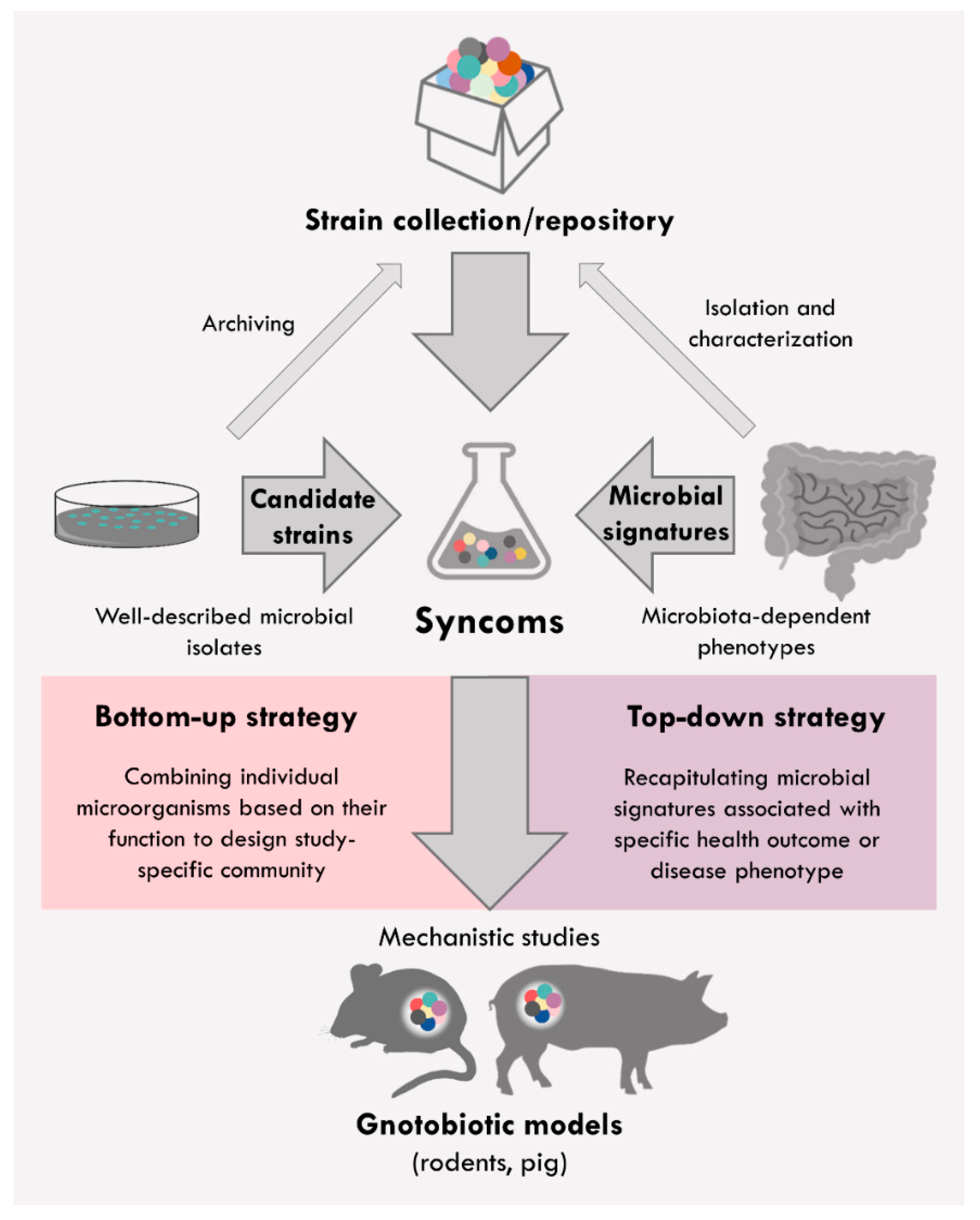{kind=link}
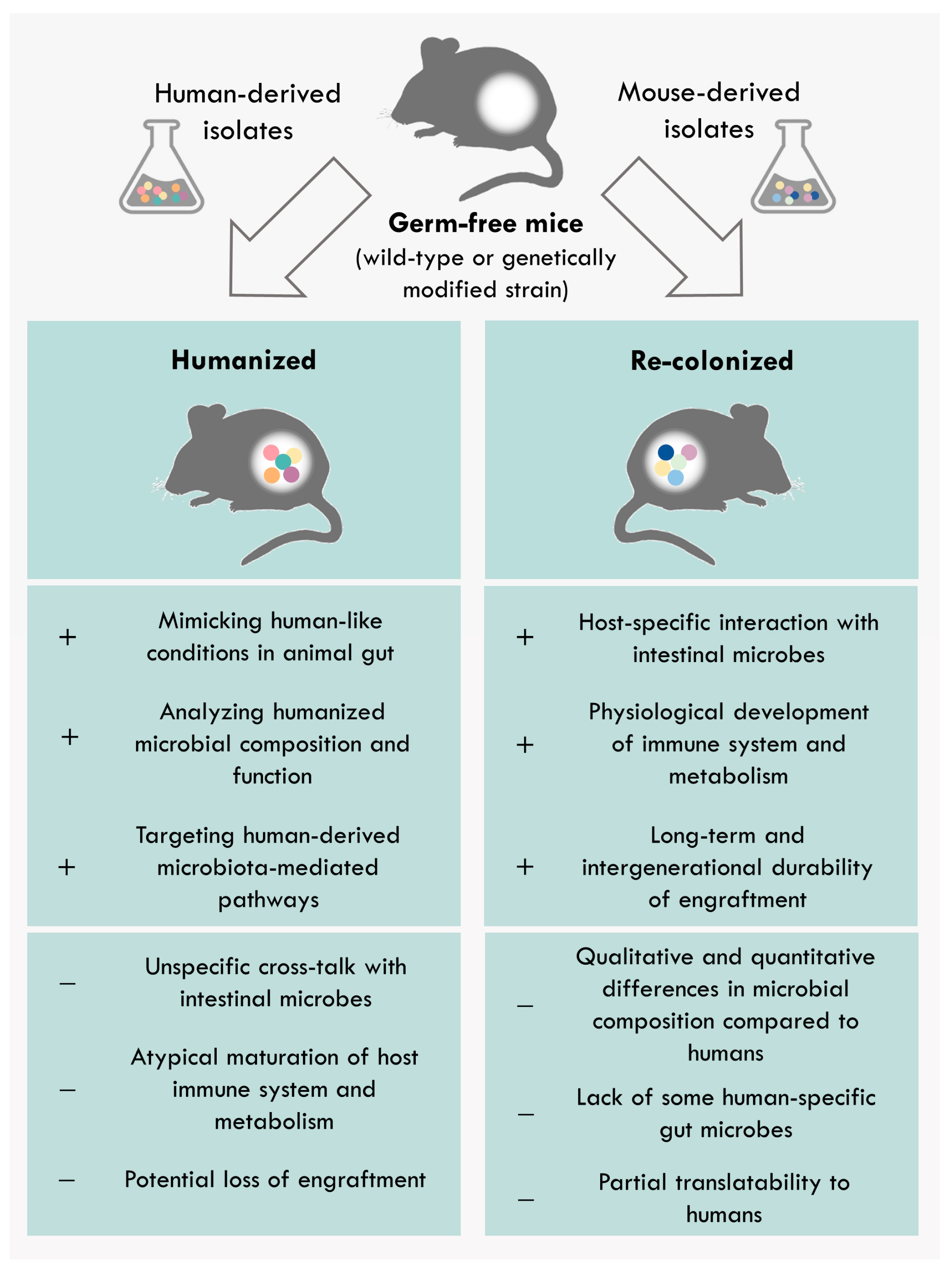{kind=link}
| Syncom | Composition | Application/Research Question | Ref. |
|---|---|---|---|
| ASF | Clostridium sp. ASF356, L. acidophilus ASF360, L. murinus ASF361, M. schaedleri ASF457, E. plexicaudatum ASF492, Pseudoflavonifractor sp. ASF500, S. arabinosiphila ASF502, P. goldsteinii ASF519 | Standardization of laboratory rodent husbandry | [21] |
| Impact of host-mediated factors on chronic inflammation | [32] | ||
| +E. coli strains | Impact of microbiota on intestinal inflammation | [33] | |
| +Murine norovirus +Segmented filamentous bacteria | [23,34] | ||
| L. acidophilus ASF360, L. murinus ASF361, M. schaedleri ASF457, P. goldsteinii ASF519, B. fibrisolvens | Diet-related microbial protection against colorectal cancer | [35] | |
| GM15 | L. johnsonii, L. murinus, L. reuteri, P. goldsteinii, B. acidifaciens, 3× Lachnospiraceae sp. strains, B. caecimuris, S. arabinosiphila ASF502, Clostridium sp. ASF356, E clostridioformis YL32, C. cocleatum, E. coli, A. colihominis | Standardization of laboratory rodent husbandry | [27] |
| OMM12 | A. muciniphila YL44, B. caecimuris I48, M. intestinale YL27, T. muris YL45, B. longum subsp. animalis YL2, E. faecalis KB1, A. muris KB18, E. clostridioformis YL32, B. coccoides YL58, F. plautii YL31, L. reuteri I49, C. innocuum I46 | Mechanisms of colonization resistance against enteric pathogens | [29] |
| +E. muris | Host–microbe metabolic cross-talk and bile metabolism | [31] | |
| +C. scindens | Mechanisms of colonization resistance against C. difficile | [36] | |
| +Murine norovirus +Segmented filamentous bacteria | Impact of microbiota on intestinal inflammation | [23] | |
| +A. finegoldii | Host–microbe metabolic cross-talk and diet impact on inflammation | [37] | |
| +M. schaedleri | Impact of microbiota on Salmonella-induced intestinal inflammation | [38] |
| Syncom | Composition | Application/Research Question | Ref. |
|---|---|---|---|
| SIHUMI | A. caccae, B. producta, C. ramosum, L. plantarum, B. Theta1, B. longum, E. coli K-12 | Host–bacteria interactions | [40] |
| SIHUMIx | A. caccae, B. producta, C. ramosum, L. plantarum, B. theta1, B. longum, E. coli K-12, C. butyricum | Host–microbe metabolic cross-talk and impact of microbiota on intestinal inflammation | [40] |
| Diet-related microbial effect on obesity | [48,49] | ||
| +A. muciniphila | Impact of microbiota on intestinal inflammation | [50] | |
| Impact of microbiota on Salmonella-induced intestinal inflammation | [51] | ||
| SIHUMI | E. faecalis, R. gnavus, F. prausnitzii, L. plantarum, B. vulgatus, E. coli, B. longum subsp. longum | Impact of IBD-related microbiota on intestinal inflammation | [52] |
| −E. faecalis | [53] | ||
| SIM | E. hallii, E. rectale, B. adolescentis, C. aerofaciens, D. piger, R. inulinivorans, R. bromii, B. theta, P. copri, A. muciniphila | Microbe–diet interaction and impact on metabolism | [41] |
| MET-1 | 4× Bifidobacterium species, C. aerofaciens, B. ovatus, P. distasonis, 2× Lactobacillus species, S. mitis, F. prausnitzii, C. cocleatum, A. intestine, Blautia sp., 2× Dorea species, L. pectinoshiza, 2× Roseburia species, 4× Ruminococcus species, 7× Eubacterium species, E. coli, Raoultella sp. | Host–microbe interaction and protection against systemic disease | [54] |
| B. theta1 , B. vulgatus, B. longum, B. hansenii, C. scindens, C. aerofaciens, E. coli, E. ventriosum, F. prausnitzii, L. rhamnosus | Host–microbe metabolic cross-talk and impact on metabolism | [42] | |
| E. coli K-12, L. johnsonii, B. longum, E. coli Nissle | Microbial cooperation and competition | [45] | |
| B. ovatus, B. vulgatos, B. theta1 | Microbial cooperation | [46] | |
| B. longum subsp. infantis, B. breve, B. bifidum, B. dentium | Host–microbe interaction and microbial impact on nervous system | [55] | |
| R. gnavus, B. theta, C. hathewayi, C. orbiscindens | Mechanisms of colonization resistance against C. perfringens | [56] | |
| C. ramosum, C. asparagiforme, C. indolis, C. hathewayi, C. bolteae, Clostridiales 1_7_47FAA, 2× Clostridium species, C. scindens, Clostridiaceae JC13, 3× Lachnospiraceae species, B. producta, E. fissicatena, Ruminococcus sp. ID8, A. colihominis | Impact of microbiota on chronic intestinal inflammation | [57] | |
| B. caccae, B. ovatus, B. theta1, B. uniformis, B. vulgatus, B. cellulosilyticus WH2, C. scindens, C. spiroforme, C. aerofaciens, D. longicatena, E. rectale, F. prausnitzii, P. distasonis, R. obeum, R. torques | Microbe–diet interaction | [43] | |
| Microbe–microbe interactions | [44] | ||
| −E. rectale, −F. prausnitzii, −R. torques | Microbe–diet interaction | [58] | |
| B. caccae, B. ovatus, B. theta1,E. rectale, C. aerofaciens, C. symbiosum, E. coli, M. formatexigens | Microbe–diet interaction and impact on metabolism | [59] | |
| + D. piger, + E. rectale, + B. hydrogenotrophica | [60] | ||
| B4PC2 | B. uniformis, B. vulgatus, B. producta, P. distasonis, C. hylemonae, C. hiranonis | Host–microbe metabolic cross-talk and bile acid metabolism | [61,62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bolsega, S.; Bleich, A.; Basic, M. Synthetic Microbiomes on the Rise—Application in Deciphering the Role of Microbes in Host Health and Disease. Nutrients 2021, 13, 4173. https://doi.org/10.3390/nu13114173
Bolsega S, Bleich A, Basic M. Synthetic Microbiomes on the Rise—Application in Deciphering the Role of Microbes in Host Health and Disease. Nutrients. 2021; 13(11):4173. https://doi.org/10.3390/nu13114173
Chicago/Turabian StyleBolsega, Silvia, André Bleich, and Marijana Basic. 2021. "Synthetic Microbiomes on the Rise—Application in Deciphering the Role of Microbes in Host Health and Disease" Nutrients 13, no. 11: 4173. https://doi.org/10.3390/nu13114173
APA StyleBolsega, S., Bleich, A., & Basic, M. (2021). Synthetic Microbiomes on the Rise—Application in Deciphering the Role of Microbes in Host Health and Disease. Nutrients, 13(11), 4173. https://doi.org/10.3390/nu13114173