
Gut Microbial Metabolite-Mediated Regulation of the Intestinal Barrier in the Pathogenesis of Inflammatory Bowel Disease


Abstract
:1. Introduction
2. Epithelial Barrier in Inflammatory Bowel Disease
2.1. Structure and Function of the Epithelial Barrier
2.2. Dysregulation of the Gut Barrier in IBD
3. Gut Metabolites in Inflammatory Bowel Disease
3.1. Why Study Gut Metabolites in IBD?
3.2. Metabolomics Techniques to Study Gut Metabolites
3.2.1. Targeted and Untargeted Approaches
3.2.2. Techniques
3.2.3. Sample Types and Sample Processing
3.2.4. Data Pre-Processing and Analysis
3.3. Gut Metabolome Changes in IBD
3.3.1. Overview of Clinical Studies
3.3.2. Faecal Metabolomics as a Diagnostic or Prognostic Tool for IBD

{kind=link}
| Study | Patient Groups | Metabolomics Technique | Upregulated Metabolites | Downregulated Metabolites |
|---|---|---|---|---|
| Le Gall et al., 2011 [52] | UC (n = 13), IBS (n = 10), Healthy (n = 22) | 1H NMR | In UC: Taurine, Cadaverine, Glucose and Choline | In UC: 2-methylbutyrate |
| Walton et al., 2013 [55] | Healthy (n = 19), CD (n = 22) and UC (n = 20) | GC-MS | In CD versus healthy: Butanoic aid, 1-propanol, propanoic acid, butanoic acid, indole | |
| Bjerrum et al., 2015 [69] | UC (n = 48), CD (n = 44), Healthy (n = 21) | 1H NMR | In active CD and active UC: Amino acids In active UC: Lactate, Taurine | In active CD: Butyrate and Propionate |
| De Preter et al., 2015 [71] | Healthy (n = 40), CD (n = 83), UC (n = 68) | GC-MS | In CD: 1-ethyl3-methylbenzene, benzene acetaldehyde, phenol, 2-methyl propanal, carbon disulfide and 1-methoxy-4-methylbenzene In UC: cyclohexane, 3-methyl butanal and pyrrole | In CD and UC: Medium chain fatty acids, Protein fermentation metabolites In UC only: Furan, 5-methyl-2-furancarboxaldehyde and 3,4-dimethylthiophene |
| Jacobs et al., 2016 [74] | Paediatric IBD patients (n = 36), Healthy family (n = 56) | UPLC- ToFMS | Amino acid derivatives and bile acids | Stercobilin, Acetyl-glutamic acid and boldione |
| Ahmed et al., 2016 [72] | Healthy (n = 109), CD (n = 117) and UC (n = 100) | GC-MS | In active CD: 1-octen-3-ol, heptanal, propanal, benzeneacetaldehyde, 6-methyl-2-heptanone and decane | In active CD: Pentanoic acid, 2-methyl butanoic acid, methanethiol, 3-methylphenol |
| Santoru et al., 2017 [62] | Healthy (n = 51), UC (n = 82) and CD (n = 50) | 1H NMR, GC-MS and LC-QToF-MS | In CD: Alanine, phenylacetic acid, glyceric acid, phenylethylamine, putrescine and cadaverine, diacylglycerols In UC: Cadaverine, alanine, 4-hydroxyphenylacetic acid, 4-aminovaleric acid, TMAO, diacylglycerols | In CD and UC: Vitamins, 3-methyladipic acid, 5β-coprostanol, 3-hydroxybutyric acid, 2-hydroxy-3-methyvaleric acid and hydrocinnamic acid, urobilinogen |
| Kolho et al., 2017 [70] | Healthy (n = 14), IBD (n = 23) | UPLC-MS/MS | In UC: Amino acids, citrulline, ornithine, creatinine, choline, kynurenine, taurine, normetanephrin | In UC: Cytosine |
| Alghamdi et al., 2018 [56] | Healthy (n = 11) and CD (n = 11) | LC-MS | C20 Sphingenine, octadecenoylsphingenine, Sphingomyelins, LCFAs | Ornithine isomer, Tyrosine |
| Weng et al., 2019 [73] | Healthy (n = 42), UC (n= 107) and CD (n = 173) | GC/MS, LC-NEG/MS, and LC-POS/MS | In UC and CD: LCFAs, MCFAs, bile acids, and vitamins In UC versus healthy: Glycochenodeoxycholate and glycolithocholic acid | |
| Franzosa et al., 2019 [63] | Healthy (n = 34), CD (n = 68), UC (n = 53) | LC-MS (combination of 4 techniques) | In CD versus healthy: Sphingolipids, carboximidic acids, bile acids, lactate | In UC and CD: Triterpenoids and LCFAs, triacylglycerols, pantothenate |
| Lloyd Price et al., 2019 [64] | 132 subjects (non-IBD, UC and CD) | LC-MS (combination of 4 techniques) | In CD versus non-IBD: Polyunsaturated fatty acids, Nicotinuric acid, Bile acids, acylcarnitine | Vitamins, Lithocholate and deoxycholate, SCFAs |
4. Gut Metabolites as Key Regulators of Epithelial Barrier Function in IBD
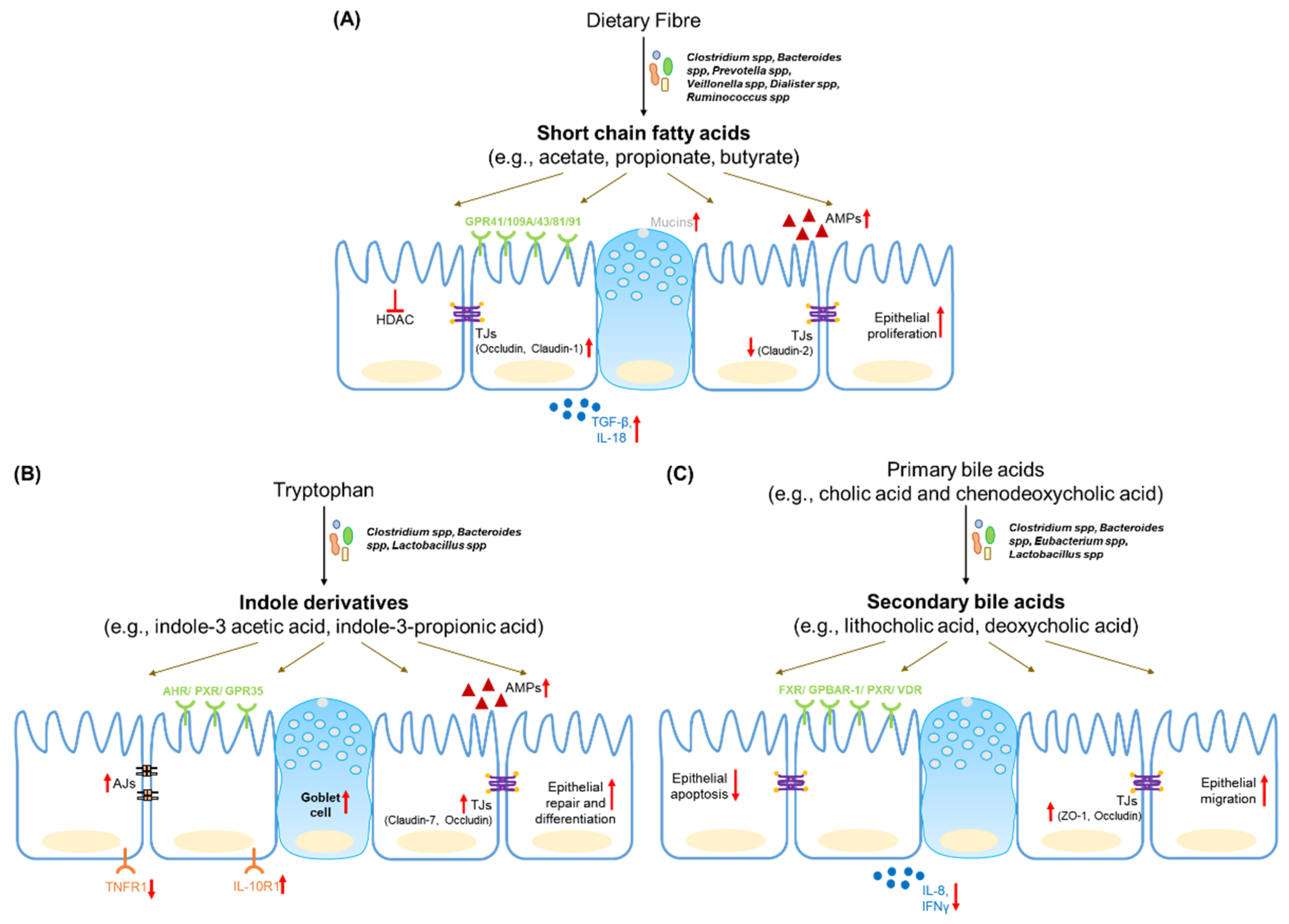
4.1. Short Chain Fatty Acids
4.2. Tryptophan Metabolites
4.3. Bile Acid Metabolites
4.4. Vitamins
| Metabolite | Microbial Source in the Gut | Mammalian Receptors | Effect on the Intestinal Epithelial Barrier |
|---|---|---|---|
| Short chain Fatty acids Butyrate, Acetate, Propionate, Lactate, Succinate, Valerate, etc. | Butyrate: Clostridium clusters I, III, IV, XI, XIVa, XV, and XVI [90] Acetate: Bacteroides spp. and Prevotella spp. Propionate: Bacteroides spp., Veillonella spp., Dialister spp. or Ruminococcus spp. [91] | Butyrate: GPR41, GPR109A, GPR65 (predicted) Acetate: GPR43 Propionate: GPR 41, GPR43 Lactate: GPR81 Succinate: GPR91 [92] |
|
| Bile acids Cholic acid, Lithocholic acid (LCA), Deoxycholic acid (DCA), Ursodeoxycholic acid (UDCA) etc. | Bacteroides spp., Eubacterium spp., Lactobacillus spp. and Clostridium spp. [115] | Farnesoid X receptor (FXR), GPBAR-1/TGR5, Pregnane X receptor (PXR), Vitamin D receptor (VDR), [115] |
|
| Tryptophan metabolites Kynurenic acid, hydroxytryptamine, Indole derivatives | Lactobacillus spp., Clostridium spp. and Bacteroides spp. | GPR35 (predicted), Aryl hydrocarbon receptor (AHR), Pregnane X receptor (PXR) [106,111] |
|
4.5. Other Metabolites
4.5.1. Medium and Long Chain Fatty Acids
4.5.2. Sulfur-Containing Metabolites
4.5.3. Polyamines
4.5.4. Polyphenol Metabolites
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Collaborators GBDIBD. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Ramos, G.P.; Papadakis, K.A. Mechanisms of Disease: Inflammatory Bowel Diseases. Mayo Clin. Proc. 2019, 94, 155–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchesi, J.R.; Holmes, E.; Khan, F.; Kochhar, S.; Scanlan, P.; Shanahan, F.; Wilson, I.D.; Wang, Y. Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J. Proteome Res. 2007, 6, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Brant, S.R. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology 2011, 140, 1704–1712. [Google Scholar] [CrossRef] [Green Version]
- Uhlig, H.H.; Schwerd, T.; Koletzko, S.; Shah, N.; Kammermeier, J.; Elkadri, A.; Ouahed, J.; Wilson, D.C.; Travis, S.P.; Turner, D.; et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology 2014, 147, 990–1007.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suri, K.; Bubier, J.A.; Wiles, M.V.; Shultz, L.D.; Amiji, M.M.; Hosur, V. Role of MicroRNA in Inflammatory Bowel Disease: Clinical Evidence and the Development of Preclinical Animal Models. Cells 2021, 10, 2204. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wu, G.D.; Albenberg, L.; Tomov, V.T. Gut microbiota and IBD: Causation or correlation? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 573–584. [Google Scholar] [CrossRef] [Green Version]
- De Preter, V. Metabolomics in the Clinical Diagnosis of Inflammatory Bowel Disease. Dig. Dis. 2015, 33 (Suppl. 1), 2–10. [Google Scholar] [CrossRef]
- Martini, E.; Krug, S.M.; Siegmund, B.; Neurath, M.F.; Becker, C. Mend Your Fences: The Epithelial Barrier and its Relationship With Mucosal Immunity in Inflammatory Bowel Disease. Cell Mol. Gastroenterol. Hepatol. 2017, 4, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Whitley, C.S.; Haribabu, B.; Jala, V.R. Regulation of Intestinal Barrier Function by Microbial Metabolites. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 1463–1482. [Google Scholar] [CrossRef]
- Ulluwishewa, D.; Anderson, R.C.; McNabb, W.C.; Moughan, P.J.; Wells, J.M.; Roy, N.C. Regulation of tight junction permeability by intestinal bacteria and dietary components. J. Nutr. 2011, 141, 769–776. [Google Scholar] [CrossRef] [Green Version]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21. [Google Scholar] [CrossRef]
- Fasano, A. Intestinal zonulin: Open sesame! Gut 2001, 49, 159–162. [Google Scholar] [CrossRef] [Green Version]
- Cario, E.; Gerken, G.; Podolsky, D.K. Toll-like receptor 2.enhances ZO-1-associated intestinal epithelial barrier integrity via protein kinase C. Gastroenterology 2004, 127, 224–238. [Google Scholar] [CrossRef]
- Darwich, A.S.; Aslam, U.; Ashcroft, D.M.; Rostami-Hodjegan, A. Meta-analysis of the turnover of intestinal epithelia in preclinical animal species and humans. Drug Metab. Dispos. 2014, 42, 2016–2022. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.M.; Duckworth, C.A.; Watson, A.J.; Frey, M.R.; Miguel, J.C.; Burkitt, M.D.; Sutton, R.; Hughes, K.R.; Hall, L.J.; Caamano, J.H.; et al. A mouse model of pathological small intestinal epithelial cell apoptosis and shedding induced by systemic administration of lipopolysaccharide. Dis. Model. Mech. 2013, 6, 1388–1399. [Google Scholar] [CrossRef] [Green Version]
- Hughes, K.R.; Schofield, Z.; Dalby, M.J.; Caim, S.; Chalklen, L.; Bernuzzi, F.; Alcon-Giner, C.; Le Gall, G.; Watson, A.J.M.; Hall, L.J.; et al. The early life microbiota protects neonatal mice from pathological small intestinal epithelial cell shedding. FASEB J. 2020, 34, 7075–7088. [Google Scholar] [CrossRef] [Green Version]
- Van der Sluis, M.; De Koning, B.A.; De Bruijn, A.C.; Velcich, A.; Meijerink, J.P.; Van Goudoever, J.B.; Buller, H.A.; Dekker, J.; Van Seuningen, I.; Renes, I.B.; et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef]
- Morampudi, V.; Dalwadi, U.; Bhinder, G.; Sham, H.P.; Gill, S.K.; Chan, J.; Bergstrom, K.S.; Huang, T.; Ma, C.; Jacobson, K.; et al. The goblet cell-derived mediator RELM-beta drives spontaneous colitis in Muc2-deficient mice by promoting commensal microbial dysbiosis. Mucosal. Immunol. 2016, 9, 1218–1233. [Google Scholar] [CrossRef] [Green Version]
- Ayabe, T.; Satchell, D.P.; Wilson, C.L.; Parks, W.C.; Selsted, M.E.; Ouellette, A.J. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat. Immunol. 2000, 1, 113–118. [Google Scholar] [CrossRef]
- Shimizu, Y.; Nakamura, K.; Yoshii, A.; Yokoi, Y.; Kikuchi, M.; Shinozaki, R.; Nakamura, S.; Ohira, S.; Sugimoto, R.; Ayabe, T. Paneth cell alpha-defensin misfolding correlates with dysbiosis and ileitis in Crohn’s disease model mice. Life Sci. Alliance 2020, 3. [Google Scholar] [CrossRef]
- Aldhous, M.C.; Noble, C.L.; Satsangi, J. Dysregulation of human beta-defensin-2 protein in inflammatory bowel disease. PLoS ONE 2009, 4, e6285. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Leong, R.W.; Wasinger, V.C.; Ip, M.; Yang, M.; Phan, T.G. Impaired Intestinal Permeability Contributes to Ongoing Bowel Symptoms in Patients with Inflammatory Bowel Disease and Mucosal Healing. Gastroenterology 2017, 153, 723–731.e1. [Google Scholar] [CrossRef]
- Szymanska, E.; Wierzbicka, A.; Dadalski, M.; Kierkus, J. Fecal Zonulin as a Noninvasive Biomarker of Intestinal Permeability in Pediatric Patients with Inflammatory Bowel Diseases-Correlation with Disease Activity and Fecal Calprotectin. J. Clin. Med. 2021, 10, 3905. [Google Scholar] [CrossRef]
- Caviglia, G.P.; Dughera, F.; Ribaldone, D.G.; Rosso, C.; Abate, M.L.; Pellicano, R.; Bresso, F.; Smedile, A.; Saracco, G.M.; Astegiano, M. Serum zonulin in patients with inflammatory bowel disease: A pilot study. Minerva. Med. 2019, 110, 95–100. [Google Scholar] [CrossRef]
- Arrieta, M.C.; Madsen, K.; Doyle, J.; Meddings, J. Reducing small intestinal permeability attenuates colitis in the IL10 gene-deficient mouse. Gut 2009, 58, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Resta-Lenert, S.; Smitham, J.; Barrett, K.E. Epithelial dysfunction associated with the development of colitis in conventionally housed mdr1a-/- mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G153–G162. [Google Scholar] [CrossRef] [Green Version]
- Laukoetter, M.G.; Nava, P.; Lee, W.Y.; Severson, E.A.; Capaldo, C.T.; Babbin, B.A.; Williams, I.R.; Koval, M.; Peatman, E.; Campbell, J.A.; et al. JAM-A regulates permeability and inflammation in the intestine in vivo. J. Exp. Med. 2007, 204, 3067–3076. [Google Scholar] [CrossRef] [PubMed]
- Kuo, W.T.; Zuo, L.; Odenwald, M.A.; Madha, S.; Singh, G.; Gurniak, C.B.; Abraham, C.; Turner, J.R. The Tight Junction Protein ZO-1 Is Dispensable for Barrier Function but Critical for Effective Mucosal Repair. Gastroenterology 2021, 161, 1924–1939. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Chaturvedi, R.; Olivares-Villagomez, D.; Habib, T.; Asim, M.; Shivesh, P.; Polk, D.B.; Wilson, K.T.; Washington, M.K.; Van Kaer, L.; et al. Targeted colonic claudin-2 expression renders resistance to epithelial injury, induces immune suppression, and protects from colitis. Mucosal. Immunol. 2014, 7, 1340–1353. [Google Scholar] [CrossRef] [PubMed]
- Turpin, W.; Espin-Garcia, O.; Bedrani, L.; Madsen, K.; Meddings, J.B.; Raygoza Garay, J.A.; Silverberg, M.S.; Smith, M.I.; Griffiths, A.M.; Moayyedi, P.; et al. Analysis of Genetic Association of Intestinal Permeability in Healthy First-degree Relatives of Patients with Crohn’s Disease. Inflamm. Bowel. Dis. 2019, 25, 1796–1804. [Google Scholar] [CrossRef]
- Boirivant, M.; Amendola, A.; Butera, A.; Sanchez, M.; Xu, L.; Marinaro, M.; Kitani, A.; Di Giacinto, C.; Strober, W.; Fuss, I.J. A transient breach in the epithelial barrier leads to regulatory T-cell generation and resistance to experimental colitis. Gastroenterology 2008, 135, 1612–1623.e5. [Google Scholar] [CrossRef]
- Aldars-Garcia, L.; Marin, A.C.; Chaparro, M.; Gisbert, J.P. The Interplay between Immune System and Microbiota in Inflammatory Bowel Disease: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 3076. [Google Scholar] [CrossRef]
- Imdad, A.; Nicholson, M.R.; Tanner-Smith, E.E.; Zackular, J.P.; Gomez-Duarte, O.G.; Beaulieu, D.B.; Acra, S. Fecal transplantation for treatment of inflammatory bowel disease. Cochrane Database Syst. Rev. 2018, 11, CD012774. [Google Scholar] [CrossRef]
- Sokol, H.; Landman, C.; Seksik, P.; Berard, L.; Montil, M.; Nion-Larmurier, I.; Bourrier, A.; Le Gall, G.; Lalande, V.; De Rougemont, A.; et al. Fecal microbiota transplantation to maintain remission in Crohn’s disease: A pilot randomized controlled study. Microbiome 2020, 8, 12. [Google Scholar] [CrossRef]
- Uchimura, Y.; Fuhrer, T.; Li, H.; Lawson, M.A.; Zimmermann, M.; Yilmaz, B.; Zindel, J.; Ronchi, F.; Sorribas, M.; Hapfelmeier, S.; et al. Antibodies Set Boundaries Limiting Microbial Metabolite Penetration and the Resultant Mammalian Host Response. Immunity 2018, 49, 545–559.e5. [Google Scholar] [CrossRef] [Green Version]
- Lavelle, A.; Sokol, H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [Green Version]
- Lamb, C.A.; Kennedy, N.A.; Raine, T.; Hendy, P.A.; Smith, P.J.; Limdi, J.K.; Hayee, B.; Lomer, M.C.E.; Parkes, G.C.; Selinger, C.; et al. British Society of Gastroenterology consensus guidelines on the management of inflammatory bowel disease in adults. Gut 2019, 68 (Suppl. 3), s1–s106. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, K.; Catesson, A.; Griffin, J.L.; Holmes, E.; Williams, H.R.T. Metabolomic Analysis in Inflammatory Bowel Disease: A Systematic Review. J. Crohns Colitis. 2021, 15, 813–826. [Google Scholar] [CrossRef]
- Bjerrum, J.T.; Wang, Y.L.; Seidelin, J.B.; Nielsen, O.H. IBD metabonomics predicts phenotype, disease course, and treatment response. EBioMedicine 2021, 71, 103551. [Google Scholar] [CrossRef] [PubMed]
- Bauermeister, A.; Mannochio-Russo, H.; Costa-Lotufo, L.V.; Jarmusch, A.K.; Dorrestein, P.C. Mass spectrometry-based metabolomics in microbiome investigations. Nat. Rev. Microbiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, G.; Radcenco, A.L.; Evaristo, J.; Monnerat, G. Novel strategies for clinical investigation and biomarker discovery: A guide to applied metabolomics. Horm. Mol. Biol. Clin. Investig. 2019, 38, 20180045. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.X.; Wang, S.Y.; Kuo, C.H.; Tsai, I.L. Metabolome analysis for investigating host-gut microbiota interactions. J. Formos. Med. Assoc. 2019, 118 (Suppl. 1), S10–S22. [Google Scholar] [CrossRef]
- Smirnov, K.S.; Maier, T.V.; Walker, A.; Heinzmann, S.S.; Forcisi, S.; Martinez, I.; Walter, J.; Schmitt-Kopplin, P. Challenges of metabolomics in human gut microbiota research. Int. J. Med. Microbiol. 2016, 306, 266–279. [Google Scholar] [CrossRef]
- Krautkramer, K.A.; Fan, J.; Backhed, F. Gut microbial metabolites as multi-kingdom intermediates. Nat. Rev. Microbiol. 2021, 19, 77–94. [Google Scholar] [CrossRef]
- Kozik, A.J.; Nakatsu, C.H.; Chun, H.; Jones-Hall, Y.L. Comparison of the fecal, cecal, and mucus microbiome in male and female mice after TNBS-induced colitis. PLoS ONE 2019, 14, e0225079. [Google Scholar] [CrossRef]
- Yuan, C.; Graham, M.; Staley, C.; Subramanian, S. Mucosal Microbiota and Metabolome along the Intestinal Tract Reveal a Location-Specific Relationship. mSystems 2020, 5. [Google Scholar] [CrossRef]
- Wang, Y.; Holmes, E.; Comelli, E.M.; Fotopoulos, G.; Dorta, G.; Tang, H.; Rantalainen, M.J.; Lindon, J.C.; Corthesy-Theulaz, I.E.; Fay, L.B.; et al. Topographical variation in metabolic signatures of human gastrointestinal biopsies revealed by high-resolution magic-angle spinning 1H NMR spectroscopy. J. Proteome Res. 2007, 6, 3944–3951. [Google Scholar] [CrossRef]
- Ruppin, H.; Bar-Meir, S.; Soergel, K.H.; Wood, C.M.; Schmitt, M.G., Jr. Absorption of short-chain fatty acids by the colon. Gastroenterology 1980, 78, 1500–1507. [Google Scholar] [CrossRef]
- Mourad, F.H.; Barada, K.A.; Saade, N.E. Impairment of Small Intestinal Function in Ulcerative Colitis: Role of Enteric Innervation. J. Crohns Colitis. 2017, 11, 369–377. [Google Scholar] [CrossRef] [Green Version]
- Le Gall, G.; Noor, S.O.; Ridgway, K.; Scovell, L.; Jamieson, C.; Johnson, I.T.; Colquhoun, I.J.; Kemsley, E.K.; Narbad, A. Metabolomics of fecal extracts detects altered metabolic activity of gut microbiota in ulcerative colitis and irritable bowel syndrome. J. Proteome Res. 2011, 10, 4208–4218. [Google Scholar] [CrossRef]
- Williams, H.R.; Cox, I.J.; Walker, D.G.; Cobbold, J.F.; Taylor-Robinson, S.D.; Marshall, S.E.; Orchard, T.R. Differences in gut microbial metabolism are responsible for reduced hippurate synthesis in Crohn’s disease. BMC Gastroenterol. 2010, 10, 108. [Google Scholar] [CrossRef] [Green Version]
- Vernocchi, P.; Del Chierico, F.; Putignani, L. Gut Microbiota Profiling: Metabolomics Based Approach to Unravel Compounds Affecting Human Health. Front. Microbiol. 2016, 7, 1144. [Google Scholar] [CrossRef]
- Walton, C.; Fowler, D.P.; Turner, C.; Jia, W.; Whitehead, R.N.; Griffiths, L.; Dawson, C.; Waring, R.H.; Ramsden, D.B.; Cole, J.A.; et al. Analysis of volatile organic compounds of bacterial origin in chronic gastrointestinal diseases. Inflamm. Bowel. Dis. 2013, 19, 2069–2078. [Google Scholar] [CrossRef]
- Alghamdi, A.; Gerasimidis, K.; Blackburn, G.; Akinci, D.; Edwards, C.; Russell, R.K.; Watson, D.G. Untargeted Metabolomics of Extracts from Faecal Samples Demonstrates Distinct Differences between Paediatric Crohn’s Disease Patients and Healthy Controls but No Significant Changes Resulting from Exclusive Enteral Nutrition Treatment. Metabolites 2018, 8, 82. [Google Scholar] [CrossRef] [Green Version]
- Scoville, E.A.; Allaman, M.M.; Brown, C.T.; Motley, A.K.; Horst, S.N.; Williams, C.S.; Koyama, T.; Zhao, Z.; Adams, D.W.; Beaulieu, D.B.; et al. Alterations in Lipid, Amino Acid, and Energy Metabolism Distinguish Crohn’s Disease from Ulcerative Colitis and Control Subjects by Serum Metabolomic Profiling. Metabolomics 2018, 14, 17. [Google Scholar] [CrossRef]
- Ooi, M.; Nishiumi, S.; Yoshie, T.; Shiomi, Y.; Kohashi, M.; Fukunaga, K.; Nakamura, S.; Matsumoto, T.; Hatano, N.; Shinohara, M.; et al. GC/MS-based profiling of amino acids and TCA cycle-related molecules in ulcerative colitis. Inflamm. Res. 2011, 60, 831–840. [Google Scholar] [CrossRef]
- Hisamatsu, T.; Ono, N.; Imaizumi, A.; Mori, M.; Suzuki, H.; Uo, M.; Hashimoto, M.; Naganuma, M.; Matsuoka, K.; Mizuno, S.; et al. Decreased Plasma Histidine Level Predicts Risk of Relapse in Patients with Ulcerative Colitis in Remission. PLoS ONE 2015, 10, e0140716. [Google Scholar] [CrossRef]
- Nikolaus, S.; Schulte, B.; Al-Massad, N.; Thieme, F.; Schulte, D.M.; Bethge, J.; Rehman, A.; Tran, F.; Aden, K.; Hasler, R.; et al. Increased Tryptophan Metabolism Is Associated with Activity of Inflammatory Bowel Diseases. Gastroenterology 2017, 153, 1504–1516.e2. [Google Scholar] [CrossRef] [Green Version]
- Williams, H.R.; Cox, I.J.; Walker, D.G.; North, B.V.; Patel, V.M.; Marshall, S.E.; Jewell, D.P.; Ghosh, S.; Thomas, H.J.; Teare, J.P.; et al. Characterization of inflammatory bowel disease with urinary metabolic profiling. Am. J. Gastroenterol. 2009, 104, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Santoru, M.L.; Piras, C.; Murgia, A.; Palmas, V.; Camboni, T.; Liggi, S.; Ibba, I.; Lai, M.A.; Orru, S.; Blois, S.; et al. Cross sectional evaluation of the gut-microbiome metabolome axis in an Italian cohort of IBD patients. Sci. Rep. 2017, 7, 9523. [Google Scholar] [CrossRef] [PubMed]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2013, 62, 531–539. [Google Scholar] [CrossRef]
- Jansson, J.; Willing, B.; Lucio, M.; Fekete, A.; Dicksved, J.; Halfvarson, J.; Tysk, C.; Schmitt-Kopplin, P. Metabolomics reveals metabolic biomarkers of Crohn’s disease. PLoS ONE 2009, 4, e6386. [Google Scholar] [CrossRef] [Green Version]
- Santoru, M.L.; Piras, C.; Murgia, F.; Leoni, V.P.; Spada, M.; Murgia, A.; Liggi, S.; Lai, M.A.; Usai, P.; Caboni, P.; et al. Metabolic Alteration in Plasma and Biopsies from Patients With IBD. Inflamm. Bowel. Dis. 2021, 27, 1335–1345. [Google Scholar] [CrossRef]
- Bezabeh, T.; Somorjai, R.L.; Smith, I.C.; Nikulin, A.E.; Dolenko, B.; Bernstein, C.N. The use of 1H magnetic resonance spectroscopy in inflammatory bowel diseases: Distinguishing ulcerative colitis from Crohn’s disease. Am. J. Gastroenterol. 2001, 96, 442–448. [Google Scholar] [CrossRef]
- Bjerrum, J.T.; Wang, Y.; Hao, F.; Coskun, M.; Ludwig, C.; Gunther, U.; Nielsen, O.H. Metabonomics of human fecal extracts characterize ulcerative colitis, Crohn’s disease and healthy individuals. Metabolomics 2015, 11, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Kolho, K.L.; Pessia, A.; Jaakkola, T.; de Vos, W.M.; Velagapudi, V. Faecal and Serum Metabolomics in Paediatric Inflammatory Bowel Disease. J. Crohns Colitis. 2017, 11, 321–334. [Google Scholar] [CrossRef]
- De Preter, V.; Machiels, K.; Joossens, M.; Arijs, I.; Matthys, C.; Vermeire, S.; Rutgeerts, P.; Verbeke, K. Faecal metabolite profiling identifies medium-chain fatty acids as discriminating compounds in IBD. Gut 2015, 64, 447–458. [Google Scholar] [CrossRef]
- Ahmed, I.; Greenwood, R.; Costello, B.; Ratcliffe, N.; Probert, C.S. Investigation of faecal volatile organic metabolites as novel diagnostic biomarkers in inflammatory bowel disease. Aliment. Pharmacol Ther. 2016, 43, 596–611. [Google Scholar] [CrossRef] [Green Version]
- Weng, Y.J.; Gan, H.Y.; Li, X.; Huang, Y.; Li, Z.C.; Deng, H.M.; Chen, S.Z.; Zhou, Y.; Wang, L.S.; Han, Y.P.; et al. Correlation of diet, microbiota and metabolite networks in inflammatory bowel disease. J. Dig. Dis. 2019, 20, 447–459. [Google Scholar] [CrossRef]
- Jacobs, J.P.; Goudarzi, M.; Singh, N.; Tong, M.; McHardy, I.H.; Ruegger, P.; Asadourian, M.; Moon, B.H.; Ayson, A.; Borneman, J.; et al. A Disease-Associated Microbial and Metabolomics State in Relatives of Pediatric Inflammatory Bowel Disease Patients. Cell Mol. Gastroenterol. Hepatol. 2016, 2, 750–766. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Du, B.; Shi, Y.; Lu, Y.; Zhou, Y.; Liu, B. Combined Signature of the Fecal Microbiome and Plasma Metabolome in Patients with Ulcerative Colitis. Med. Sci. Monit. 2019, 25, 3303–3315. [Google Scholar] [CrossRef]
- Ding, N.S.; Hart, A.; De Cruz, P. Systematic review: Predicting and optimising response to anti-TNF therapy in Crohn’s disease—Algorithm for practical management. Aliment. Pharmacol. Ther. 2016, 43, 30–51. [Google Scholar] [CrossRef]
- Taylor, H.; Serrano-Contreras, J.I.; McDonald, J.A.K.; Epstein, J.; Fell, J.M.; Seoane, R.C.; Li, J.V.; Marchesi, J.R.; Hart, A.L. Multiomic features associated with mucosal healing and inflammation in paediatric Crohn’s disease. Aliment. Pharmacol. Ther. 2020, 52, 1491–1502. [Google Scholar] [CrossRef]
- Aden, K.; Rehman, A.; Waschina, S.; Pan, W.H.; Walker, A.; Lucio, M.; Nunez, A.M.; Bharti, R.; Zimmerman, J.; Bethge, J.; et al. Metabolic Functions of Gut Microbes Associate with Efficacy of Tumor Necrosis Factor Antagonists in Patients with Inflammatory Bowel Diseases. Gastroenterology 2019, 157, 1279–1292.e11. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Gao, X.; Zhang, X.; Xiao, F.; Hu, H.; Li, X.; Dong, F.; Sun, M.; Xiao, Y.; Ge, T.; et al. Microbial and metabolic features associated with outcome of infliximab therapy in pediatric Crohn’s disease. Gut Microbes. 2021, 13, 1–18. [Google Scholar] [CrossRef]
- Ding, N.S.; McDonald, J.A.K.; Perdones-Montero, A.; Rees, D.N.; Adegbola, S.O.; Misra, R.; Hendy, P.; Penez, L.; Marchesi, J.R.; Holmes, E.; et al. Metabonomics and the Gut Microbiome Associated with Primary Response to Anti-TNF Therapy in Crohn’s Disease. J. Crohns Colitis. 2020, 14, 1090–1102. [Google Scholar] [CrossRef]
- Lee, J.W.J.; Plichta, D.; Hogstrom, L.; Borren, N.Z.; Lau, H.; Gregory, S.M.; Tan, W.; Khalili, H.; Clish, C.; Vlamakis, H.; et al. Multi-omics reveal microbial determinants impacting responses to biologic therapies in inflammatory bowel disease. Cell Host Microbe 2021, 29, 1294–1304.e4. [Google Scholar] [CrossRef] [PubMed]
- Connors, J.; Dunn, K.A.; Allott, J.; Bandsma, R.; Rashid, M.; Otley, A.R.; Bielawski, J.P.; Van Limbergen, J. The relationship between fecal bile acids and microbiome community structure in pediatric Crohn’s disease. ISME J. 2020, 14, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Magro, F.; Rodrigues-Pinto, E.; Santos-Antunes, J.; Vilas-Boas, F.; Lopes, S.; Nunes, A.; Camila-Dias, C.; Macedo, G. High C-reactive protein in Crohn’s disease patients predicts nonresponse to infliximab treatment. J. Crohns Colitis. 2014, 8, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Roager, H.M.; Vogt, J.K.; Kristensen, M.; Hansen, L.B.S.; Ibrugger, S.; Maerkedahl, R.B.; Bahl, M.I.; Lind, M.V.; Nielsen, R.L.; Frokiaer, H.; et al. Whole grain-rich diet reduces body weight and systemic low-grade inflammation without inducing major changes of the gut microbiome: A randomised cross-over trial. Gut 2019, 68, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Ott, S.J.; Waetzig, G.H.; Rehman, A.; Moltzau-Anderson, J.; Bharti, R.; Grasis, J.A.; Cassidy, L.; Tholey, A.; Fickenscher, H.; Seegert, D.; et al. Efficacy of Sterile Fecal Filtrate Transfer for Treating Patients with Clostridium difficile Infection. Gastroenterology 2017, 152, 799–811.e7. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, A. Animal models of inflammatory bowel disease. Prog. Mol. Biol. Transl. Sci. 2012, 105, 263–320. [Google Scholar] [CrossRef]
- Hormannsperger, G.; Schaubeck, M.; Haller, D. Intestinal Microbiota in Animal Models of Inflammatory Diseases. ILAR J. 2015, 56, 179–191. [Google Scholar] [CrossRef]
- Montenegro-Burke, J.R.; Kok, B.P.; Guijas, C.; Domingo-Almenara, X.; Moon, C.; Galmozzi, A.; Kitamura, S.; Eckmann, L.; Saez, E.; Siuzdak, G.E.; et al. Metabolomics activity screening of T cell-induced colitis reveals anti-inflammatory metabolites. Sci. Signal. 2021, 14, eabf6584. [Google Scholar] [CrossRef]
- Knudsen, L.A.; Desdorf, R.; Moller, S.; Sorensen, S.B.; Hansen, A.K.; Andersen, V. Translational Potential of Metabolomics on Animal Models of Inflammatory Bowel Disease-A Systematic Critical Review. Int. J. Mol. Sci. 2020, 21, 3856. [Google Scholar] [CrossRef]
- van der Hee, B.; Wells, J.M. Microbial Regulation of Host Physiology by Short-chain Fatty Acids. Trends Microbiol. 2021, 29, 700–712. [Google Scholar] [CrossRef]
- Mayorgas, A.; Dotti, I.; Salas, A. Microbial Metabolites, Postbiotics, and Intestinal Epithelial Function. Mol. Nutr. Food Res. 2021, 65, e2000188. [Google Scholar] [CrossRef]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; Gonzalez, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Chen, F.; Wu, W.; Sun, M.; Bilotta, A.J.; Yao, S.; Xiao, Y.; Huang, X.; Eaves-Pyles, T.D.; Golovko, G.; et al. GPR43 mediates microbiota metabolite SCFA regulation of antimicrobial peptide expression in intestinal epithelial cells via activation of mTOR and STAT3. Mucosal. Immunol. 2018, 11, 752–762. [Google Scholar] [CrossRef] [Green Version]
- Melhem, H.; Kaya, B.; Ayata, C.K.; Hruz, P.; Niess, J.H. Metabolite-Sensing G Protein-Coupled Receptors Connect the Diet-Microbiota-Metabolites Axis to Inflammatory Bowel Disease. Cells 2019, 8, 450. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Herring, C.A.; Chen, B.; Kim, H.; Simmons, A.J.; Southard-Smith, A.N.; Allaman, M.M.; White, J.R.; Macedonia, M.C.; McKinley, E.T.; et al. Succinate Produced by Intestinal Microbes Promotes Specification of Tuft Cells to Suppress Ileal Inflammation. Gastroenterology 2020, 159, 2101–2115.e5. [Google Scholar] [CrossRef]
- Peng, L.; Li, Z.R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef]
- Wang, R.X.; Lee, J.S.; Campbell, E.L.; Colgan, S.P. Microbiota-derived butyrate dynamically regulates intestinal homeostasis through regulation of actin-associated protein synaptopodin. Proc. Natl. Acad. Sci. USA 2020, 117, 11648–11657. [Google Scholar] [CrossRef]
- Hatayama, H.; Iwashita, J.; Kuwajima, A.; Abe, T. The short chain fatty acid, butyrate, stimulates MUC2 mucin production in the human colon cancer cell line, LS174T. Biochem. Biophys Res. Commun. 2007, 356, 599–603. [Google Scholar] [CrossRef]
- Gaudier, E.; Jarry, A.; Blottiere, H.M.; de Coppet, P.; Buisine, M.P.; Aubert, J.P.; Laboisse, C.; Cherbut, C.; Hoebler, C. Butyrate specifically modulates MUC gene expression in intestinal epithelial goblet cells deprived of glucose. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1168–G1174. [Google Scholar] [CrossRef] [Green Version]
- Augenlicht, L.; Shi, L.; Mariadason, J.; Laboisse, C.; Velcich, A. Repression of MUC2 gene expression by butyrate, a physiological regulator of intestinal cell maturation. Oncogene 2003, 22, 4983–4992. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Oshima, T.; Tomita, T.; Fukui, H.; Miwa, H. Butyrate Alleviates Cytokine-Induced Barrier Dysfunction by Modifying Claudin-2 Levels. Biology 2021, 10, 205. [Google Scholar] [CrossRef]
- Vancamelbeke, M.; Laeremans, T.; Vanhove, W.; Arnauts, K.; Ramalho, A.S.; Farre, R.; Cleynen, I.; Ferrante, M.; Vermeire, S. Butyrate Does Not Protect Against Inflammation-induced Loss of Epithelial Barrier Function and Cytokine Production in Primary Cell Monolayers from Patients with Ulcerative Colitis. J. Crohns Colitis. 2019, 13, 1351–1361. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Picon, E.; Dotti, I.; Corraliza, A.M.; Mayorgas, A.; Esteller, M.; Perales, J.C.; Ricart, E.; Masamunt, M.C.; Carrasco, A.; Tristan, E.; et al. Intestinal Inflammation Modulates the Epithelial Response to Butyrate in Patients with Inflammatory Bowel Disease. Inflamm. Bowel. Dis. 2020, 26, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, Z.H.; Zabed, H.M.; Yun, J.; Zhang, G.; Qi, X. An Insight into the Roles of Dietary Tryptophan and Its Metabolites in Intestinal Inflammation and Inflammatory Bowel Disease. Mol. Nutr. Food Res. 2021, 65, e2000461. [Google Scholar] [CrossRef] [PubMed]
- Huhn, M.; Juan, M.H.S.; Melcher, B.; Dreis, C.; Schmidt, K.G.; Schwiebs, A.; Collins, J.; Pfeilschifter, J.M.; Vieth, M.; Stein, J.; et al. Inflammation-Induced Mucosal KYNU Expression Identifies Human Ileal Crohn’s Disease. J. Clin. Med. 2020, 9, 1360. [Google Scholar] [CrossRef] [PubMed]
- Heath-Pagliuso, S.; Rogers, W.J.; Tullis, K.; Seidel, S.D.; Cenijn, P.H.; Brouwer, A.; Denison, M.S. Activation of the Ah receptor by tryptophan and tryptophan metabolites. Biochemistry 1998, 37, 11508–11515. [Google Scholar] [CrossRef] [PubMed]
- Stockinger, B.; Shah, K.; Wincent, E. AHR in the intestinal microenvironment: Safeguarding barrier function. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Parks, O.B.; Pociask, D.A.; Hodzic, Z.; Kolls, J.K.; Good, M. Interleukin-22 Signaling in the Regulation of Intestinal Health and Disease. Front. Cell Dev. Biol. 2015, 3, 85. [Google Scholar] [CrossRef] [Green Version]
- Metidji, A.; Omenetti, S.; Crotta, S.; Li, Y.; Nye, E.; Ross, E.; Li, V.; Maradana, M.R.; Schiering, C.; Stockinger, B. The Environmental Sensor AHR Protects from Inflammatory Damage by Maintaining Intestinal Stem Cell Homeostasis and Barrier Integrity. Immunity 2018, 49, 353–362.e5. [Google Scholar] [CrossRef] [Green Version]
- Alexeev, E.E.; Lanis, J.M.; Kao, D.J.; Campbell, E.L.; Kelly, C.J.; Battista, K.D.; Gerich, M.E.; Jenkins, B.R.; Walk, S.T.; Kominsky, D.J.; et al. Microbiota-Derived Indole Metabolites Promote Human and Murine Intestinal Homeostasis through Regulation of Interleukin-10 Receptor. Am. J. Pathol. 2018, 188, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S.; et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef] [Green Version]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Powell, D.N.; Swimm, A.; Sonowal, R.; Bretin, A.; Gewirtz, A.T.; Jones, R.M.; Kalman, D. Indoles from the commensal microbiota act via the AHR and IL-10 to tune the cellular composition of the colonic epithelium during aging. Proc. Natl. Acad. Sci. USA 2020, 117, 21519–21526. [Google Scholar] [CrossRef]
- Scott, S.A.; Fu, J.; Chang, P.V. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. USA 2020, 117, 19376–19387. [Google Scholar] [CrossRef]
- Fiorucci, S.; Carino, A.; Baldoni, M.; Santucci, L.; Costanzi, E.; Graziosi, L.; Distrutti, E.; Biagioli, M. Bile Acid Signaling in Inflammatory Bowel Diseases. Dig. Dis. Sci. 2021, 66, 674–693. [Google Scholar] [CrossRef]
- Wilson, A.; Wang, Q.; Almousa, A.A.; Jansen, L.E.; Choi, Y.H.; Schwarz, U.I.; Kim, R.B. Genetic variation in the farnesoid X-receptor predicts Crohn’s disease severity in female patients. Sci. Rep. 2020, 10, 11725. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.; Renooij, W.; Murzilli, S.; Klomp, L.W.; Siersema, P.D.; Schipper, M.E.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef]
- Cipriani, S.; Mencarelli, A.; Chini, M.G.; Distrutti, E.; Renga, B.; Bifulco, G.; Baldelli, F.; Donini, A.; Fiorucci, S. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS ONE 2011, 6, e25637. [Google Scholar] [CrossRef]
- Alemi, F.; Poole, D.P.; Chiu, J.; Schoonjans, K.; Cattaruzza, F.; Grider, J.R.; Bunnett, N.W.; Corvera, C.U. The receptor TGR5 mediates the prokinetic actions of intestinal bile acids and is required for normal defecation in mice. Gastroenterology 2013, 144, 145–154. [Google Scholar] [CrossRef]
- Liu, W.; Chen, Y.; Golan, M.A.; Annunziata, M.L.; Du, J.; Dougherty, U.; Kong, J.; Musch, M.; Huang, Y.; Pekow, J.; et al. Intestinal epithelial vitamin D receptor signaling inhibits experimental colitis. J. Clin. Investig. 2013, 123, 3983–3996. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Cui, X.; Sun, Y.; Zhao, Q.; Liu, T. Intestinal vitamin D receptor signaling ameliorates dextran sulfate sodium-induced colitis by suppressing necroptosis of intestinal epithelial cells. FASEB J. 2020, 34, 13494–13506. [Google Scholar] [CrossRef]
- Lajczak-McGinley, N.K.; Porru, E.; Fallon, C.M.; Smyth, J.; Curley, C.; McCarron, P.A.; Tambuwala, M.M.; Roda, A.; Keely, S.J. The secondary bile acids, ursodeoxycholic acid and lithocholic acid, protect against intestinal inflammation by inhibition of epithelial apoptosis. Physiol. Rep. 2020, 8, e14456. [Google Scholar] [CrossRef]
- Ward, J.B.J.; Lajczak, N.K.; Kelly, O.B.; O’Dwyer, A.M.; Giddam, A.K.; Ni Gabhann, J.; Franco, P.; Tambuwala, M.M.; Jefferies, C.A.; Keely, S.; et al. Ursodeoxycholic acid and lithocholic acid exert anti-inflammatory actions in the colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G550–G558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golden, J.M.; Escobar, O.H.; Nguyen, M.V.L.; Mallicote, M.U.; Kavarian, P.; Frey, M.R.; Gayer, C.P. Ursodeoxycholic acid protects against intestinal barrier breakdown by promoting enterocyte migration via EGFR- and COX-2-dependent mechanisms. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G259–G271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caffaratti, C.; Plazy, C.; Mery, G.; Tidjani, A.R.; Fiorini, F.; Thiroux, S.; Toussaint, B.; Hannani, D.; Le Gouellec, A. What We Know So Far about the Metabolite-Mediated Microbiota-Intestinal Immunity Dialogue and How to Hear the Sound of This Crosstalk. Metabolites 2021, 11, 406. [Google Scholar] [CrossRef]
- Yoshii, K.; Hosomi, K.; Sawane, K.; Kunisawa, J. Metabolism of Dietary and Microbial Vitamin B Family in the Regulation of Host Immunity. Front. Nutr. 2019, 6, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levit, R.; Savoy de Giori, G.; de Moreno de LeBlanc, A.; LeBlanc, J.G. Effect of riboflavin-producing bacteria against chemically induced colitis in mice. J. Appl. Microbiol. 2018, 124, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Santoru, M.L.; Piras, C.; Murgia, F.; Spada, M.; Tronci, L.; Leoni, V.P.; Serreli, G.; Deiana, M.; Atzori, L. Modulatory Effect of Nicotinic Acid on the Metabolism of Caco-2 Cells Exposed to IL-1beta and LPS. Metabolites 2020, 10, 204. [Google Scholar] [CrossRef]
- Li, J.; Kong, D.; Wang, Q.; Wu, W.; Tang, Y.; Bai, T.; Guo, L.; Wei, L.; Zhang, Q.; Yu, Y.; et al. Niacin ameliorates ulcerative colitis via prostaglandin D2-mediated D prostanoid receptor 1 activation. EMBO Mol. Med. 2017, 9, 571–588. [Google Scholar] [CrossRef]
- Berruyer, C.; Pouyet, L.; Millet, V.; Martin, F.M.; LeGoffic, A.; Canonici, A.; Garcia, S.; Bagnis, C.; Naquet, P.; Galland, F. Vanin-1 licenses inflammatory mediator production by gut epithelial cells and controls colitis by antagonizing peroxisome proliferator-activated receptor gamma activity. J. Exp. Med. 2006, 203, 2817–2827. [Google Scholar] [CrossRef]
- Sugahara, H.; Odamaki, T.; Fukuda, S.; Kato, T.; Xiao, J.Z.; Abe, F.; Kikuchi, J.; Ohno, H. Probiotic Bifidobacterium longum alters gut luminal metabolism through modification of the gut microbial community. Sci. Rep. 2015, 5, 13548. [Google Scholar] [CrossRef] [Green Version]
- Skupsky, J.; Sabui, S.; Hwang, M.; Nakasaki, M.; Cahalan, M.D.; Said, H.M. Biotin Supplementation Ameliorates Murine Colitis by Preventing NF-kappaB Activation. Cell Mol. Gastroenterol. Hepatol. 2020, 9, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Ghosal, A.; Lambrecht, N.; Subramanya, S.B.; Kapadia, R.; Said, H.M. Conditional knockout of the Slc5a6 gene in mouse intestine impairs biotin absorption. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G64–G71. [Google Scholar] [CrossRef] [Green Version]
- Beane, K.E.; Redding, M.C.; Wang, X.; Hoon Pan, J.; Le, B.; Cicalo, C.; Jeon, S.; Kim, Y.J.; Lee, J.H.; Shin, E.C.; et al. Effects of dietary fibers, micronutrients, and phytonutrients on gut microbiome: A review. Appl. Biol. Chem. 2021, 64. [Google Scholar] [CrossRef]
- Lurz, E.; Horne, R.G.; Maattanen, P.; Wu, R.Y.; Botts, S.R.; Li, B.; Rossi, L.; Johnson-Henry, K.C.; Pierro, A.; Surette, M.G.; et al. Vitamin B12 Deficiency Alters the Gut Microbiota in a Murine Model of Colitis. Front. Nutr. 2020, 7, 83. [Google Scholar] [CrossRef]
- MacFarlane, A.J.; Behan, N.A.; Matias, F.M.; Green, J.; Caldwell, D.; Brooks, S.P. Dietary folate does not significantly affect the intestinal microbiome, inflammation or tumorigenesis in azoxymethane-dextran sodium sulphate-treated mice. Br. J. Nutr. 2013, 109, 630–638. [Google Scholar] [CrossRef] [Green Version]
- Karl, J.P.; Meydani, M.; Barnett, J.B.; Vanegas, S.M.; Barger, K.; Fu, X.; Goldin, B.; Kane, A.; Rasmussen, H.; Vangay, P.; et al. Fecal concentrations of bacterially derived vitamin K forms are associated with gut microbiota composition but not plasma or fecal cytokine concentrations in healthy adults. Am. J. Clin. Nutr. 2017, 106, 1052–1061. [Google Scholar] [CrossRef] [Green Version]
- Wagatsuma, K.; Yamada, S.; Ao, M.; Matsuura, M.; Tsuji, H.; Iida, T.; Miyamoto, K.; Oka, K.; Takahashi, M.; Tanaka, K.; et al. Diversity of Gut Microbiota Affecting Serum Level of Undercarboxylated Osteocalcin in Patients with Crohn’s Disease. Nutrients 2019, 11, 1541. [Google Scholar] [CrossRef] [Green Version]
- Iyer, N.; Grizotte-Lake, M.; Duncan, K.; Gordon, S.R.; Palmer, A.C.S.; Calvin, C.; Zhong, G.; Isoherranen, N.; Vaishnava, S. Epithelium intrinsic vitamin A signaling co-ordinates pathogen clearance in the gut via IL-18. PLoS Pathog. 2020, 16, e1008360. [Google Scholar] [CrossRef] [Green Version]
- Woo, V.; Eshleman, E.M.; Hashimoto-Hill, S.; Whitt, J.; Wu, S.E.; Engleman, L.; Rice, T.; Karns, R.; Qualls, J.E.; Haslam, D.B.; et al. Commensal segmented filamentous bacteria-derived retinoic acid primes host defense to intestinal infection. Cell Host Microbe. 2021, S1931-3128, 426-1. [Google Scholar] [CrossRef] [PubMed]
- Andoh, A.; Takaya, H.; Araki, Y.; Tsujikawa, T.; Fujiyama, Y.; Bamba, T. Medium- and long-chain fatty acids differentially modulate interleukin-8 secretion in human fetal intestinal epithelial cells. J. Nutr. 2000, 130, 2636–2640. [Google Scholar] [CrossRef]
- Suzuki, T. Regulation of the intestinal barrier by nutrients: The role of tight junctions. Anim. Sci. J. 2020, 91, e13357. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic. Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Walker, A.; Schmitt-Kopplin, P. The role of fecal sulfur metabolome in inflammatory bowel diseases. Int. J. Med. Microbiol. 2021, 311, 151513. [Google Scholar] [CrossRef]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalova, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef] [Green Version]
- Wen, C.; Guo, Q.; Wang, W.; Duan, Y.; Zhang, L.; Li, J.; He, S.; Chen, W.; Li, F. Taurine Alleviates Intestinal Injury by Mediating Tight Junction Barriers in Diquat-Challenged Piglet Models. Front. Physiol. 2020, 11, 449. [Google Scholar] [CrossRef]
- Singh, S.B.; Lin, H.C. Hydrogen Sulfide in Physiology and Diseases of the Digestive Tract. Microorganisms 2015, 3, 866–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Ni, Y.; Hu, L.; Zhao, Y.; Zheng, L.; Yang, S.; Ni, L.; Fu, Z. Spermidine ameliorates high-fat diet-induced hepatic steatosis and adipose tissue inflammation in preexisting obese mice. Life Sci. 2021, 265, 118739. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Ni, Y.; Wang, Z.; Tu, W.; Ni, L.; Zhuge, F.; Zheng, A.; Hu, L.; Zhao, Y.; Zheng, L.; et al. Spermidine improves gut barrier integrity and gut microbiota function in diet-induced obese mice. Gut Microbes. 2020, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Kurihara, S.; Takahashi, D.; Ohashi, W.; Nakamura, Y.; Kimura, S.; Onuki, M.; Kume, A.; Sasazawa, Y.; Furusawa, Y.; et al. Symbiotic polyamine metabolism regulates epithelial proliferation and macrophage differentiation in the colon. Nat. Commun. 2021, 12, 2105. [Google Scholar] [CrossRef]
- Bernardi, S.; Del Bo, C.; Marino, M.; Gargari, G.; Cherubini, A.; Andres-Lacueva, C.; Hidalgo-Liberona, N.; Peron, G.; Gonzalez-Dominguez, R.; Kroon, P.; et al. Polyphenols and Intestinal Permeability: Rationale and Future Perspectives. J. Agric. Food Chem. 2020, 68, 1816–1829. [Google Scholar] [CrossRef]
- Peisl, B.Y.L.; Schymanski, E.L.; Wilmes, P. Dark matter in host-microbiome metabolomics: Tackling the unknowns—A review. Anal. Chim Acta. 2018, 1037, 13–27. [Google Scholar] [CrossRef]
- Heinken, A.; Hertel, J.; Thiele, I. Metabolic modelling reveals broad changes in gut microbial metabolism in inflammatory bowel disease patients with dysbiosis. NPJ Syst. Biol. Appl. 2021, 7, 19. [Google Scholar] [CrossRef]
- Sahoo, D.; Swanson, L.; Sayed, I.M.; Katkar, G.D.; Ibeawuchi, S.R.; Mittal, Y.; Pranadinata, R.F.; Tindle, C.; Fuller, M.; Stec, D.L.; et al. Artificial intelligence guided discovery of a barrier-protective therapy in inflammatory bowel disease. Nat. Commun. 2021, 12, 4246. [Google Scholar] [CrossRef]
- Chen, H.; Nwe, P.K.; Yang, Y.; Rosen, C.E.; Bielecka, A.A.; Kuchroo, M.; Cline, G.W.; Kruse, A.C.; Ring, A.M.; Crawford, J.M.; et al. A Forward Chemical Genetic Screen Reveals Gut Microbiota Metabolites That Modulate Host Physiology. Cell 2019, 177, 1217–1231.e18. [Google Scholar] [CrossRef]
- Colosimo, D.A.; Kohn, J.A.; Luo, P.M.; Piscotta, F.J.; Han, S.M.; Pickard, A.J.; Rao, A.; Cross, J.R.; Cohen, L.J.; Brady, S.F. Mapping Interactions of Microbial Metabolites with Human G-Protein-Coupled Receptors. Cell Host Microbe. 2019, 26, 273–282.e7. [Google Scholar] [CrossRef] [Green Version]
- Grosheva, I.; Zheng, D.; Levy, M.; Polansky, O.; Lichtenstein, A.; Golani, O.; Dori-Bachash, M.; Moresi, C.; Shapiro, H.; Del Mare-Roumani, S.; et al. High-Throughput Screen Identifies Host and Microbiota Regulators of Intestinal Barrier Function. Gastroenterology 2020, 159, 1807–1823. [Google Scholar] [CrossRef]
- Liang, Y.; Liu, M.; Pu, J.; Zhu, Z.; Gao, Z.; Zhou, Q.; Gu, Q.; Li, P. Probiotics and Their Metabolites Ameliorate Inflammatory Bowel Disease: A Critical Review. Infect. Microbes Dis. 2021, 3, 4–13. [Google Scholar] [CrossRef]
- Shamoon, M.; Martin, N.M.; O’Brien, C.L. Recent advances in gut Microbiota mediated therapeutic targets in inflammatory bowel diseases: Emerging modalities for future pharmacological implications. Pharmacol. Res. 2019, 148, 104344. [Google Scholar] [CrossRef]
- Jadhav, P.; Jiang, Y.; Jarr, K.; Layton, C.; Ashouri, J.F.; Sinha, S.R. Efficacy of Dietary Supplements in Inflammatory Bowel Disease and Related Autoimmune Diseases. Nutrients 2020, 12, 2156. [Google Scholar] [CrossRef] [PubMed]
- Jakubczyk, D.; Leszczynska, K.; Gorska, S. The Effectiveness of Probiotics in the Treatment of Inflammatory Bowel Disease (IBD)-A Critical Review. Nutrients 2020, 12, 1973. [Google Scholar] [CrossRef] [PubMed]
- Ghouri, Y.A.; Richards, D.M.; Rahimi, E.F.; Krill, J.T.; Jelinek, K.A.; DuPont, A.W. Systematic review of randomized controlled trials of probiotics, prebiotics, and synbiotics in inflammatory bowel disease. Clin. Exp. Gastroenterol. 2014, 7, 473–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tursi, A.; Brandimarte, G.; Papa, A.; Giglio, A.; Elisei, W.; Giorgetti, G.M.; Forti, G.; Morini, S.; Hassan, C.; Pistoia, M.A.; et al. Treatment of relapsing mild-to-moderate ulcerative colitis with the probiotic VSL#3 as adjunctive to a standard pharmaceutical treatment: A double-blind, randomized, placebo-controlled study. Am. J. Gastroenterol. 2010, 105, 2218–2227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sood, A.; Midha, V.; Makharia, G.K.; Ahuja, V.; Singal, D.; Goswami, P.; Tandon, R.K. The probiotic preparation, VSL#3 induces remission in patients with mild-to-moderately active ulcerative colitis. Clin. Gastroenterol. Hepatol. 2009, 7, 1202–1209.e1. [Google Scholar] [CrossRef] [PubMed]
- Zocco, M.A.; dal Verme, L.Z.; Cremonini, F.; Piscaglia, A.C.; Nista, E.C.; Candelli, M.; Novi, M.; Rigante, D.; Cazzato, I.A.; Ojetti, V.; et al. Efficacy of Lactobacillus GG in maintaining remission of ulcerative colitis. Aliment. Pharmacol. Ther. 2006, 23, 1567–1574. [Google Scholar] [CrossRef] [PubMed]
- Kruis, W.; Fric, P.; Pokrotnieks, J.; Lukas, M.; Fixa, B.; Kascak, M.; Kamm, M.A.; Weismueller, J.; Beglinger, C.; Stolte, M.; et al. Maintaining remission of ulcerative colitis with the probiotic Escherichia coli Nissle 1917 is as effective as with standard mesalazine. Gut 2004, 53, 1617–1623. [Google Scholar] [CrossRef]
- Darb Emamie, A.; Rajabpour, M.; Ghanavati, R.; Asadolahi, P.; Farzi, S.; Sobouti, B.; Darbandi, A. The effects of probiotics, prebiotics and synbiotics on the reduction of IBD complications, a periodic review during 2009–2020. J. Appl. Microbiol. 2021, 130, 1823–1838. [Google Scholar] [CrossRef]
- Ye, J.; Erland, L.A.E.; Gill, S.K.; Bishop, S.L.; Verdugo-Meza, A.; Murch, S.J.; Gibson, D.L. Metabolomics-Guided Hypothesis Generation for Mechanisms of Intestinal Protection by Live Biotherapeutic Products. Biomolecules 2021, 11, 738. [Google Scholar] [CrossRef]
- Salminen, S.; Collado, M.C.; Endo, A.; Hill, C.; Lebeer, S.; Quigley, E.M.M.; Sanders, M.E.; Shamir, R.; Swann, J.R.; Szajewska, H.; et al. The International Scientific Association of Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of postbiotics. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 649–667. [Google Scholar] [CrossRef]
- Puccetti, M.; Xiroudaki, S.; Ricci, M.; Giovagnoli, S. Postbiotic-Enabled Targeting of the Host-Microbiota-Pathogen Interface: Hints of Antibiotic Decline? Pharmaceutics 2020, 12, 624. [Google Scholar] [CrossRef]
- Nataraj, B.H.; Ali, S.A.; Behare, P.V.; Yadav, H. Postbiotics-parabiotics: The new horizons in microbial biotherapy and functional foods. Microb. Cell Fact. 2020, 19, 168. [Google Scholar] [CrossRef]
- Ewaschuk, J.B.; Diaz, H.; Meddings, L.; Diederichs, B.; Dmytrash, A.; Backer, J.; Looijer-van Langen, M.; Madsen, K.L. Secreted bioactive factors from Bifidobacterium infantis enhance epithelial cell barrier function. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G1025–G1034. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Yu, Z.; Tian, F.; Zhao, J.; Zhang, H.; Zhai, Q.; Chen, W. Surface components and metabolites of probiotics for regulation of intestinal epithelial barrier. Microb. Cell Fact. 2020, 19, 23. [Google Scholar] [CrossRef]
- Kumar, M.; Garand, M.; Al Khodor, S. Integrating omics for a better understanding of Inflammatory Bowel Disease: A step towards personalized medicine. J. Transl. Med. 2019, 17, 419. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iyer, N.; Corr, S.C. Gut Microbial Metabolite-Mediated Regulation of the Intestinal Barrier in the Pathogenesis of Inflammatory Bowel Disease. Nutrients 2021, 13, 4259. https://doi.org/10.3390/nu13124259
Iyer N, Corr SC. Gut Microbial Metabolite-Mediated Regulation of the Intestinal Barrier in the Pathogenesis of Inflammatory Bowel Disease. Nutrients. 2021; 13(12):4259. https://doi.org/10.3390/nu13124259
Chicago/Turabian StyleIyer, Namrata, and Sinéad C. Corr. 2021. "Gut Microbial Metabolite-Mediated Regulation of the Intestinal Barrier in the Pathogenesis of Inflammatory Bowel Disease" Nutrients 13, no. 12: 4259. https://doi.org/10.3390/nu13124259
APA StyleIyer, N., & Corr, S. C. (2021). Gut Microbial Metabolite-Mediated Regulation of the Intestinal Barrier in the Pathogenesis of Inflammatory Bowel Disease. Nutrients, 13(12), 4259. https://doi.org/10.3390/nu13124259