
Flavonoid-Modifying Capabilities of the Human Gut Microbiome—An In Silico Study



Abstract
:1. Introduction
2. Results and Discussion
2.1. Distribution of Flavonoid-Modifying Bacterial Species across Human Gut MAGs
2.2. Screening the MAGs Amino Acid Sequence Database for Potential Flavonoid-Modifying Enzymes
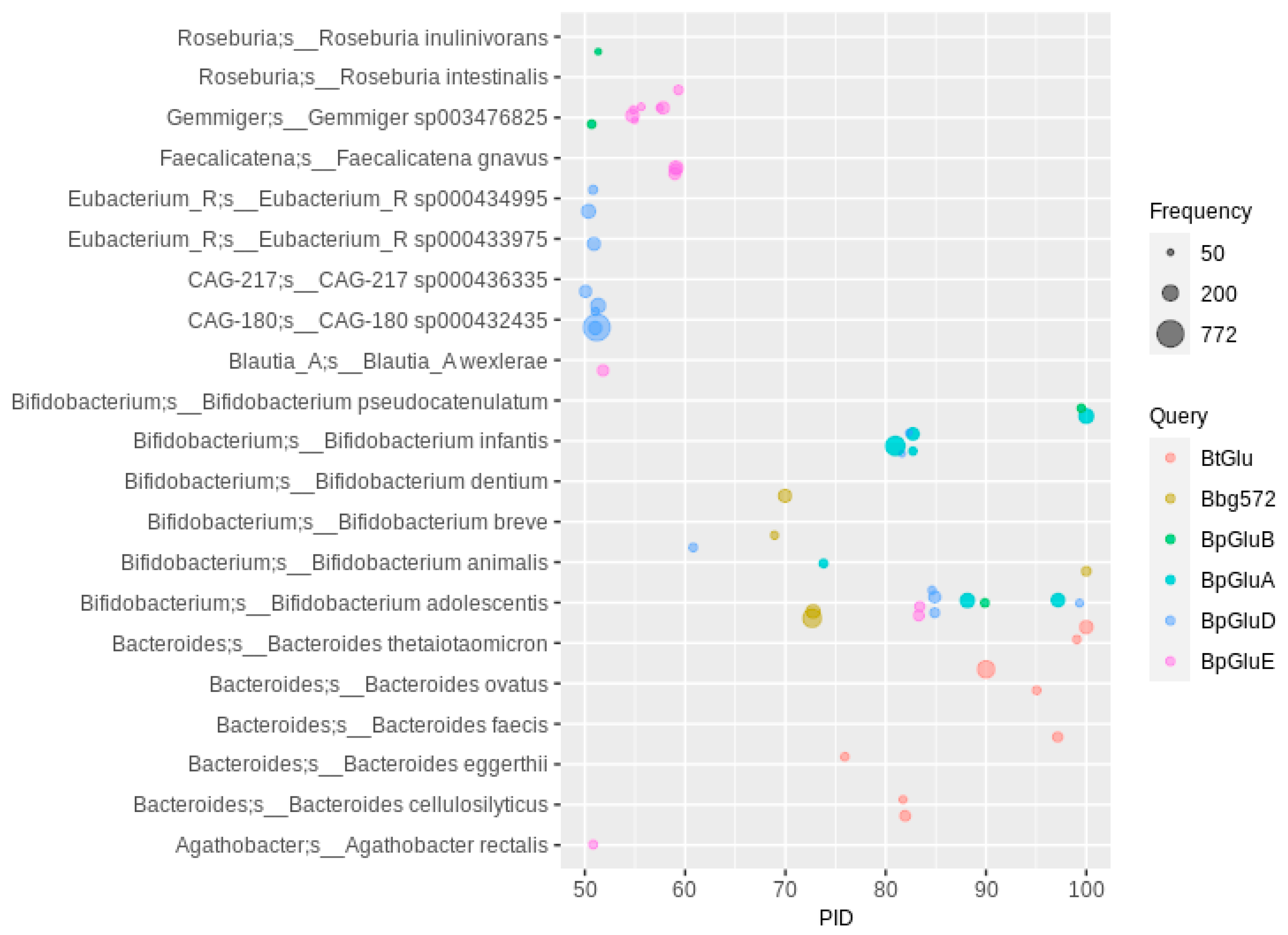
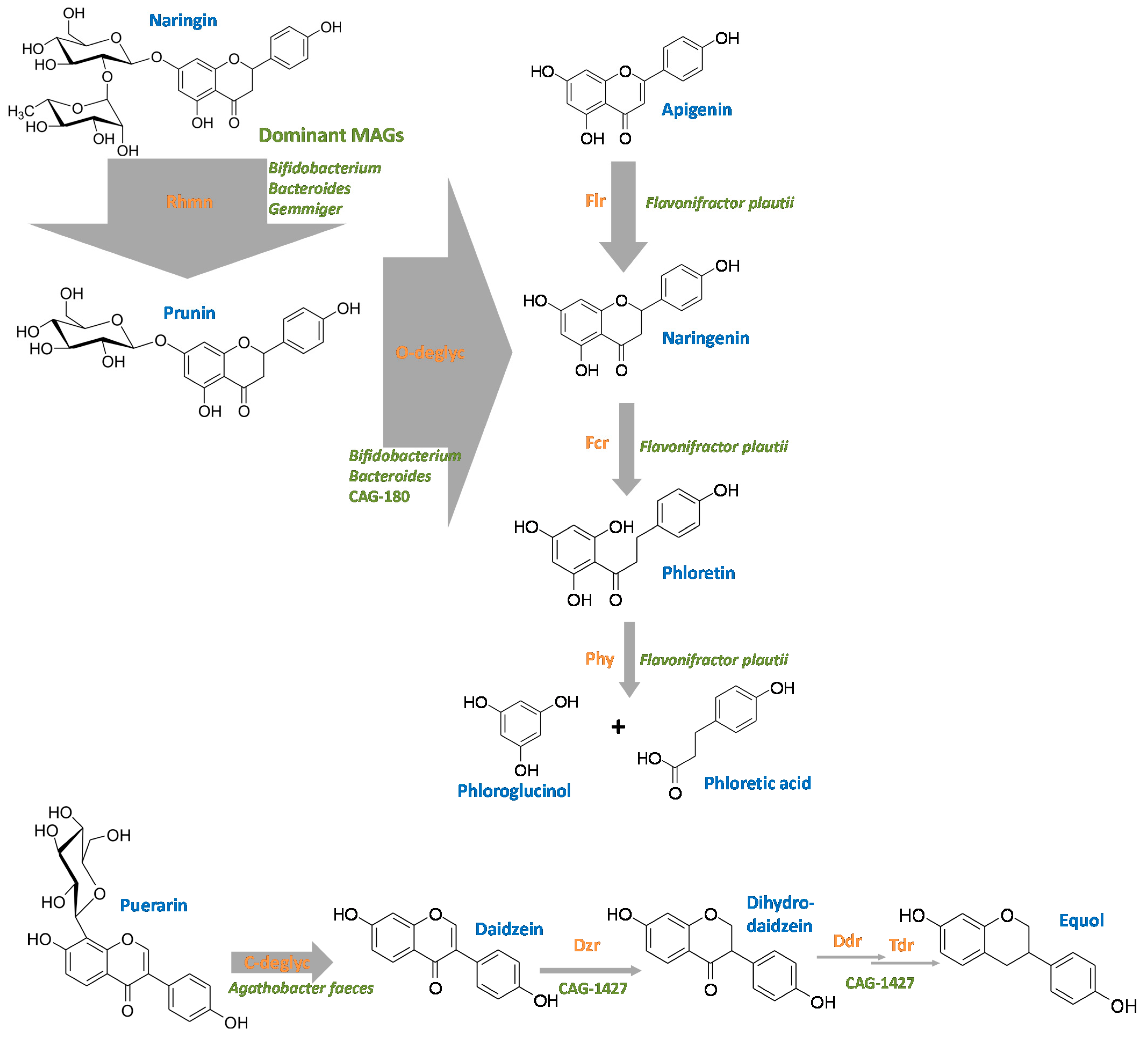
2.3. O-Deglycosylation of Flavonoids
2.4. Derhamnosylation of Flavonoids
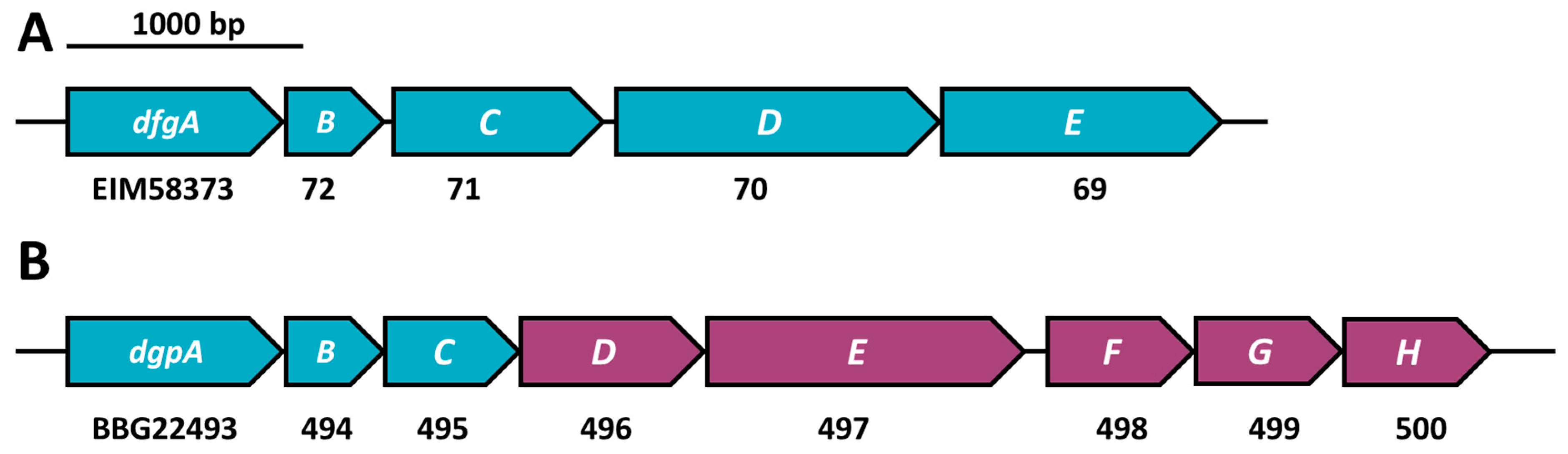
2.5. C-Deglycosylation of Flavonoids
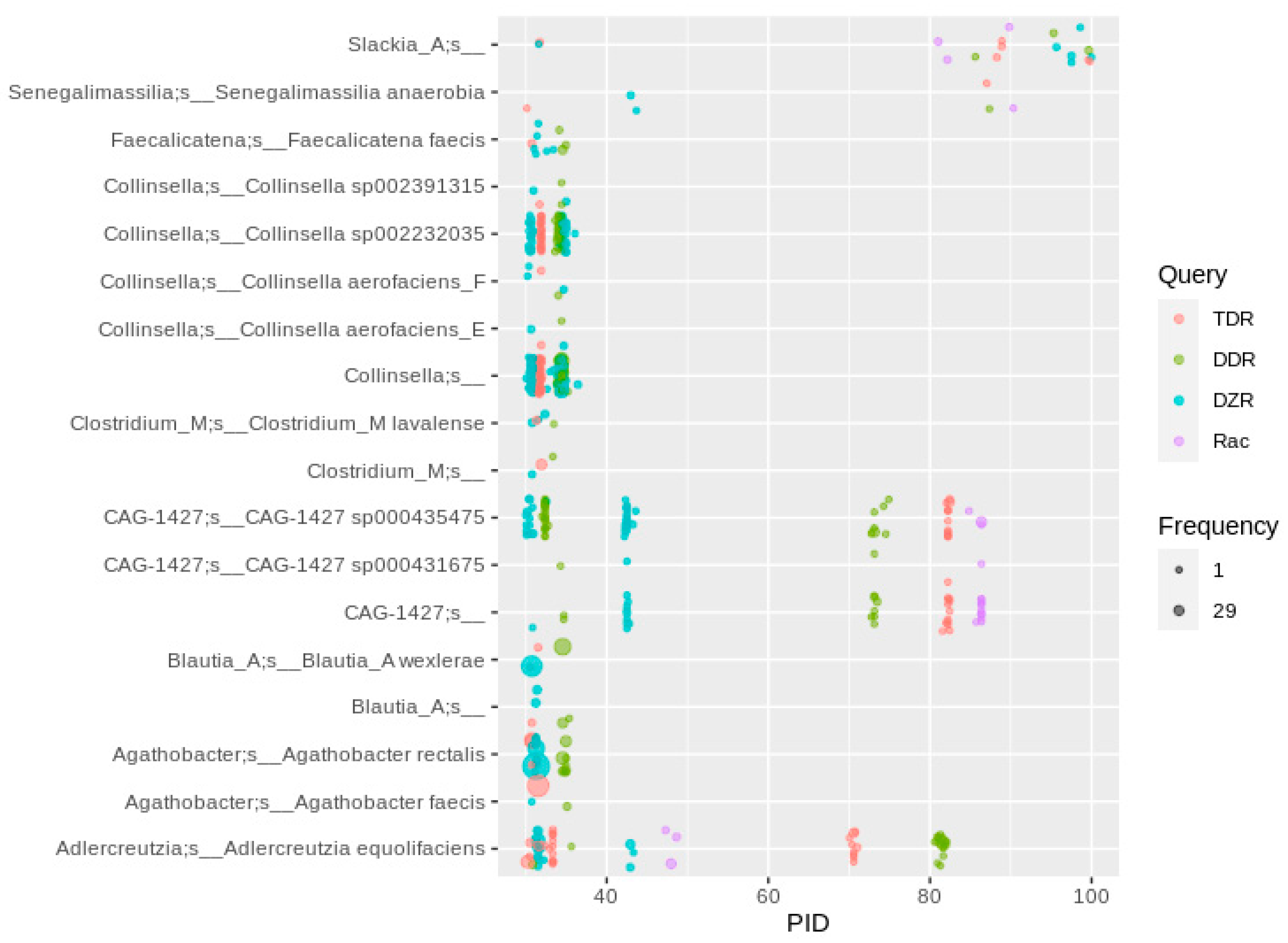
2.6. Daidzein-to-Equol Conversion
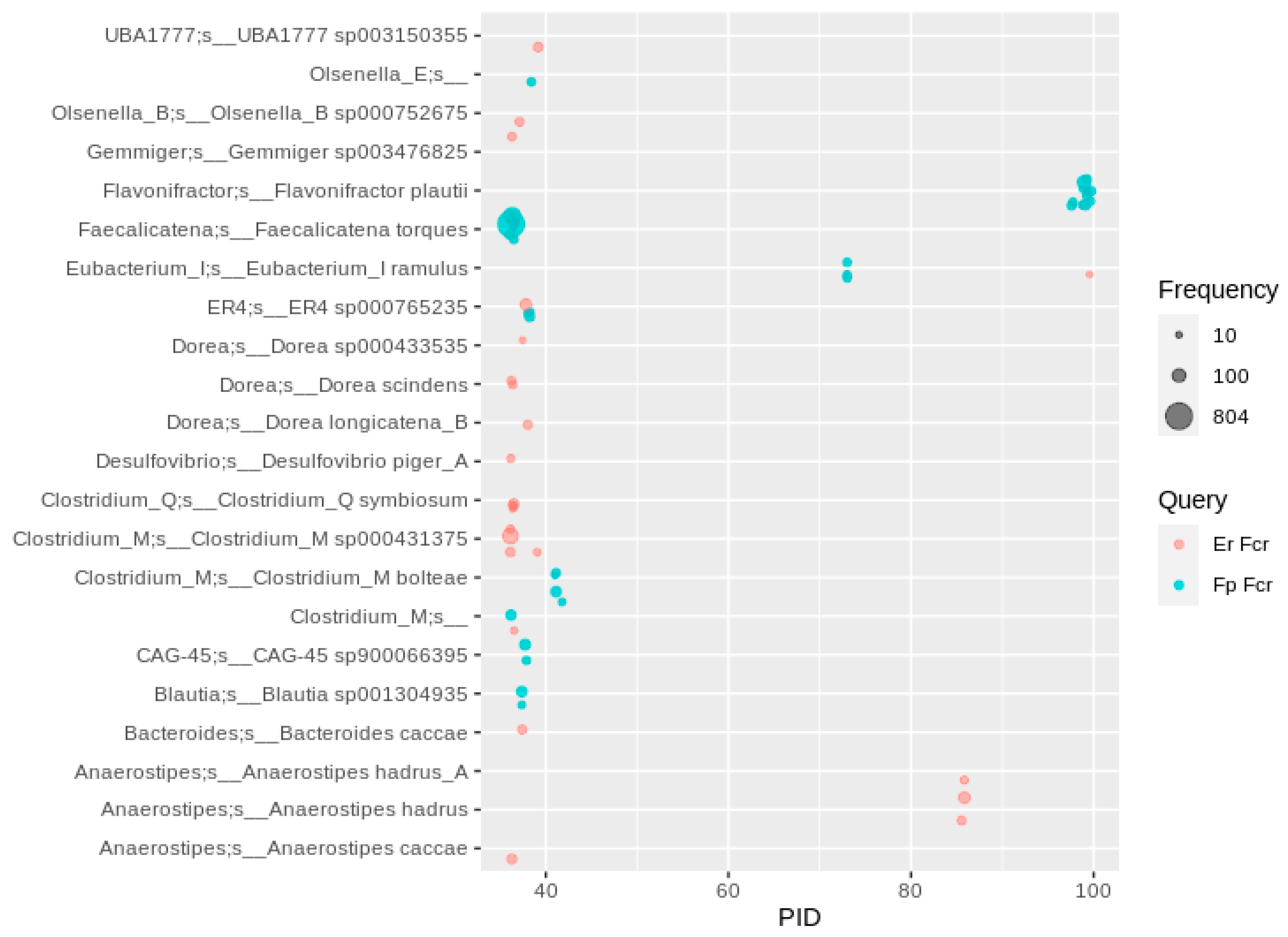
2.7. Flavone/Flavonol Reduction, Flavanone/Flavanonol Ring Cleavage, Chalcone Isomerization and Phloretin Hydrolysis
2.8. O-Demethylation of Flavonoids
3. Conclusions
4. Methods
4.1. Retrieval of Query Enzyme Sequences, MAGs Data and Set-Up of UHGP BLAST Database
4.2. BLAST Searches, Data Mangling and Quantification
4.3. MAGs Gene Cluster Analysis
4.4. Construction of Phylogenetic Trees
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Vargas, F.; Romecin, P.; Garcia-Guillen, A.I.; Wangesteen, R.; Vargas-Tendero, P.; Paredes, M.D.; Atucha, N.M.; Garcia-Estan, J. Flavonoids in Kidney Health and Disease. Front. Physiol. 2018, 9, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef] [Green Version]
- Pei, R.; Liu, X.; Bolling, B. Flavonoids and gut health. Curr. Opin. Biotechnol. 2020, 61, 153–159. [Google Scholar] [CrossRef]
- Farzaei, M.H.; Singh, A.K.; Kumar, R.; Croley, C.R.; Pandey, A.K.; Coy-Barrera, E.; Kumar Patra, J.; Das, G.; Kerry, R.G.; Annunziata, G.; et al. Targeting Inflammation by Flavonoids: Novel Therapeutic Strategy for Metabolic Disorders. Int. J. Mol. Sci. 2019, 20, 4957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caro-Ordieres, T.; Marin-Royo, G.; Opazo-Rios, L.; Jimenez-Castilla, L.; Moreno, J.A.; Gomez-Guerrero, C.; Egido, J. The Coming Age of Flavonoids in the Treatment of Diabetic Complications. J. Clin. Med. 2020, 9, 346. [Google Scholar] [CrossRef] [Green Version]
- Bondonno, N.P.; Dalgaard, F.; Kyro, C.; Murray, K.; Bondonno, C.P.; Lewis, J.R.; Croft, K.D.; Gislason, G.; Scalbert, A.; Cassidy, A.; et al. Flavonoid intake is associated with lower mortality in the Danish Diet Cancer and Health Cohort. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Bondonno, N.P.; Lewis, J.R.; Blekkenhorst, L.C.; Bondonno, C.P.; Shin, J.H.; Croft, K.D.; Woodman, R.J.; Wong, G.; Lim, W.H.; Gopinath, B.; et al. Association of flavonoids and flavonoid-rich foods with all-cause mortality: The Blue Mountains Eye Study. Clin. Nutr. 2020, 39, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, A.; Minihane, A.M. The role of metabolism (and the microbiome) in defining the clinical efficacy of dietary flavonoids. Am. J. Clin. Nutr. 2017, 105, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zheng, S.; Li, L.; Jiang, H. Metabolism of flavonoids in human: A comprehensive review. Curr. Drug. Metab. 2014, 15, 48–61. [Google Scholar] [CrossRef]
- Rodriguez-Mateos, A.; Vauzour, D.; Krueger, C.G.; Shanmuganayagam, D.; Reed, J.; Calani, L.; Mena, P.; Del Rio, D.; Crozier, A. Bioavailability, bioactivity and impact on health of dietary flavonoids and related compounds: An update. Arch. Toxicol. 2014, 88, 1803–1853. [Google Scholar] [CrossRef]
- Braune, A.; Blaut, M. Bacterial species involved in the conversion of dietary flavonoids in the human gut. Gut Microbes 2016, 7, 216–234. [Google Scholar] [CrossRef] [Green Version]
- Duda-Chodak, A.; Tarko, T.; Satora, P.; Sroka, P. Interaction of dietary compounds, especially polyphenols, with the intestinal microbiota: A review. Eur. J. Nutr. 2015, 54, 325–341. [Google Scholar] [CrossRef] [Green Version]
- Rafii, F. The role of colonic bacteria in the metabolism of the natural isoflavone daidzin to equol. Metabolites 2015, 5, 56–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayo, B.; Vazquez, L.; Florez, A.B. Equol: A Bacterial Metabolite from The Daidzein Isoflavone and Its Presumed Beneficial Health Effects. Nutrients 2019, 11, 2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guadamuro, L.; Florez, A.B.; Alegria, A.; Vazquez, L.; Mayo, B. Characterization of four beta-glucosidases acting on isoflavone-glycosides from Bifidobacterium pseudocatenulatum IPLA 36007. Food Res. Int. 2017, 100, 522–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youn, S.Y.; Park, M.S.; Ji, G.E. Identification of the beta-glucosidase gene from Bifidobacterium animalis subsp. lactis and its expression in B. bifidum BGN4. J. Microbiol. Biotechnol. 2012, 22, 1714–1723. [Google Scholar] [CrossRef]
- Byun, D.H.; Choi, H.J.; Lee, H.W.; Jeon, H.Y.; Choung, W.J.; Shim, J.H. Properties and applications of O-glycosidase from Bacteroides thetaiotaomicron that specifically hydrolyses isoflavone glycosides. Int. J. Food Sci. Technol. 2015, 50, 1405–1412. [Google Scholar] [CrossRef]
- Yadav, V.; Yadav, P.K.; Yadav, S.; Yadav, K.D.S. Alpha-L-Rhamnosidase: A review. Process. Biochem. 2010, 45, 1226–1235. [Google Scholar] [CrossRef]
- Bang, S.H.; Hyun, Y.J.; Shim, J.; Hong, S.W.; Kim, D.H. Metabolism of rutin and poncirin by human intestinal microbiota and cloning of their metabolizing alpha-L-rhamnosidase from Bifidobacterium dentium. J. Microbiol. Biotechnol. 2015, 25, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Zhang, B.L.; Xie, T.; Li, G.C.; Tuo, Y.; Xiang, Y.T. Biotransformation of rutin to isoquercitrin using recombinant alpha-L-rhamnosidase from Bifidobacterium breve. Biotechnol. Lett. 2015, 37, 1257–1264. [Google Scholar] [CrossRef]
- Wu, T.; Pei, J.; Ge, L.; Wang, Z.; Ding, G.; Xiao, W.; Zhao, L. Characterization of a alpha-l-rhamnosidase from Bacteroides thetaiotaomicron with high catalytic efficiency of epimedin C. Bioorg. Chem. 2018, 81, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Beekwilder, J.; Marcozzi, D.; Vecchi, S.; de Vos, R.; Janssen, P.; Francke, C.; van Hylckama Vlieg, J.; Hall, R.D. Characterization of Rhamnosidases from Lactobacillus plantarum and Lactobacillus acidophilus. Appl. Environ. Microbiol. 2009, 75, 3447–3454. [Google Scholar] [CrossRef] [Green Version]
- Avila, M.; Jaquet, M.; Moine, D.; Requena, T.; Pelaez, C.; Arigoni, F.; Jankovic, I. Physiological and biochemical characterization of the two alpha-L-rhamnosidases of Lactobacillus plantarum NCC245. Microbiology 2009, 155, 2739–2749. [Google Scholar] [CrossRef] [Green Version]
- Li, B.C.; Zhang, T.; Li, Y.Q.; Ding, G.B. Target Discovery of Novel alpha-L-Rhamnosidases from Human Fecal Metagenome and Application for Biotransformation of Natural Flavonoid Glycosides. Appl. Biochem. Biotechnol. 2019, 189, 1245–1261. [Google Scholar] [CrossRef]
- Braune, A.; Engst, W.; Blaut, M. Identification and functional expression of genes encoding flavonoid O- and C-glycosidases in intestinal bacteria. Environ. Microbiol. 2016, 18, 2117–2129. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Zhu, S.; Komatsu, K.; Hattori, M.; Iwashima, M. Expression and Characterization of the Human Intestinal Bacterial Enzyme Which Cleaves the C-Glycosidic Bond in 3″-Oxo-puerarin. Biol. Pharm. Bull. 2019, 42, 417–423. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Zhu, S.; Komatsu, K.; Hattori, M.; Iwashima, M. Deglycosylation of the Isoflavone C-Glucoside Puerarin by a Combination of Two Recombinant Bacterial Enzymes and 3-Oxo-Glucose. Appl. Environ. Microbiol. 2020, 86, e00607-20. [Google Scholar] [CrossRef] [PubMed]
- Han, J.T.; Zhang, S.P.; Jia, W.J.; Zhang, Z.; Wang, Y.; He, Y.X. Discovery and structural analysis of a phloretin hydrolase from the opportunistic human pathogen Mycobacterium abscessus. FEBS J. 2019, 286, 1959–1971. [Google Scholar] [CrossRef] [PubMed]
- Schoefer, L.; Braune, A.; Blaut, M. Cloning and expression of a phloretin hydrolase gene from Eubacterium ramulus and characterization of the recombinant enzyme. Appl. Environ. Microbiol. 2004, 70, 6131–6137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schröder, C.; Matthies, A.; Engst, W.; Blaut, M.; Braune, A. Identification and expression of genes involved in the conversion of daidzein and genistein by the equol-forming bacterium Slackia isoflavoniconvertens. Appl. Environ. Microbiol. 2013, 79, 3494–3502. [Google Scholar] [CrossRef] [Green Version]
- Shimada, Y.; Takahashi, M.; Miyazawa, N.; Abiru, Y.; Uchiyama, S.; Hishigaki, H. Identification of a novel dihydrodaidzein racemase essential for biosynthesis of equol from daidzein in Lactococcus sp. strain 20-92. Appl. Environ. Microbiol. 2012, 78, 4902–4907. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, H.; Moriyama, K.; Nomoto, K.; Akaza, H. Identification of an enzyme system for daidzein-to-equol conversion in Slackia sp. strain NATTS. Appl. Environ. Microbiol. 2012, 78, 1228–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, H.H.; Hsieh, Y.C.; Chen, Y.H.; Wang, H.Y.; Lu, C.Y.; Chen, C.J.; Li, Y.K. Three important amino acids control the regioselectivity of flavonoid glucosidation in glycosyltransferase-1 from Bacillus cereus. Appl. Microbiol. Biotechnol. 2016, 100, 8411–8424. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, T.; Kaufmann, F.; Diekert, G. Isolation and characterization of a veratrol:corrinoid protein methyl transferase from Acetobacterium dehalogenans. Arch. Microbiol. 2001, 175, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Studenik, S.; Vogel, M.; Diekert, G. Characterization of an O-demethylase of Desulfitobacterium hafniense DCB-2. J. Bacteriol. 2012, 194, 3317–3326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paraiso, I.L.; Plagmann, L.S.; Yang, L.; Zielke, R.; Gombart, A.F.; Maier, C.S.; Sikora, A.E.; Blakemore, P.R.; Stevens, J.F. Reductive Metabolism of Xanthohumol and 8-Prenylnaringenin by the Intestinal Bacterium Eubacterium ramulus. Mol. Nutr. Food Res. 2019, 63, e1800923. [Google Scholar] [CrossRef]
- Possemiers, S.; Rabot, S.; Espin, J.C.; Bruneau, A.; Philippe, C.; Gonzalez-Sarrias, A.; Heyerick, A.; Tomas-Barberan, F.A.; De Keukeleire, D.; Verstraete, W. Eubacterium limosum activates isoxanthohumol from hops (Humulus lupulus L.) into the potent phytoestrogen 8-prenylnaringenin in vitro and in rat intestine. J. Nutr. 2008, 138, 1310–1316. [Google Scholar] [CrossRef]
- Kawada, Y.; Yokoyama, S.; Yanase, E.; Niwa, T.; Suzuki, T. The production of S-equol from daidzein is associated with a cluster of three genes in Eggerthella sp. YY7918. Biosci. Microbiota Food Health 2016, 35, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Shimada, Y.; Yasuda, S.; Takahashi, M.; Hayashi, T.; Miyazawa, N.; Sato, I.; Abiru, Y.; Uchiyama, S.; Hishigaki, H. Cloning and expression of a novel NADP(H)-dependent daidzein reductase, an enzyme involved in the metabolism of daidzein, from equol-producing Lactococcus strain 20–92. Appl. Environ. Microbiol. 2010, 76, 5892–5901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iino, C.; Shimoyama, T.; Iino, K.; Yokoyama, Y.; Chinda, D.; Sakuraba, H.; Fukuda, S.; Nakaji, S. Daidzein Intake Is Associated with Equol Producing Status through an Increase in the Intestinal Bacteria Responsible for Equol Production. Nutrients 2019, 11, 433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.B.; Atkinson, C.; Frankenfeld, C.L.; Jokela, T.; Wahala, K.; Thomas, W.K.; Lampe, J.W. Prevalence of daidzein-metabolizing phenotypes differs between Caucasian and Korean American women and girls. J. Nutr. 2006, 136, 1347–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankenfeld, C.L.; Atkinson, C.; Thomas, W.K.; Gonzalez, A.; Jokela, T.; Wahala, K.; Schwartz, S.M.; Li, S.S.; Lampe, J.W. High concordance of daidzein-metabolizing phenotypes in individuals measured 1 to 3 years apart. Br. J. Nutr. 2005, 94, 873–876. [Google Scholar] [CrossRef] [PubMed]
- Frankenfeld, C.L. O-desmethylangolensin: The importance of equol’s lesser known cousin to human health. Adv. Nutr. 2011, 2, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Schneider, H.; Blaut, M. Anaerobic degradation of flavonoids by Eubacterium ramulus. Arch. Microbiol. 2000, 173, 71–75. [Google Scholar] [CrossRef]
- Braune, A.; Gutschow, M.; Blaut, M. An NADH-Dependent Reductase from Eubacterium ramulus Catalyzes the Stereospecific Heteroring Cleavage of Flavanones and Flavanonols. Appl. Environ. Microbiol. 2019, 85, e01233-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gall, M.; Thomsen, M.; Peters, C.; Pavlidis, I.V.; Jonczyk, P.; Grunert, P.P.; Beutel, S.; Scheper, T.; Gross, E.; Backes, M.; et al. Enzymatic conversion of flavonoids using bacterial chalcone isomerase and enoate reductase. Angew. Chem. Int. Ed. Engl. 2014, 53, 1439–1442. [Google Scholar] [CrossRef]
- Braune, A.; Engst, W.; Elsinghorst, P.W.; Furtmann, N.; Bajorath, J.; Gutschow, M.; Blaut, M. Chalcone Isomerase from Eubacterium ramulus Catalyzes the Ring Contraction of Flavanonols. J. Bacteriol. 2016, 198, 2965–2974. [Google Scholar] [CrossRef] [Green Version]
- Herles, C.; Braune, A.; Blaut, M. First bacterial chalcone isomerase isolated from Eubacterium ramulus. Arch. Microbiol. 2004, 181, 428–434. [Google Scholar] [CrossRef]
- Thomsen, M.; Tuukkanen, A.; Dickerhoff, J.; Palm, G.J.; Kratzat, H.; Svergun, D.I.; Weisz, K.; Bornscheuer, U.T.; Hinrichs, W. Structure and catalytic mechanism of the evolutionarily unique bacterial chalcone isomerase. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 907–917. [Google Scholar] [CrossRef]
- Yang, G.; Hong, S.; Yang, P.; Sun, Y.; Wang, Y.; Zhang, P.; Jiang, W.; Gu, Y. Discovery of an ene-reductase for initiating flavone and flavonol catabolism in gut bacteria. Nat. Commun. 2021, 12, 790. [Google Scholar] [CrossRef]
- Shimada, Y.; Takahashi, M.; Miyazawa, N.; Ohtani, T.; Abiru, Y.; Uchiyama, S.; Hishigaki, H. Identification of two novel reductases involved in equol biosynthesis in Lactococcus strain 20–92. J. Mol. Microbiol. Biotechnol. 2011, 21, 160–172. [Google Scholar] [CrossRef]
- Braune, A.; Blaut, M. Evaluation of inter-individual differences in gut bacterial isoflavone bioactivation in humans by PCR-based targeting of genes involved in equol formation. J. Appl. Microbiol. 2018, 124, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, H.B.; Almeida, M.; Juncker, A.S.; Rasmussen, S.; Li, J.H.; Sunagawa, S.; Plichta, D.R.; Gautier, L.; Pedersen, A.G.; Le Chatelier, E.; et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 2014, 32, 822–828. [Google Scholar] [CrossRef]
- Almeida, A.; Mitchell, A.L.; Boland, M.; Forster, S.C.; Gloor, G.B.; Tarkowska, A.; Lawley, T.D.; Finn, R.D. A new genomic blueprint of the human gut microbiota. Nature 2019, 568, 499–504. [Google Scholar] [CrossRef] [Green Version]
- Nayfach, S.; Shi, Z.J.; Seshadri, R.; Pollard, K.S.; Kyrpides, N.C. New insights from uncultivated genomes of the global human gut microbiome. Nature 2019, 568, 505–510. [Google Scholar] [CrossRef] [Green Version]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; Ghensi, P.; et al. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 2019, 176, 649–662.e620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hur, H.G.; Lay, J.O., Jr.; Beger, R.D.; Freeman, J.P.; Rafii, F. Isolation of human intestinal bacteria metabolizing the natural isoflavone glycosides daidzin and genistin. Arch. Microbiol. 2000, 174, 422–428. [Google Scholar] [CrossRef]
- Mattarelli, P.; Bonaparte, C.; Pot, B.; Biavati, B. Proposal to reclassify the three biotypes of Bifidobacterium longum as three subspecies: Bifidobacterium longum subsp. longum subsp. nov., Bifidobacterium longum subsp. infantis comb. nov. and Bifidobacterium longum subsp. suis comb. nov. Int. J. Syst. Evol. Microbiol. 2008, 58, 767–772. [Google Scholar] [CrossRef] [Green Version]
- Ivey, K.L.; Chan, A.T.; Izard, J.; Cassidy, A.; Rogers, G.B.; Rimm, E.B. Role of Dietary Flavonoid Compounds in Driving Patterns of Microbial Community Assembly. mBio 2019, 10, e01205–e01219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, A.; Nayfach, S.; Boland, M.; Strozzi, F.; Beracochea, M.; Shi, Z.J.; Pollard, K.S.; Sakharova, E.; Parks, D.H.; Hugenholtz, P.; et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 2021, 39, 105–114. [Google Scholar] [CrossRef]
- Shimojo, Y.; Ozawa, Y.; Toda, T.; Igami, K.; Shimizu, T. Probiotic Lactobacillus paracasei A221 improves the functionality and bioavailability of kaempferol-glucoside in kale by its glucosidase activity. Sci. Rep. 2018, 8, 9239. [Google Scholar] [CrossRef]
- Gaya, P.; Peiroten, A.; Alvarez, I.; Medina, M.; Landete, J.M. Productionof the bioactive isoflavone O-desmethylangolensin by Enterococcus faecium INIA P553 with high efficiency. J. Funct. Foods 2018, 40, 180–186. [Google Scholar] [CrossRef]
- Gaya, P.; Peiroten, A.; Medina, M.; Landete, J.M. Isoflavone metabolism by a collection of lactic acid bacteria and bifidobacteria with biotechnological interest. Int. J. Food Sci. Nutr. 2016, 67, 117–124. [Google Scholar] [CrossRef]
- Kim, C.C.; Healey, G.R.; Kelly, W.J.; Patchett, M.L.; Jordens, Z.; Tannock, G.W.; Sims, I.M.; Bell, T.J.; Hedderley, D.; Henrissat, B.; et al. Genomic insights from Monoglobus pectinilyticus: A pectin-degrading specialist bacterium in the human colon. ISME J. 2019, 13, 1437–1456. [Google Scholar] [CrossRef]
- Kim, C.C.; Kelly, W.J.; Patchett, M.L.; Tannock, G.W.; Jordens, Z.; Stoklosinski, H.M.; Taylor, J.W.; Sims, I.M.; Bell, T.J.; Rosendale, D.I. Monoglobus pectinilyticus gen. nov., sp. nov., a pectinolytic bacterium isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2017, 67, 4992–4998. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Nakatsu, C.H.; Armstrong, A.; Clavijo, A.P.; Martin, B.R.; Barnes, S.; Weaver, C.M. Fecal bacterial community changes associated with isoflavone metabolites in postmenopausal women after soy bar consumption. PLoS ONE 2014, 9, e108924. [Google Scholar] [CrossRef]
- Guadamuro, L.; Dohrmann, A.B.; Tebbe, C.C.; Mayo, B.; Delgado, S. Bacterial communities and metabolic activity of faecal cultures from equol producer and non-producer menopausal women under treatment with soy isoflavones. BMC Microbiol. 2017, 17, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guadamuro, L.; Azcarate-Peril, M.A.; Tojo, R.; Mayo, B.; Delgado, S. Use of high throughput amplicon sequencing and ethidium monoazide dye to track microbiota changes in an equol-producing menopausal woman receiving a long-term isoflavones treatment. AIMS Microbiol. 2019, 5, 102–116. [Google Scholar] [CrossRef]
- Schoefer, L.; Mohan, R.; Schwiertz, A.; Braune, A.; Blaut, M. Anaerobic degradation of flavonoids by Clostridium orbiscindens. Appl. Environ. Microbiol. 2003, 69, 5849–5854. [Google Scholar] [CrossRef] [Green Version]
- Allen-Vercoe, E.; Daigneault, M.; White, A.; Panaccione, R.; Duncan, S.H.; Flint, H.J.; O’Neal, L.; Lawson, P.A. Anaerostipes hadrus comb. nov., a dominant species within the human colonic microbiota; reclassification of Eubacterium hadrum Moore et al. 1976. Anaerobe 2012, 18, 523–529. [Google Scholar] [CrossRef]
- Gupta, A.; Dhakan, D.B.; Maji, A.; Saxena, R.; Prasoodanan, P.K.V.; Mahajan, S.; Pulikkan, J.; Kurian, J.; Gomez, A.M.; Scaria, J.; et al. Association of Flavonifractor plautii, a Flavonoid-Degrading Bacterium, with the Gut Microbiome of Colorectal Cancer Patients in India. mSystems 2019, 4, e00438-19. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag: New York, NY, USA; Available online: https://ggplot2.tidyverse.org (accessed on 1 June 2021).
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Steenwyk, J.L.; Buida, T.J., 3rd; Li, Y.; Shen, X.X.; Rokas, A. ClipKIT: A multiple sequence alignment trimming software for accurate phylogenomic inference. PLoS Biol. 2020, 18, e3001007. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, 293–296. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession No. | Enzyme | Reaction | Source Bacterium | Reference |
|---|---|---|---|---|
| KEF29323.1 | Beta-glucosidase BpBluA | 7-O-deglycosylation of isoflavones | Bifidobacterium pseudocatenulatum IPLA36007 | [15] |
| KEF27912.1 | Beta-glucosidase BpGluB | 7-O-deglycosylation of isoflavones | Bifidobacterium pseudocatenulatum IPLA36007 | [15] |
| KEF28010.1 | Beta-glucosidase BpGluD | 7-O-deglycosylation of isoflavones | Bifidobacterium pseudocatenulatum IPLA36007 | [15] |
| KEF28001.1 | Beta-glucosidase BpGluE | 7-O-deglycosylation of isoflavones | Bifidobacterium pseudocatenulatum IPLA36007 | [15] |
| AFS33105.1 | Beta-glucosidase Bbg572 | 7-O-deglycosylation of isoflavones, 3-/4-/7-O-deglycosylation of quercetin glycosides | Bifidobacterium animalis ssp. lactis SH5 | [16] |
| AAO76887.1 | Beta-glucosidase BtGlu | 7-O-deglycosylation of isoflavones | Bacteroides thetaiotaomicron VPI-5482 | [17] |
| AGS77942.1 | Alpha-L-Rhamnosidase BdR | Derhamnosylation of 1→2 and 1→6 O-glycosidic bond of rutinosylated flavonoids | Bifidobacterium dentium K13 | [19] |
| AHJ22585.1 | Alpha-L-Rhamnosidase BbR | Derhamnosylation of 1→6 O-glycosidic bond of rutin | Bifidobacterium breve 689b | [20] |
| WP_011107561 | Alpha-L-Rhamnosidase BtR | Derhamnosylation of 1→2 O-glycosidic bonds of rutinosylated flavonoids | Bacteroides thetaiotaomicron VPI-5482 | [21] |
| QBM20340.1 | Alpha-L-Rhamnosidase HFM-RhaA (MGR1) | Derhamnosylation of 1→2 and 1→6 O-glycosidic bond of rutinosylated flavonoids | Human gut metagenome | [24] |
| QBM20341.1 | Alpha-L-Rhamnosidase HFM-RhaB (MGR2) | Derhamnosylation of 1→2 O-glycosidic bond of rutinosylated flavonoids | Human gut metagenome | [24] |
| QBM20342.1 | Alpha-L-Rhamnosidase HFM-RhaC (MGR3) | Derhamnosylation of 1→2 and 1→6 O-glycosidic bond of rutinosylated flavonoids | Human gut metagenome | [24] |
| CCC80440.1 | Alpha-L-Rhamnosidase LpR1 | Derhamnosylation of 1→6 O-glycosidic bond of rutinosylated flavonoids | Lactobacillus plantarum WCFS1 * | [22] |
| CCC80442.1 | Alpha-L-Rhamnosidase LpR2 | Derhamnosylation of 1→6 O-glycosidic bond of rutinosylated flavonoids | Lactobacillus plantarum WCFS1 * | [22] |
| AAV43293.1 | Alpha-L-Rhamnosidase LaR | Derhamnosylation of 1→2 and 1→6 O-glycosidic bond of rutinosylated flavonoids | Lactobacillus acidophilus NCFM | [22] |
| EUA80835.1 | Phloretin hydrolase | Phloretin hydrolysis | Mycobacteroides abscessus ssp. bolletii 103 | [28] |
| AAQ12341.1 | Phloretin hydrolase | Phloretin hydrolysis | Eubacterium ramulus DSM 16296 | [29] |
| AKC35075.1 AKC35076.1 | DfgCD | 7-O-deglycosylation of isoflavones and flavones | Catenibacillus scindens DSM 106146 | [25] |
| EIM58373.1 EIM58372.1 EIM58371.1 EIM58370.1 EIM58369.1 | DfgABCDE | C-deglycosylation of flavones | Eubacterium cellulosolvens ATCC 43171 | [25] |
| BBG22493.1 BBG22494.1 BBG22495.1 | DgpABC | C-deglycosylation of isoflavones | Dorea sp. strain PUE | [26] |
| ANU40626.1 | Flavone/flavonol reductase (Flr) | C-ring double bond reduction | Flavonifractor plautii DSM 6740 | [50] |
| ADK16070.1 | Flavone/flavonol reductase (Flr) | C-ring double bond reduction | Clostridium ljungdahlii DSM 13528 | [50] |
| AGS82961.1 | Flavanone/flavanonol-cleaving reductase (Fcr) | Reductive ring cleavage of flavanones and flavanonols | Eubacterium ramulus DSM 16296 | [45] |
| WP_154024723.1 | Flavanone/flavanonol-cleaving reductase (Fcr) | Reductive ring cleavage of flavonones and flavanonols | Flavonifractor plautii DSM 6740 | [45] |
| AIS36173.1 | Chalcone isomerase (CHI) | Isomerization of chalcones and flavanonols | Eubacterium ramulus DSM 16296 | [46,47,49] |
| EHM54434.1 | Chalcone isomerase (CHI) | Isomerization of chalcones | Flavonifractor plautii ATCC 29863 (formerly Clostridium orbiscindens) | [46,47,49] |
| AFV15450.1 | Tetrahydrodaidzein reductase (TDR) | Reduction of tetrahydrodaidzein | Slackia isoflavoniconvertens DSM 22006 | [30] |
| AFV15451.1 | Dihydrodaidzein reductase (DDR) | Reduction of dihydrodaidzein | Slackia isoflavoniconvertens DSM 22006 | [30] |
| AFV15453.1 | Daidzein reductase (DZR) | Reduction of daidzein | Slackia isoflavoniconvertens DSM 22006 | [30] |
| BAL46928.1 | Tetrahydrodaidzein reductase (TDR) | Reduction of tetrahydrodaidzein | Slackia sp. NATTS | [32] |
| BAL46929.1 | Dihydrodaidzein reductase | Reduction of dihidydrodaidzein | Slackia sp. NATTS | [32] |
| BAL46930.1 | Daidzein reductase | Reduction of daidzein | Slackia sp. NATTS | [32] |
| BAM25050.1 | Dihydrodaidzein Racemase | Racemization of dihydrodaidzein | Lactococcus garviae 20-92 | [31] |
| BAJ72744.1 | Tetrahydrodaidzein reductase | Reduction of tetrahydrodaidzein | Lactococcus garviae 20-92 | [51] |
| BAJ72745.1 | Dihydrodaidzein reductase | Reduction of didydrodaidzein | Lactococcus garviae 20-92 | [51] |
| BAJ22678.1 | Daidzein reductase | Reduction of daidzein | Lactococcus garviae 20-92 | [39] |
| WP_013979960.1 | Tetrahydrodaidzein reductase | Reduction of tetrahydrodaidzein | Eggerthella sp. YY7918 | [38] |
| WP_013979959.1 | Dihydrodaidzein reductase | Reduction of dihidydrodaidzein | Eggerthella sp. YY7918 | [38] |
| WP_013979957.1 | Daidzein reductase | Reduction of daidzein | Eggerthella sp. YY7918 | [38] |
| ANI69959.1 | O demethylase (ODem) activating enzyme (AE) | Activation of CP | Eubacterium limosum ZL-II | [33] |
| ANI69960.1 | ODem Methyltransferase (MT) 1 | O-Demethylation of secoisolariciresinol | Eubacterium limosum ZL-II | [33] |
| ANI69961.1 | ODem Corrinoid protein (CP) | Methyl transfer | Eubacterium limosum ZL-II | [33] |
| ANI69962.1 | ODem MT2 | Methyl transfer to CP | Eubacterium limosum ZL-II | [33] |
| Species | Flavonoid Class | Prevalence |
|---|---|---|
| O-Deglycosylation | ||
| Bacteroides ovatus | Flavonols | 3.1 |
| Bacteroides uniformis | Flavonols, flavanones | 18.5 |
| Bacteroides thetaiotaomicron1 | Isoflavones | 1.9 |
| Bifidobacterium adolescentis | Flavanones, isoflavones | 7.4 |
| Bifidobacterium angulatum | Isoflavones | 0.3 |
| Bifidobacterium animalis | Anthocyanidins, isoflavones | 0.3 |
| Bifidobacterium bifidum | Flavanones, isoflavones | 3.3 |
| Bifidobacterium breve | Flavonols, isoflavones, flavanones | 1.5 |
| Bifidobacterium catenulatum | Flavonols, isoflavones, flavanones | 0.8 |
| Bifidobacterium dentium | Flavonols, flavanones | 0.3 |
| Bifidobacterium infantis | Flavonols, isoflavones, flavanones | 8.4 |
| Bifidobacterium longum | Isoflavones | 0 |
| Bifidobacterium pseudocatenulatum | Flavonols, flavanones | 3.5 |
| Bifidobacterium pseudolongum | Isoflavones | <0.1 |
| Blautia producta (MRG-PMF1, 99%) | Flavonols, flavones, flavanones, isoflavones | <0.1 |
| Catenibacillus scindens | Flavones, isoflavones | n.d. (<0.1) |
| Enterobacter cloacae | Flavanones | 0.1 |
| Enterococcus avium | Flavonols | 0.1 |
| Enterococcus casseliflavus | Flavonols | <0.1 |
| Enterococcus faecalis | Flavanones | 2.9 |
| Eubacterium cellulosolvens | Flavones, isoflavones | 0 |
| Eubacterium ramulus | Flavonols, flavones, dihydrochalcones, isoflavones | 0.5 |
| Escherichia sp. HGH21 (99% to E. coli (MIDI *) | Isoflavones | (28) |
| Escherichia sp. 4 (E. fergusonii) | Flavones | <0.1 |
| Parabacteroides distasonis | Flavonols, flavanones | 14.5 |
| Lactobacillus acidophilus | Flavanones | <0.1 |
| Lactobacillus buchneri | Flavanones | <0.1 |
| Lactobacillus casei | Flavanones, anthocyanidins | 0 |
| Lactobacillus leichmanii | Flavanones | n.d. |
| Lactobacillus plantarum | Flavanones, anthocyanidins | 0.2 |
| Lactococcus lactis | Flavonols, isoflavones, flavanones | 0.2 |
| Lactococcus paracasei [62] | Isoflavones, flavonols | 0.2 |
| C-Deglycosylation | ||
| Eubacterium cellulosolvens | Flavones, isoflavones | 0 |
| Strain PUE (1346 nt), Dorea longicatena (98%) | Isoflavones | n.d. (2.9) |
| Catenibacillus scindens | Flavones, isoflavones | n.d. |
| Enterococcus casseliflavus (sp. 45, 99%) | Flavones | <0.1 |
| Enterococcus faecium (MRG-IFC-2, 99%) | Isoflavones | 1.8 |
| Lactococcus lactis (MRG-IFC-1, 99%) | Isoflavones | 0.2 |
| C-Ring cleavage | ||
| Flavonifractor plautii (formerly C. orbiscindens) | Flavonols/flavanonols, flavones/flavanones | 2.1 |
| Catenibacillus scindens | Flavones | n.d. |
| Eubacterium ramulus | Flavonols/flavanonols, flavones/flavanones, isoflavones | 0.5 1 |
| Clostridium butyricum | Flavanones | 0.3 |
| Lactobacillus plantarum | Flavan-3-ols | 0.2 |
| Strain SY8519 (Eubacterium_I sp000270305) | Isoflavones | n.d. |
| Adlercreutzia equolifaciens | Flavan-3-ols | 0.8 |
| Eggerthella lenta | Flavanonols (SDG-2), flavan-3-ols | 0.9 |
| Enterococcus faecium [63] | Isoflavones | 1.8 |
| Reduction | ||
| Bifidobacterium animalis | Isoflavones | 0.3 |
| Bifidobacterium longum | Isoflavones | 0 |
| Bifidobacterium pseudolongum | Isoflavones | <0.1 |
| Adlercreutzia equolifaciens | Isoflavones | 0.8 |
| Lactococcus garvieae | Isoflavones | <0.1 |
| Slackia sp. NATTS | Isoflavones | n.d. |
| Slackia isoflavoniconvertens | Isoflavones | n.d. |
| Lactobacillus rhamnosus [64] | Isoflavones | 0.4 |
| Enterococcus faecalis [64] | Isoflavones | 2.9 |
| Enterococcus faecium [63] | Isoflavones | 1.8 |
| Dehydroxylation | ||
| Eschericha sp. 4 (E. fergusonii) | Flavones | <0.1 |
| Adlercreutzia equolifaciens | Flavan-3-ols | 0.8 |
| Eggerthella lenta (SDG-2, 99%) | Flavan-3-ols | 0.9 |
| O-Demethylation | ||
| Blautia producta (MRG-PMF1, 99%) | Flavonols, flavones, flavanones, isoflavones | <0.1 |
| Eubacterium limosum (strains DSM 20543T and LMG P23546) | Isoflavones, flavanones | <0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goris, T.; Cuadrat, R.R.C.; Braune, A. Flavonoid-Modifying Capabilities of the Human Gut Microbiome—An In Silico Study. Nutrients 2021, 13, 2688. https://doi.org/10.3390/nu13082688
Goris T, Cuadrat RRC, Braune A. Flavonoid-Modifying Capabilities of the Human Gut Microbiome—An In Silico Study. Nutrients. 2021; 13(8):2688. https://doi.org/10.3390/nu13082688
Chicago/Turabian StyleGoris, Tobias, Rafael R. C. Cuadrat, and Annett Braune. 2021. "Flavonoid-Modifying Capabilities of the Human Gut Microbiome—An In Silico Study" Nutrients 13, no. 8: 2688. https://doi.org/10.3390/nu13082688
APA StyleGoris, T., Cuadrat, R. R. C., & Braune, A. (2021). Flavonoid-Modifying Capabilities of the Human Gut Microbiome—An In Silico Study. Nutrients, 13(8), 2688. https://doi.org/10.3390/nu13082688