
Alcohol’s Impact on the Gut and Liver


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
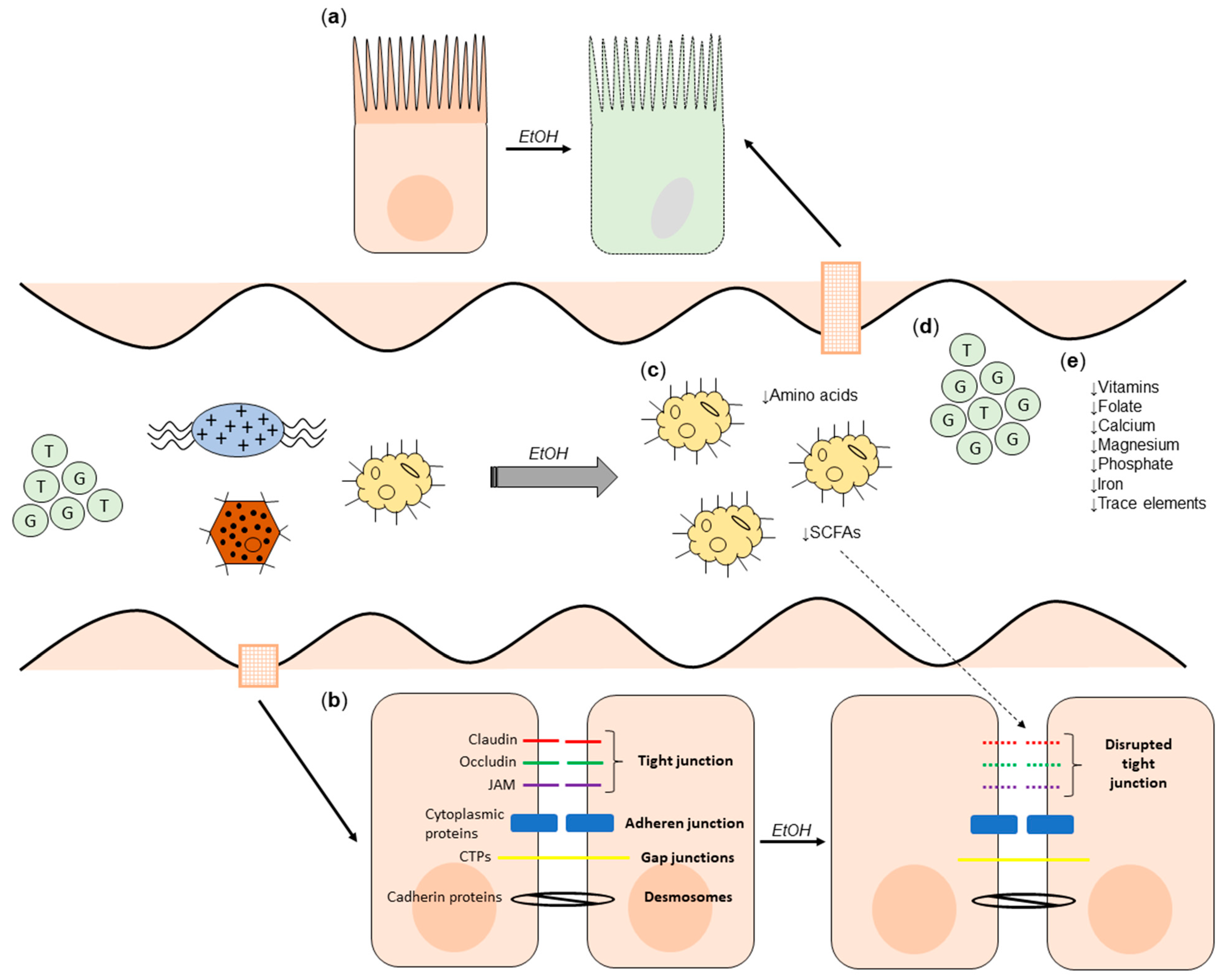
2. Alcohol and the Gut
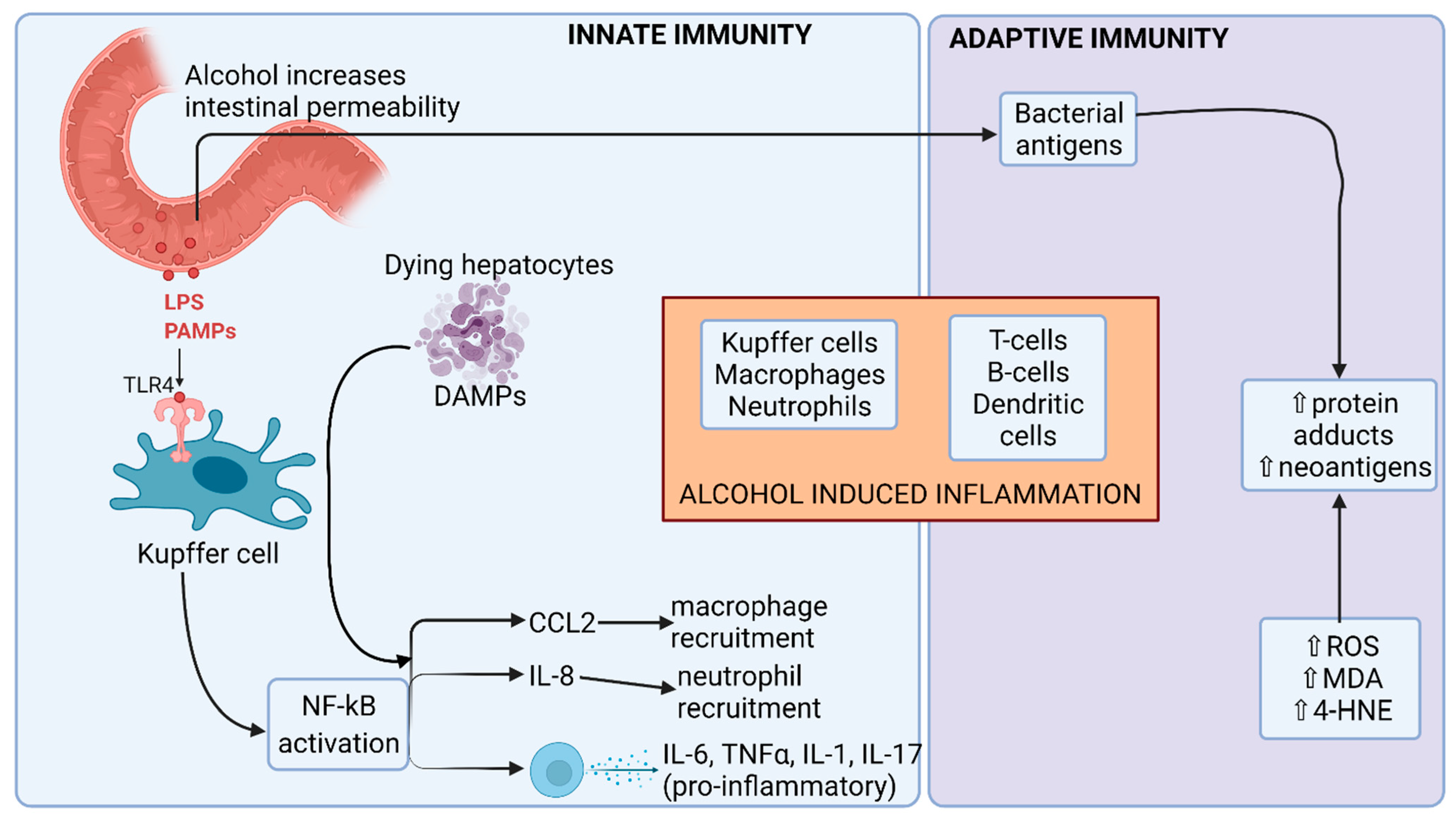
2.1. The Effect of Alcohol on Microbial Composition and Gut Barrier Function
2.2. The Effect of Alcohol on the Metabolome
2.3. The Effect of Alcohol Consumption on Nutritional Status
3. Alcohol and the Liver
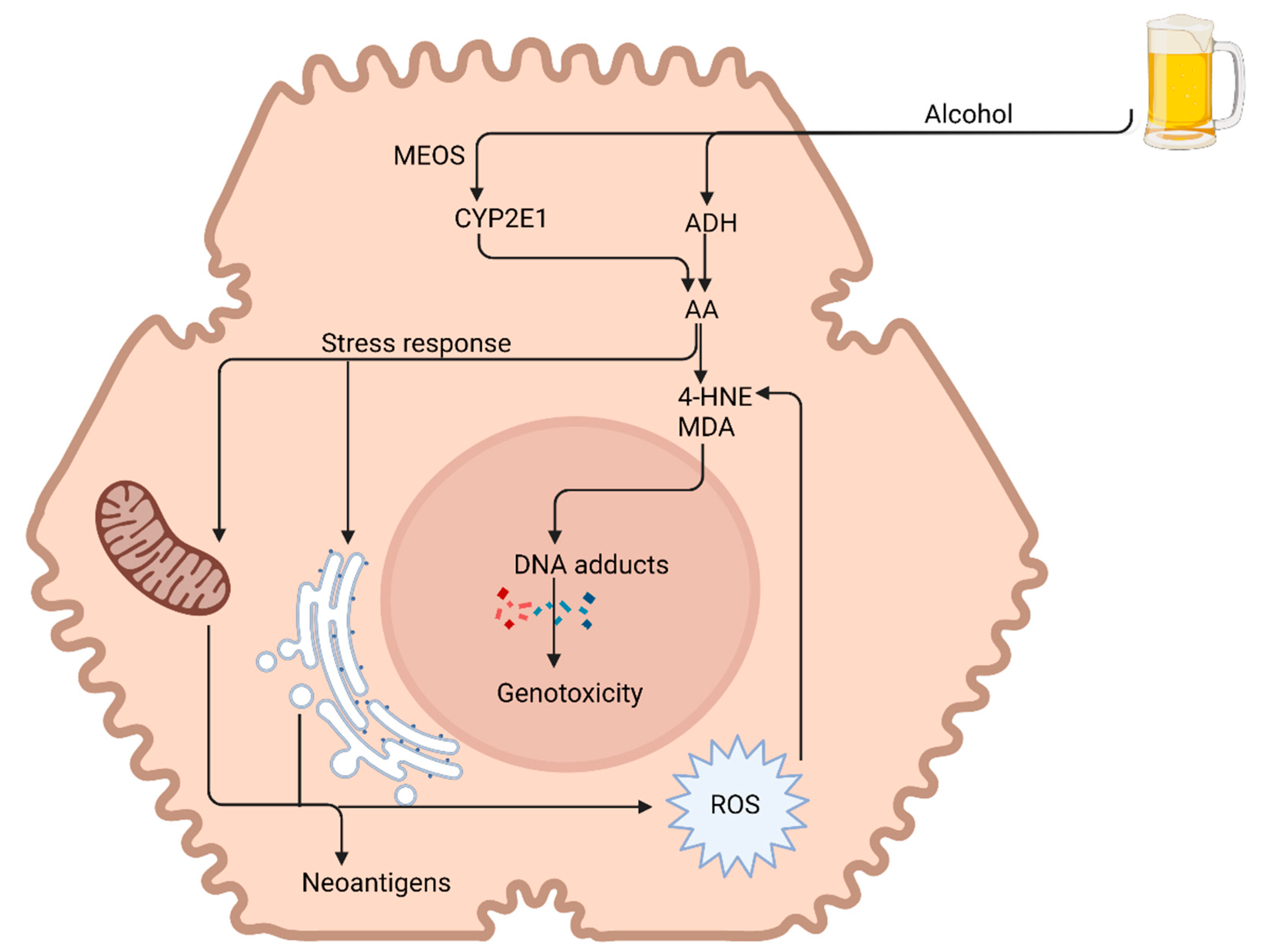
3.1. Alcohol Metabolism
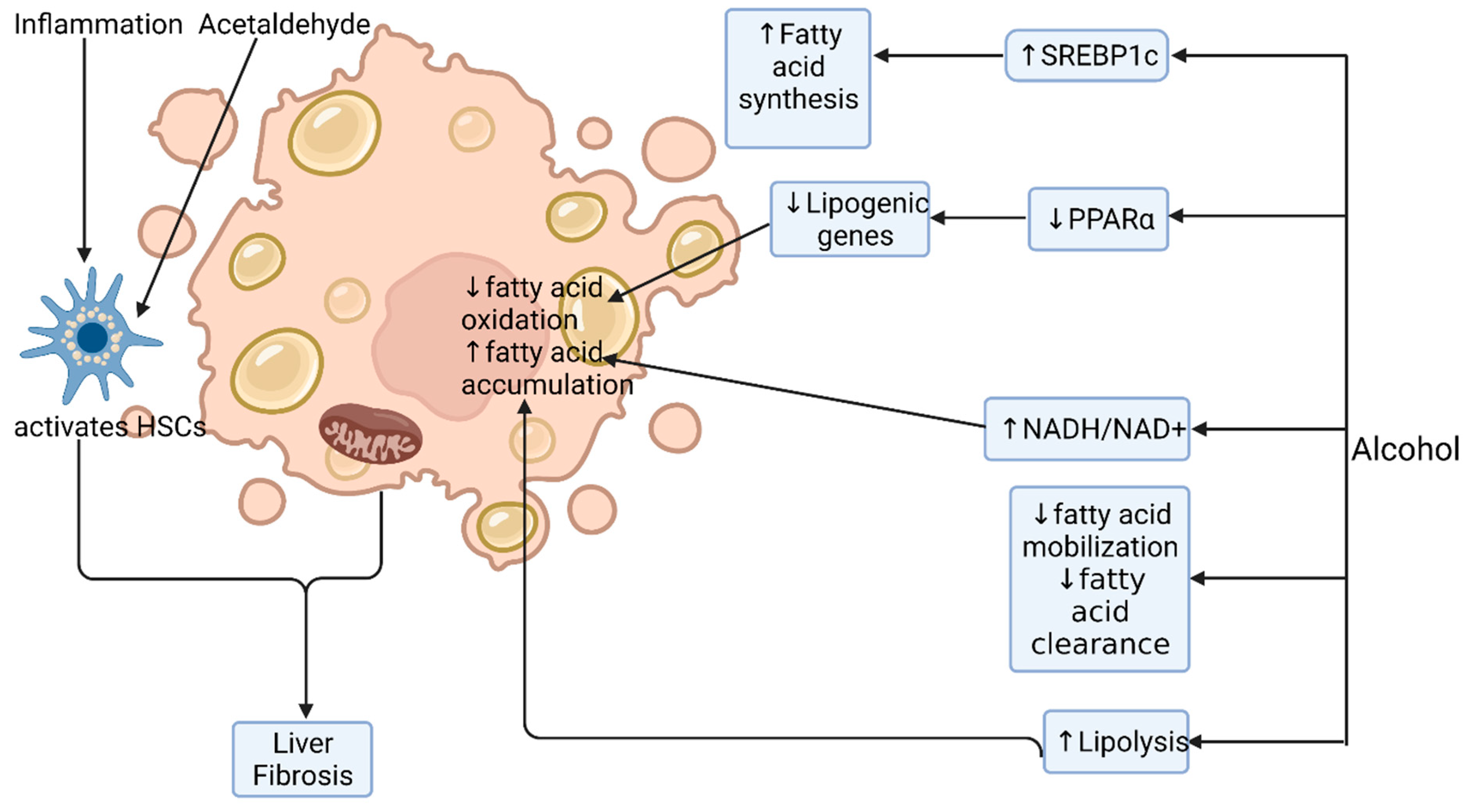
3.2. Alcohol-Related Steatosis
3.3. Alcoholic Steatohepatitis
3.4. Alcohol-Induced Fibrosis and Cirrhosis
3.5. Hepatocellular Carcinoma
4. Research Priorities and Future Perspectives
4.1. Microbiome
4.2. Short-Chain Fatty Acids
4.3. Dietary Manipulation
4.4. Micronutrient Supplementation
4.5. Immune Dysregulation
4.6. Repurposing of Existing Therapies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Paton, A. Alcohol in the body. BMJ 2005, 330, 85–87. [Google Scholar] [CrossRef]
- GBD Alcohol Drug Use Collaborators. The global burden of disease attributable to alcohol and drug use in 195 countries and territories, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Psychiatry 2018, 5, 987–1012. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Alcohol Factsheet. Available online: https://www.who.int/en/news-room/fact-sheets/detail/alcohol (accessed on 20 August 2021).
- GBD Causes of Death Collaborators. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: A systematic analysis for the global burden of disease study 2016. Lancet 2017, 390, 1151–1210. [Google Scholar] [CrossRef] [Green Version]
- Global Burden of Disease Liver Cancer Collaboration; Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; et al. The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level: Results from the global burden of disease study 2015. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar] [CrossRef]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamran, U.; Towey, J.; Khanna, A.; Chauhan, A.; Rajoriya, N.; Holt, A. Nutrition in alcohol-related liver disease: Physiopathology and management. World J. Gastroenterol. WJG 2020, 26, 2916–2930. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Relationships between nutrition, alcohol use, and liver disease. Alcohol Res. Health 2003, 27, 220–231. [Google Scholar] [PubMed]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, J.S. Alcohol, liver disease and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Engen, P.A.; Green, S.J.; Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. The gastrointestinal microbiome: Alcohol effects on the composition of intestinal microbiota. Alcohol Res. 2015, 37, 223–236. [Google Scholar] [PubMed]
- Zhong, W.; Zhou, Z. Alterations of the gut microbiome and metabolome in alcoholic liver disease. World J. Gastrointest. Pathophysiol. 2014, 5, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Litwinowicz, K.; Choroszy, M.; Waszczuk, E. Changes in the composition of the human intestinal microbiome in alcohol use disorder: A systematic review. Am. J. Drug Alcohol Abus. 2020, 46, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Keshavarzian, A.; Farhadi, A.; Forsyth, C.B.; Rangan, J.; Jakate, S.; Shaikh, M.; Banan, A.; Fields, J.Z. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J. Hepatol. 2009, 50, 538–547. [Google Scholar] [CrossRef] [Green Version]
- Shukla, P.K.; Meena, A.S.; Rao, V.; Rao, R.G.; Balazs, L.; Rao, R. Human defensin-5 blocks ethanol and colitis-induced dysbiosis, tight junction disruption and inflammation in mouse intestine. Sci. Rep. 2018, 8, 16241. [Google Scholar] [CrossRef] [Green Version]
- Starkel, P.; Leclercq, S.; de Timary, P.; Schnabl, B. Intestinal dysbiosis and permeability: The yin and yang in alcohol dependence and alcoholic liver disease. Clin. Sci. 2018, 132, 199–212. [Google Scholar] [CrossRef]
- Dhanda, A.D.; Collins, P.L. Immune dysfunction in acute alcoholic hepatitis. World J. Gastroenterol. WJG 2015, 21, 11904–11913. [Google Scholar] [CrossRef]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Couch, R.D.; Dailey, A.; Zaidi, F.; Navarro, K.; Forsyth, C.B.; Mutlu, E.; Engen, P.A.; Keshavarzian, A. Alcohol induced alterations to the human fecal VOC metabolome. PLoS ONE 2015, 10, e0119362. [Google Scholar] [CrossRef] [Green Version]
- Cresci, G.A.; Bush, K.; Nagy, L.E. Tributyrin supplementation protects mice from acute ethanol-induced gut injury. Alcohol. Clin. Exp. Res. 2014, 38, 1489–1501. [Google Scholar] [CrossRef]
- Cresci, G.A.; Glueck, B.; McMullen, M.R.; Xin, W.; Allende, D.; Nagy, L.E. Prophylactic tributyrin treatment mitigates chronic-binge ethanol-induced intestinal barrier and liver injury. J. Gastroenterol. Hepatol. 2017, 32, 1587–1597. [Google Scholar] [CrossRef]
- Jiang, X.W.; Li, Y.T.; Ye, J.Z.; Lv, L.X.; Yang, L.Y.; Bian, X.Y.; Wu, W.R.; Wu, J.J.; Shi, D.; Wang, Q.; et al. New strain of Pediococcus pentosaceus alleviates ethanol-induced liver injury by modulating the gut microbiota and short-chain fatty acid metabolism. World J. Gastroenterol. WJG 2020, 26, 6224–6240. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Jiang, S.; Zhao, X.; Li, H.; Lin, W.; Li, B.; Lu, J.; Sun, Y.; Yuan, J. Community-metabolome correlations of gut microbiota from child-turcotte-pugh of A and B patients. Front. Microbiol. 2016, 7, 1856. [Google Scholar] [CrossRef] [Green Version]
- Voutilainen, T.; Karkkainen, O. Changes in the human metabolome associated with alcohol use: A review. Alcohol Alcohol. 2019, 54, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Kakiyama, G.; Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Heuman, D.M.; Kang, D.J.; Takei, H.; Nittono, H.; Ridlon, J.M.; Fuchs, M.; et al. Colonic inflammation and secondary bile acids in alcoholic cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G929–G937. [Google Scholar] [CrossRef] [Green Version]
- Lieber, C.S. ALCOHOL: Its metabolism and interaction with nutrients. Annu. Rev. Nutr. 2000, 20, 395–430. [Google Scholar] [CrossRef]
- Bishehsari, F.; Magno, E.; Swanson, G.; Desai, V.; Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. Alcohol and gut-derived inflammation. Alcohol Res. 2017, 38, 163–171. [Google Scholar] [PubMed]
- Dhanda, A.; Atkinson, S.; Vergis, N.; Enki, D.; Fisher, A.; Clough, R.; Cramp, M.; Thursz, M. Trace element deficiency is highly prevalent and associated with infection and mortality in patients with alcoholic hepatitis. Aliment. Pharmacol. Ther. 2020, 52, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Katikireddi, S.V.; Whitley, E.; Lewsey, J.; Gray, L.; Leyland, A.H. Socioeconomic status as an effect modifier of alcohol consumption and harm: Analysis of linked cohort data. Lancet Public Health 2017, 2, e267. [Google Scholar] [CrossRef]
- Cederbaum, A.I.; Lu, Y.; Wu, D. Role of oxidative stress in alcohol-induced liver injury. Arch. Toxicol. 2009, 83, 519–548. [Google Scholar] [CrossRef]
- Mello, T.; Ceni, E.; Surrenti, C.; Galli, A. Alcohol induced hepatic fibrosis: Role of acetaldehyde. Mol. Asp. Med. 2008, 29, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Millonig, G.; Nair, J.; Patsenker, E.; Stickel, F.; Mueller, S.; Bartsch, H.; Seitz, H.K. Ethanol-induced cytochrome P4502E1 causes carcinogenic etheno-DNA lesions in alcoholic liver disease. Hepatology 2009, 50, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.K.; Yates, E.; Lilly, K.; Dhanda, A.D. Oxidative stress in alcohol-related liver disease. World J. Hepatol. 2020, 12, 332–349. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Alcoholic fatty liver: Its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 2004, 34, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.; Aithal, G.P.; Becker, U.; Gleeson, D.; Masson, S.; Wyatt, J.I.; Rowe, I.A.; WALDO Study Group. Natural history of histologically proven alcohol-related liver disease: A systematic review. J. Hepatol. 2019, 71, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Primers 2018, 4, 16. [Google Scholar] [CrossRef]
- Baraona, E.; Lieber, C.S. Effects of ethanol on lipid metabolism. J. Lipid Res. 1979, 20, 289–315. [Google Scholar] [CrossRef]
- You, M.; Fischer, M.; Deeg, M.A.; Crabb, D.W. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J. Biol. Chem. 2002, 277, 29342–29347. [Google Scholar] [CrossRef] [Green Version]
- Purohit, V.; Gao, B.; Song, B.J. Molecular mechanisms of alcoholic fatty liver. Alcohol. Clin. Exp. Res. 2009, 33, 191–205. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Shi, X.; Zhong, W.; Zhao, Y.; Tang, Y.; Sun, W.; Yin, X.; Bogdanov, B.; Kim, S.; McClain, C.; et al. Chronic alcohol exposure disturbs lipid homeostasis at the adipose tissue-liver axis in mice: Analysis of triacylglycerols using high-resolution mass spectrometry in combination with in vivo metabolite deuterium labeling. PLoS ONE 2013, 8, e55382. [Google Scholar] [CrossRef] [PubMed]
- Li, H.H.; Tyburski, J.B.; Wang, Y.W.; Strawn, S.; Moon, B.H.; Kallakury, B.V.; Gonzalez, F.J.; Fornace, A.J., Jr. Modulation of fatty acid and bile acid metabolism by peroxisome proliferator-activated receptor alpha protects against alcoholic liver disease. Alcohol. Clin. Exp. Res. 2014, 38, 1520–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, A.; Pinaire, J.; Fischer, M.; Dorris, R.; Crabb, D.W. The transcriptional and DNA binding activity of peroxisome proliferator-activated receptor alpha is inhibited by ethanol metabolism. A novel mechanism for the development of ethanol-induced fatty liver. J. Biol. Chem. 2001, 276, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, B.; Ahmad, M.F.; Nagy, L.E.; Tsukamoto, H. Inflammatory pathways in alcoholic steatohepatitis. J. Hepatol. 2019, 70, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanda, A.D.; Williams, E.L.; Yates, E.; Lait, P.J.P.; Schewitz-Bowers, L.P.; Hegazy, D.; Cramp, M.E.; Collins, P.L.; Lee, R.W.J. Intermediate monocytes in acute alcoholic hepatitis are functionally activated and induce IL-17 expression in CD4(+) T cells. J. Immunol. 2019, 203, 3190–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markwick, L.J.; Riva, A.; Ryan, J.M.; Cooksley, H.; Palma, E.; Tranah, T.H.; Manakkat Vijay, G.K.; Vergis, N.; Thursz, M.; Evans, A.; et al. Blockade of PD1 and TIM3 restores innate and adaptive immunity in patients with acute alcoholic hepatitis. Gastroenterology 2015, 148, 590–602. [Google Scholar] [CrossRef]
- Wang, S.; Pacher, P.; De Lisle, R.C.; Huang, H.; Ding, W.X. A mechanistic review of cell death in alcohol-induced liver injury. Alcohol. Clin. Exp. Res. 2016, 40, 1215–1223. [Google Scholar] [CrossRef] [Green Version]
- Feldstein, A.E.; Gores, G.J. Apoptosis in alcoholic and nonalcoholic steatohepatitis. Front. Biosci. A J. Virtual Libr. 2005, 10, 3093–3099. [Google Scholar] [CrossRef] [Green Version]
- Gaul, S.; Leszczynska, A.; Alegre, F.; Kaufmann, B.; Johnson, C.D.; Adams, L.A.; Wree, A.; Damm, G.; Seehofer, D.; Calvente, C.J.; et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J. Hepatol. 2021, 74, 156–167. [Google Scholar] [CrossRef]
- Bala, S.; Csak, T.; Saha, B.; Zatsiorsky, J.; Kodys, K.; Catalano, D.; Satishchandran, A.; Szabo, G. The pro-inflammatory effects of miR-155 promote liver fibrosis and alcohol-induced steatohepatitis. J. Hepatol. 2016, 64, 1378–1387. [Google Scholar] [CrossRef] [Green Version]
- Bala, S.; Csak, T.; Kodys, K.; Catalano, D.; Ambade, A.; Furi, I.; Lowe, P.; Cho, Y.; Iracheta-Vellve, A.; Szabo, G. Alcohol-induced miR-155 and HDAC11 inhibit negative regulators of the TLR4 pathway and lead to increased LPS responsiveness of Kupffer cells in alcoholic liver disease. J. Leukoc. Biol. 2017, 102, 487–498. [Google Scholar] [CrossRef] [Green Version]
- Saikia, P.; Bellos, D.; McMullen, M.R.; Pollard, K.A.; de la Motte, C.; Nagy, L.E. MicroRNA 181b-3p and its target importin alpha5 regulate toll-like receptor 4 signaling in Kupffer cells and liver injury in mice in response to ethanol. Hepatology 2017, 66, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Goldar, S.; Khaniani, M.S.; Derakhshan, S.M.; Baradaran, B. Molecular mechanisms of apoptosis and roles in cancer development and treatment. Asian Pac. J. Cancer Prev. 2015, 16, 2129–2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Chiba, T.; Marusawa, H.; Ushijima, T. Inflammation-associated cancer development in digestive organs: Mechanisms and roles for genetic and epigenetic modulation. Gastroenterology 2012, 143, 550–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagymasi, K.; Blazovics, A.; Lengyel, G.; Kocsis, I.; Feher, J. Oxidative damage in alcoholic liver disease. Eur. J. Gastroenterol. Hepatol. 2001, 13, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.; Wang, X.; Sun, C.; Zheng, X.; Wei, H.; Tian, Z.; Sun, R. Chronic alcohol consumption promotes diethylnitrosamine-induced hepatocarcinogenesis via immune disturbances. Sci. Rep. 2017, 7, 2567. [Google Scholar] [CrossRef]
- Sakamoto, T.; Hara, M.; Higaki, Y.; Ichiba, M.; Horita, M.; Mizuta, T.; Eguchi, Y.; Yasutake, T.; Ozaki, I.; Yamamoto, K.; et al. Influence of alcohol consumption and gene polymorphisms of ADH2 and ALDH2 on hepatocellular carcinoma in a Japanese population. Int. J. Cancer 2006, 118, 1501–1507. [Google Scholar] [CrossRef]
- Khalesi, S.; Johnson, D.W.; Campbell, K.; Williams, S.; Fenning, A.; Saluja, S.; Irwin, C. Effect of probiotics and synbiotics consumption on serum concentrations of liver function test enzymes: A systematic review and meta-analysis. Eur. J. Nutr. 2018, 57, 2037–2053. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Duan, K.; Wang, C.; McClain, C.; Feng, W. Probiotics and alcoholic liver disease: Treatment and potential mechanisms. Gastroenterol. Res. Pract. 2016, 2016, 5491465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, J.S.; Gavis, E.A.; Fagan, A.; Wade, J.B.; Thacker, L.R.; Fuchs, M.; Patel, S.; Davis, B.; Meador, J.; Puri, P.; et al. A randomized clinical trial of fecal microbiota transplant for alcohol use disorder. Hepatology 2021, 73, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Hamer, H.M.; Jonkers, D.M.; Vanhoutvin, S.A.; Troost, F.J.; Rijkers, G.; de Bruine, A.; Bast, A.; Venema, K.; Brummer, R.J. Effect of butyrate enemas on inflammation and antioxidant status in the colonic mucosa of patients with ulcerative colitis in remission. Clin. Nutr. 2010, 29, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Chen, J.; Xia, J.; Wang, B.; Liu, H.; Yang, L.; Wang, Y.; Ling, Z. Role of probiotics in the treatment of minimal hepatic encephalopathy in patients with HBV-induced liver cirrhosis. J. Int. Med. Res. 2018, 46, 3596–3604. [Google Scholar] [CrossRef] [PubMed]
- Horvath, A.; Leber, B.; Schmerboeck, B.; Tawdrous, M.; Zettel, G.; Hartl, A.; Madl, T.; Stryeck, S.; Fuchs, D.; Lemesch, S.; et al. Randomised clinical trial: The effects of a multispecies probiotic vs. placebo on innate immune function, bacterial translocation and gut permeability in patients with cirrhosis. Aliment. Pharmacol. Ther. 2016, 44, 926–935. [Google Scholar] [CrossRef]
- Morris, E.M.; Jackman, M.R.; Johnson, G.C.; Liu, T.W.; Lopez, J.L.; Kearney, M.L.; Fletcher, J.A.; Meers, G.M.; Koch, L.G.; Britton, S.L.; et al. Intrinsic aerobic capacity impacts susceptibility to acute high-fat diet-induced hepatic steatosis. American journal of physiology. Endocrinol. Metab. 2014, 307, E355–E364. [Google Scholar] [CrossRef] [Green Version]
- Panasevich, M.R.; Morris, E.M.; Chintapalli, S.V.; Wankhade, U.D.; Shankar, K.; Britton, S.L.; Koch, L.G.; Thyfault, J.P.; Rector, R.S. Gut microbiota are linked to increased susceptibility to hepatic steatosis in low-aerobic-capacity rats fed an acute high-fat diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G166–G179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajaj, J.S.; Idilman, R.; Mabudian, L.; Hood, M.; Fagan, A.; Turan, D.; White, M.B.; Karakaya, F.; Wang, J.; Atalay, R.; et al. Diet affects gut microbiota and modulates hospitalization risk differentially in an international cirrhosis cohort. Hepatology 2018, 68, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Margain, A.; Macias-Rodriguez, R.U.; Rios-Torres, S.L.; Roman-Calleja, B.M.; Mendez-Guerrero, O.; Rodriguez-Cordova, P.; Torre, A. Effect of a high-protein, high-fiber diet plus supplementation with branched-chain amino acids on the nutritional status of patients with cirrhosis. Rev. Gastroenterol. Mex. 2018, 83, 9–15. [Google Scholar] [CrossRef]
- Liu, Q.; Duan, Z.P.; Ha, D.K.; Bengmark, S.; Kurtovic, J.; Riordan, S.M. Synbiotic modulation of gut flora: Effect on minimal hepatic encephalopathy in patients with cirrhosis. Hepatology 2004, 39, 1441–1449. [Google Scholar] [CrossRef]
- Burk, R.F.; Hill, K.E.; Motley, A.K.; Byrne, D.W.; Norsworthy, B.K. Selenium deficiency occurs in some patients with moderate-to-severe cirrhosis and can be corrected by administration of selenate but not selenomethionine: A randomized controlled trial. Am. J. Clin. Nutr. 2015, 102, 1126–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maret, W. Zinc and human disease. Met. Ions Life Sci. 2013, 13, 389–414. [Google Scholar] [CrossRef]
- Nangliya, V.; Sharma, A.; Yadav, D.; Sunder, S.; Nijhawan, S.; Mishra, S. Study of trace elements in liver cirrhosis patients and their role in prognosis of disease. Biol. Trace Elem. Res. 2015, 165, 35–40. [Google Scholar] [CrossRef]
- Tan, H.K.; Streeter, A.; Cramp, M.E.; Dhanda, A.D. Effect of zinc treatment on clinical outcomes in patients with liver cirrhosis: A systematic review and meta-analysis. World J. Hepatol. 2020, 12, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, M.; Song, M.; Falkner, K.; McClain, C.; Cave, M. Zinc sulfate for alcoholic cirrhosis (ZAC) clinical trial—Interim analysis of liver injury/inflammation biomarkers. Hepatology 2014, 60, 794A. [Google Scholar]
- Otten, A.T.; Bourgonje, A.R.; Peters, V.; Alizadeh, B.Z.; Dijkstra, G.; Harmsen, H.J.M. Vitamin C supplementation in healthy individuals leads to shift of bacterial populations in the gut—A pilot study. Antioxidants 2021, 10, 1278. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Rawat, A.; Alwakeel, M.; Sharif, E.; Al Khodor, S. The potential role of vitamin D supplementation as a gut microbiota modifier in healthy individuals. Sci. Rep. 2020, 10, 21641. [Google Scholar] [CrossRef]
- Phillips, M.; Curtis, H.; Portmann, B.; Donaldson, N.; Bomford, A.; O’Grady, J. Antioxidants versus corticosteroids in the treatment of severe alcoholic hepatitis--a randomised clinical trial. J. Hepatol. 2006, 44, 784–790. [Google Scholar] [CrossRef]
- Thursz, M.R.; Richardson, P.; Allison, M.; Austin, A.; Bowers, M.; Day, C.P.; Downs, N.; Gleeson, D.; MacGilchrist, A.; Grant, A.; et al. Prednisolone or pentoxifylline for alcoholic hepatitis. N. Engl. J. Med. 2015, 372, 1619–1628. [Google Scholar] [CrossRef] [Green Version]
- Naveau, S.; Chollet-Martin, S.; Dharancy, S.; Mathurin, P.; Jouet, P.; Piquet, M.A.; Davion, T.; Oberti, F.; Broet, P.; Emilie, D.; et al. A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology 2004, 39, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Vergis, N.; Atkinson, S.R.; Knapp, S.; Maurice, J.; Allison, M.; Austin, A.; Forrest, E.H.; Masson, S.; McCune, A.; Patch, D.; et al. In patients with severe alcoholic hepatitis, prednisolone increases susceptibility to infection and infection-related mortality, and is associated with high circulating levels of bacterial DNA. Gastroenterology 2017, 152, 1068–1077.e1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, J.; Memon, R.S.; Shahid, I.; Rizwan, T.; Zaman, M.; Menezes, R.G.; Kumar, S.; Siddiqi, T.J.; Usman, M.S. Antidiabetic drugs and non-alcoholic fatty liver disease: A systematic review, meta-analysis and evidence map. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2021, 53, 44–51. [Google Scholar] [CrossRef]
- Athyros, V.G.; Alexandrides, T.K.; Bilianou, H.; Cholongitas, E.; Doumas, M.; Ganotakis, E.S.; Goudevenos, J.; Elisaf, M.S.; Germanidis, G.; Giouleme, O.; et al. The use of statins alone, or in combination with pioglitazone and other drugs, for the treatment of non-alcoholic fatty liver disease/non-alcoholic steatohepatitis and related cardiovascular risk. An Expert Panel Statement. Metab. Clin. Exp. 2017, 71, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Jalali, M.; Rahimlou, M.; Mahmoodi, M.; Moosavian, S.P.; Symonds, M.E.; Jalali, R.; Zare, M.; Imanieh, M.H.; Stasi, C. The effects of metformin administration on liver enzymes and body composition in non-diabetic patients with non-alcoholic fatty liver disease and/or non-alcoholic steatohepatitis: An up-to date systematic review and meta-analysis of randomized controlled trials. Pharmacol. Res. 2020, 159, 104799. [Google Scholar] [CrossRef]
- Weersma, R.K.; Zhernakova, A.; Fu, J. Interaction between drugs and the gut microbiome. Gut 2020, 69, 1510–1519. [Google Scholar] [CrossRef]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pohl, K.; Moodley, P.; Dhanda, A.D. Alcohol’s Impact on the Gut and Liver. Nutrients 2021, 13, 3170. https://doi.org/10.3390/nu13093170
Pohl K, Moodley P, Dhanda AD. Alcohol’s Impact on the Gut and Liver. Nutrients. 2021; 13(9):3170. https://doi.org/10.3390/nu13093170
Chicago/Turabian StylePohl, Keith, Prebashan Moodley, and Ashwin D. Dhanda. 2021. "Alcohol’s Impact on the Gut and Liver" Nutrients 13, no. 9: 3170. https://doi.org/10.3390/nu13093170
APA StylePohl, K., Moodley, P., & Dhanda, A. D. (2021). Alcohol’s Impact on the Gut and Liver. Nutrients, 13(9), 3170. https://doi.org/10.3390/nu13093170