
Vitamin D and Secondary Hyperparathyroidism in Chronic Kidney Disease: A Critical Appraisal of the Past, Present, and the Future

Abstract
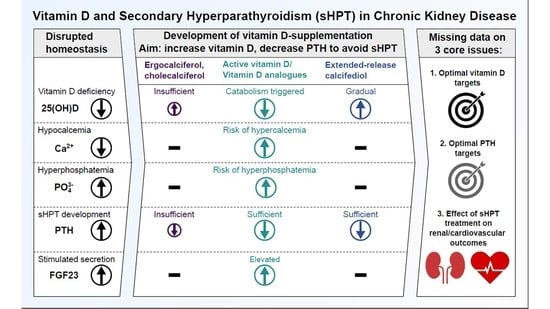
:
{kind=link}
{kind=link}
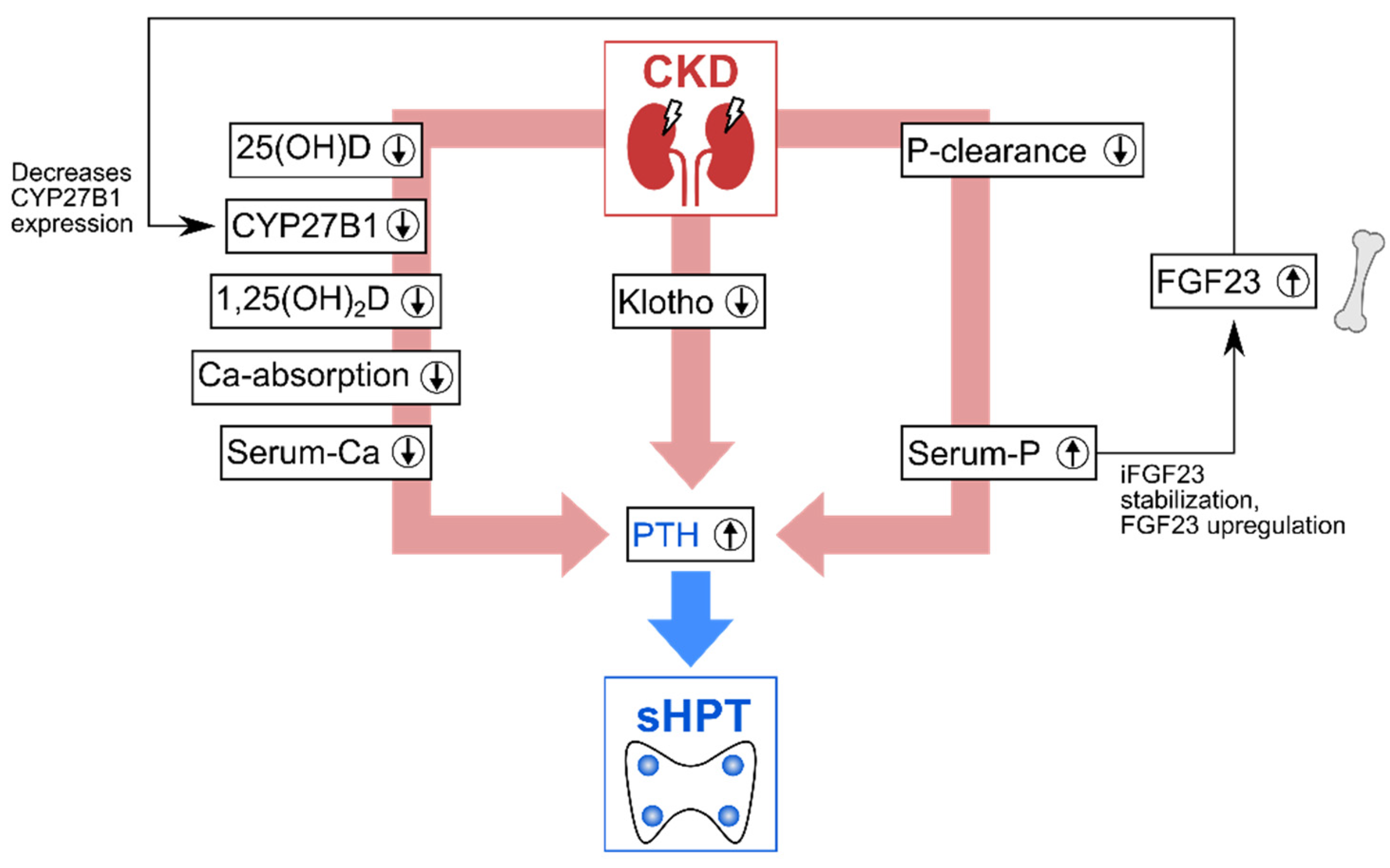{kind=link}
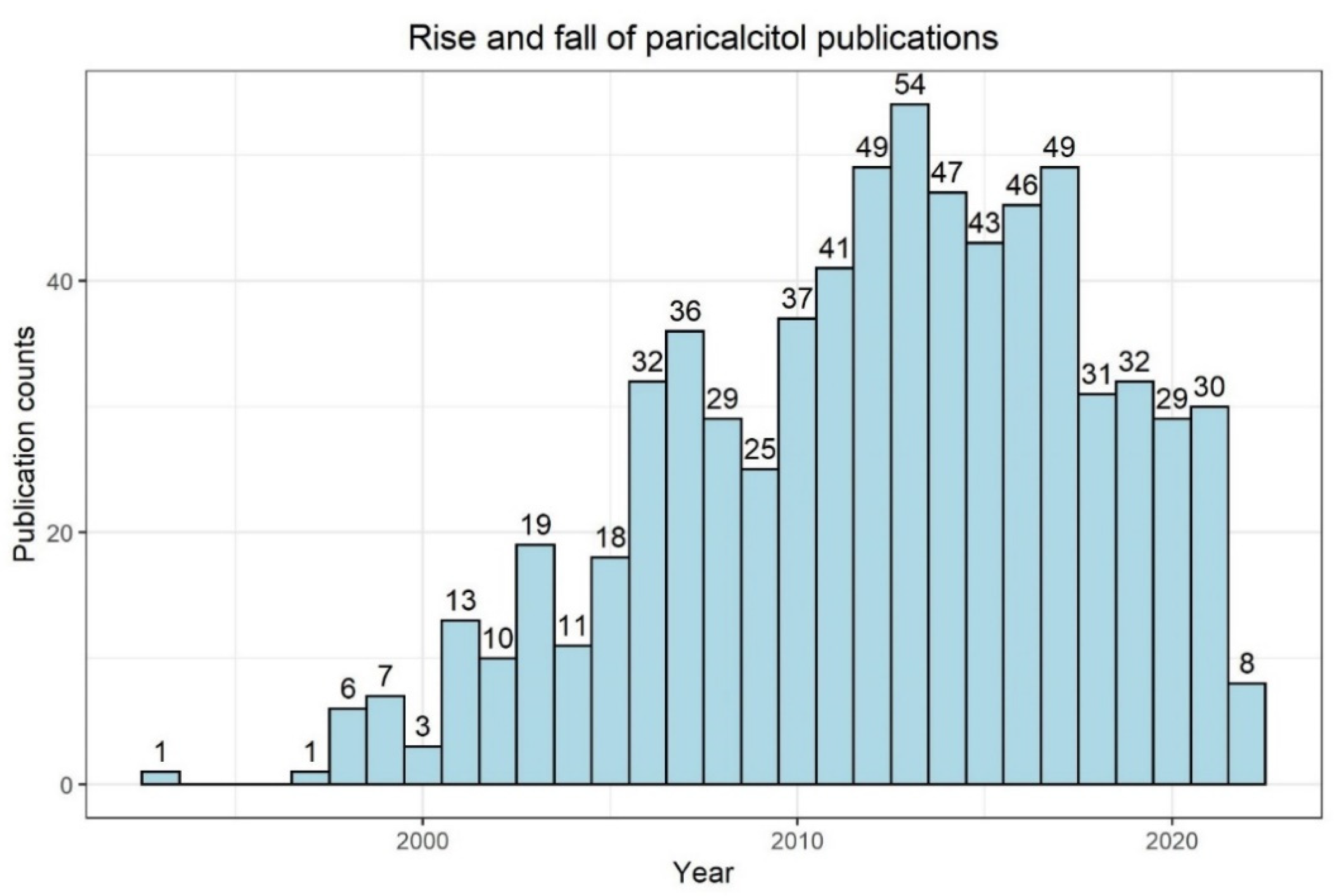{kind=link}
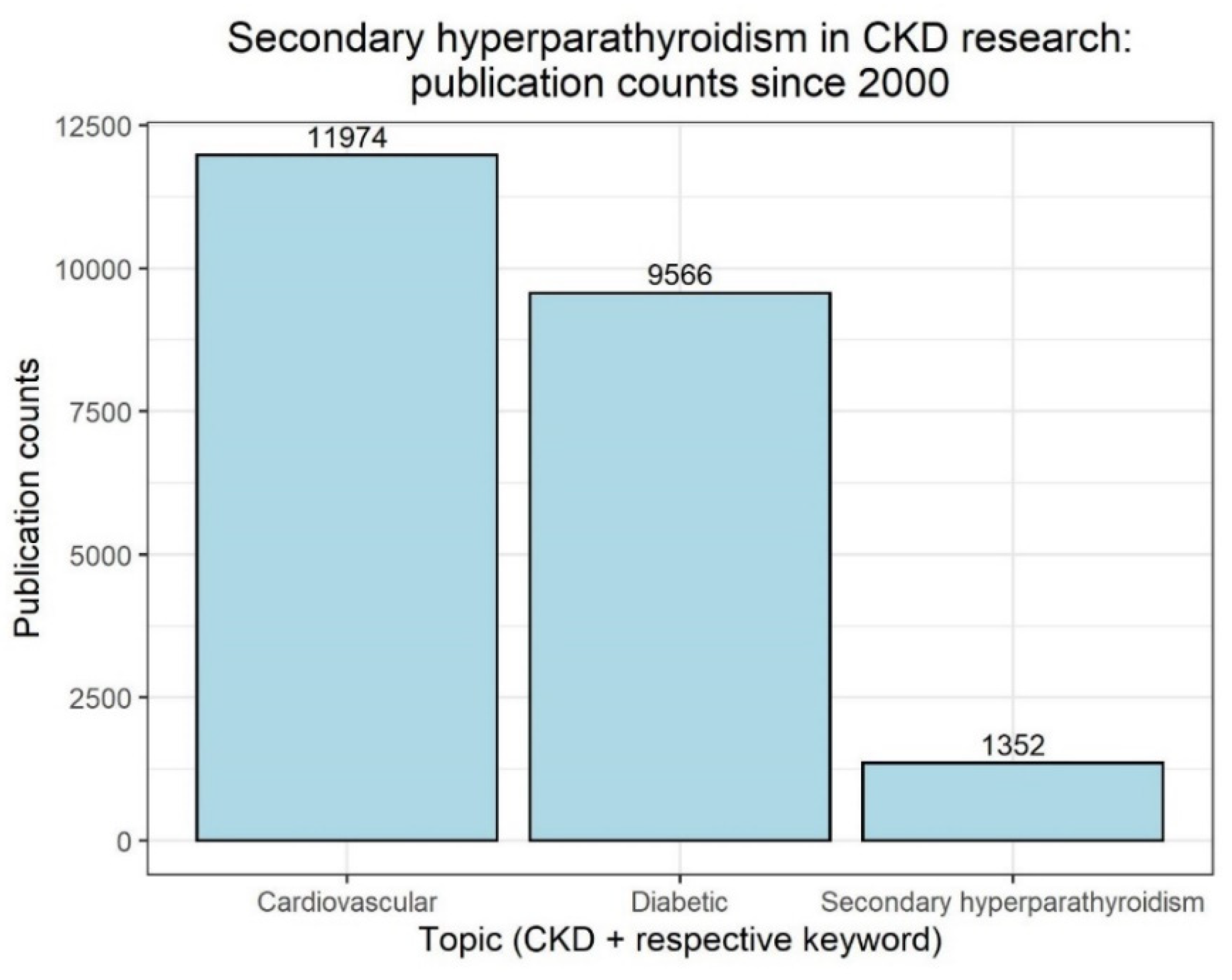{kind=link}
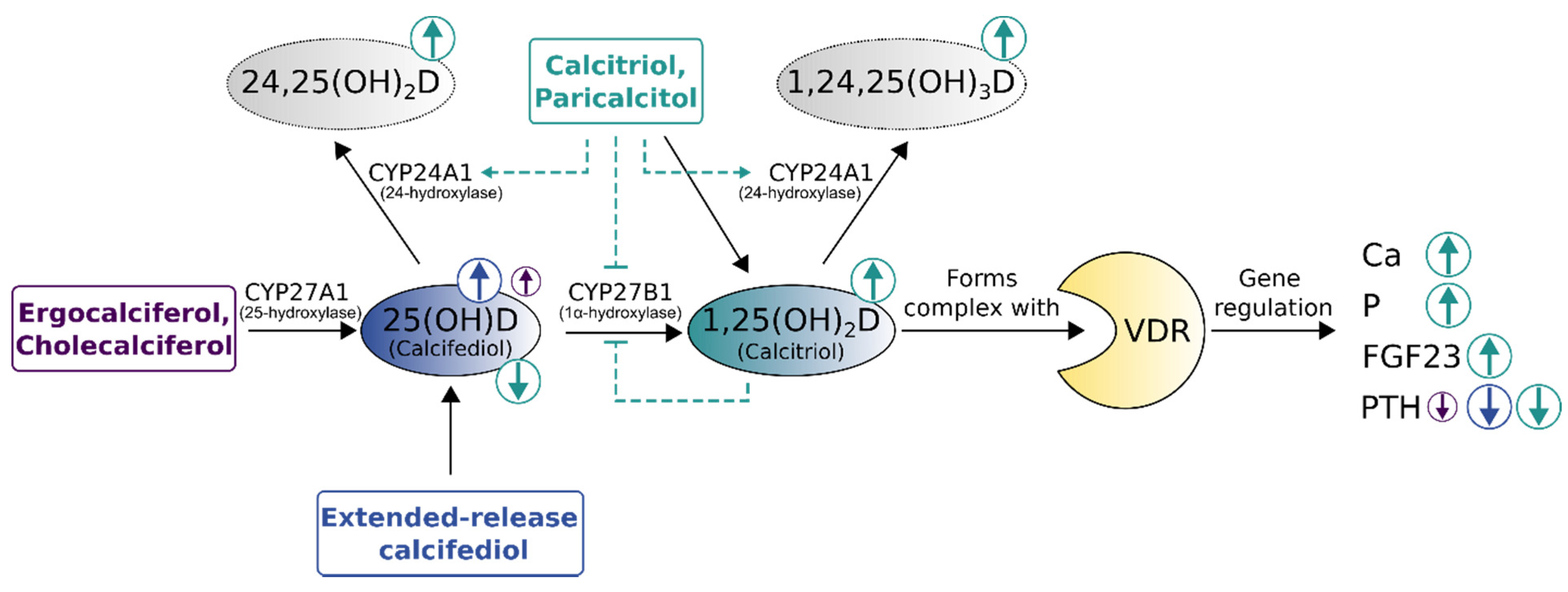{kind=link}
1. Introduction
1.1. Aetiology and Prevalence of Vitamin D in Chronic Kidney Disease
1.2. Consequences of Vitamin D Deficiency in CKD
1.3. Vitamin D Deficiency and Parathyroid Overproduction Lead to Mineral and Bone Disorders
1.4. The Ongoing Challenge of Optimal Vitamin D Supplementation in Renal Hyperparathyroidism
2. Active Vitamin D Substitution and the Unwanted Side Effects of Overtreatment
2.1. Active Vitamin D Substitution as the Beginning of Renal HPT Treatment
2.2. Active Vitamin D-Induced Hypercalcemia, Hyperphosphatemia and Vascular Calcification
2.3. Active Vitamin D-Induced Adynamic Bone Disease
3. Alternative Vitamin D Substances and Efforts to Avoid Calcium Overload
4. Extended-Release Calcifediol: A Combination of Efficacy and Safety as the Next Step in sHPT Treatment
5. Will the Usage of SGLT2-Inhibitors Facilitate CKD–MBD Research?
6. Vitamin D Treatment in Summary: A Call for Action
- What is the optimal PTH target level for CKD and dialysis patients?
- What is the optimal vitamin D level to support optimal PTH titration?
- How can HPT treatment support reduction in the occurrence of hard renal and cardiovascular events in CKD and dialysis patients?
7. Future Directions
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Franca Gois, P.H.; Wolley, M.; Ranganathan, D.; Seguro, A.C. Vitamin D Deficiency in Chronic Kidney Disease: Recent Evidence and Controversies. Int. J. Environ. Res. Public Health 2018, 15, 1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levey, A.S.; Eckardt, K.-U.; Dorman, N.M.; Christiansen, S.L.; Hoorn, E.J.; Ingelfinger, J.R.; Inker, L.A.; Levin, A.; Mehrotra, R.; Palevsky, P.M.; et al. Nomenclature for kidney function and disease: Report of a Kidney Disease: Improving Global Outcomes (KDIGO) Consensus Conference. Kidney Int. 2020, 97, 1117–1129. [Google Scholar] [CrossRef] [PubMed]
- de Boer, I.H.; Caramori, M.L.; Chan, J.C.; Heerspink, H.J.; Hurst, C.; Khunti, K.; Rossing, P. Kidney Disease: Improving Global Outcomes (KDIGO) Diabetes Work Group. KDIGO 2020 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease. Kidney Int. 2020, 98, S1–S116. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Cai, X.; Lee, J.; Mehta, R.; Zhang, X.; Yang, W.; Nessel, L.; Anderson, A.H.; Lo, J.; Porter, A.; et al. Longitudinal Evolution of Markers of Mineral Metabolism in Patients With CKD: The Chronic Renal Insufficiency Cohort (CRIC) Study. Am. J. Kidney Dis. 2020, 75, 235–244. [Google Scholar] [CrossRef]
- Li, M.; Li, Y. Prevalence and influencing factors of vitamin D deficiency in chronic kidney disease: A cross-sectional study. Int. J. Clin. Pharmacol. Ther. 2020, 58, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Coccia, P.; Blazquez, J.; Contreras, M.; Ferrais, V.; Raddavero, C.; Ghezzi, L.; Busaniche, J.; Beneitez, G.; Kozak, A.; Ferraris, J. Alta prevalencia de deficiencia de vitamina D en niños con enfermedad renal crónica y trasplante renal. Arch. Argent. Pediatr. 2017, 115, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Nigwekar, S.U.; Bhan, I.; Thadhani, R. Ergocalciferol and cholecalciferol in CKD. Am. J. Kidney Dis. 2012, 60, 139–156. [Google Scholar] [CrossRef] [Green Version]
- Nigwekar, S.U.; Tamez, H.; Thadhani, R.I. Vitamin D and chronic kidney disease-mineral bone disease (CKD-MBD). Bonekey Rep. 2014, 3, 498. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Glowacki, J. Chronic kidney disease and vitamin D metabolism in human bone marrow-derived MSCs. Ann. N. Y. Acad. Sci. 2017, 1402, 43–55. [Google Scholar] [CrossRef]
- Christensen, M.H.E.; Apalset, E.M.; Nordbø, Y.; Varhaug, J.E.; Mellgren, G.; Lien, E.A. 1,25-dihydroxyvitamin D and the vitamin D receptor gene polymorphism Apa1 influence bone mineral density in primary hyperparathyroidism. PLoS ONE 2013, 8, e56019. [Google Scholar] [CrossRef]
- Cunningham, J.; Locatelli, F.; Rodriguez, M. Secondary hyperparathyroidism: Pathogenesis, disease progression, and therapeutic options. Clin. J. Am. Soc. Nephrol. 2011, 6, 913–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galuška, D.; Pácal, L.; Kaňková, K. Pathophysiological Implication of Vitamin D in Diabetic Kidney Disease. Kidney Blood Press. Res. 2021, 46, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Germain, M. Secondary-Hyperparathyroidism-in-Non-dialysis-Chronic-Kidney-Disease. EMJ Nephrol. 2021, 9, 37–45. [Google Scholar]
- Leifheit-Nestler, M.; Haffner, D. How FGF23 shapes multiple organs in chronic kidney disease. Mol. Cell. Pediatr. 2021, 8, 12. [Google Scholar] [CrossRef]
- Centeno, P.P.; Herberger, A.; Mun, H.-C.; Tu, C.; Nemeth, E.F.; Chang, W.; Conigrave, A.D.; Ward, D.T. Phosphate acts directly on the calcium-sensing receptor to stimulate parathyroid hormone secretion. Nat. Commun. 2019, 10, 4693. [Google Scholar] [CrossRef] [PubMed]
- Mace, M.L.; Olgaard, K.; Lewin, E. New Aspects of the Kidney in the Regulation of Fibroblast Growth Factor 23 (FGF23) and Mineral Homeostasis. Int. J. Mol. Sci. 2020, 21, 8810. [Google Scholar] [CrossRef]
- Hruska, K.A.; Sugatani, T.; Agapova, O.; Fang, Y. The chronic kidney disease—Mineral bone disorder (CKD-MBD): Advances in pathophysiology. Bone 2017, 100, 80–86. [Google Scholar] [CrossRef]
- KDIGO. KDIGO 2017 Clinical Practice Guideline Update for the Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease–Mineral and Bone Disorder (CKD-MBD). Kidney Int. Suppl. 2017, 7, 1–59. [Google Scholar] [CrossRef] [Green Version]
- Moe, S.M.; Drüeke, T.; Lameire, N.; Eknoyan, G. Chronic kidney disease-mineral-bone disorder: A new paradigm. Adv. Chronic Kidney Dis. 2007, 14, 3–12. [Google Scholar] [CrossRef]
- Rodriguez, M.; Nemeth, E.; Martin, D. The calcium-sensing receptor: A key factor in the pathogenesis of secondary hyperparathyroidism. Am. J. Physiol. Ren. Physiol. 2005, 288, F253–F264. [Google Scholar] [CrossRef] [Green Version]
- Drueke, T.; Martin, D.; Rodriguez, M. Can calcimimetics inhibit parathyroid hyperplasia? Evidence from preclinical studies. Nephrol. Dial. Transpl. 2007, 22, 1828–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, S.; Giachelli, C.M. Vascular calcification in CKD-MBD: Roles for phosphate, FGF23, and Klotho. Bone 2017, 100, 87–93. [Google Scholar] [CrossRef]
- Ketteler, M.; Schlieper, G.; Floege, J. Calcification and cardiovascular health: New insights into an old phenomenon. Hypertension 2006, 47, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Górriz, J.L.; Molina, P.; Cerverón, M.J.; Vila, R.; Bover, J.; Nieto, J.; Barril, G.; Martínez-Castelao, A.; Fernández, E.; Escudero, V.; et al. Vascular calcification in patients with nondialysis CKD over 3 years. Clin. J. Am. Soc. Nephrol. 2015, 10, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slatopolsky, E.; Gonzalez, E.; Martin, K. Pathogenesis and treatment of renal osteodystrophy. Blood Purif. 2003, 21, 318–326. [Google Scholar] [CrossRef]
- Pimentel, A.; Ureña-Torres, P.; Bover, J.; Luis Fernandez-Martín, J.; Cohen-Solal, M. Bone Fragility Fractures in CKD Patients. Calcif. Tissue Int. 2021, 108, 539–550. [Google Scholar] [CrossRef]
- Naylor, K.L.; McArthur, E.; Leslie, W.D.; Fraser, L.-A.; Jamal, S.A.; Cadarette, S.M.; Pouget, J.G.; Lok, C.E.; Hodsman, A.B.; Adachi, J.D.; et al. The three-year incidence of fracture in chronic kidney disease. Kidney Int. 2014, 86, 810–818. [Google Scholar] [CrossRef] [Green Version]
- Geng, S.; Kuang, Z.; Peissig, P.L.; Page, D.; Maursetter, L.; Hansen, K.E. Parathyroid hormone independently predicts fracture, vascular events, and death in patients with stage 3 and 4 chronic kidney disease. Osteoporos Int. 2019, 30, 2019–2025. [Google Scholar] [CrossRef]
- Khan, S. Secondary hyperparathyroidism is associated with higher cost of care among chronic kidney disease patients with cardiovascular comorbidities. Nephron Clin. Pract. 2007, 105, c159–c164. [Google Scholar] [CrossRef]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int. Suppl. 2009, 113, S1–S130. [Google Scholar] [CrossRef]
- Memmos, D.E.; Eastwood, J.B.; Talner, L.B.; Gower, P.E.; Curtis, J.R.; Phillips, M.E.; Carter, G.D.; Alaghband-Zadeh, J.; Roberts, A.P.; de Wardener, H.E. Double-blind trial of oral 1,25-dihydroxy vitamin D3 versus placebo in asymptomatic hyperparathyroidism in patients receiving maintenance haemodialysis. Br. Med. J. (Clin. Res. Ed) 1981, 282, 1919–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharman, V.L.; Abrams, S.M.; Adami, S.; Cattell, W.R.; Chaput de Saintonge, D.M.; Greenwood, R.N.; Goodwin, F.J.; Hately, W.; Hattersley, L.A.; Marsh, F.P.; et al. Controlled trial of calcitriol in the prevention of bone disease in haemodialysed patients. Proc. Eur. Dial. Transpl. Assoc. 1983, 19, 287–292. [Google Scholar]
- Bordier, P.J.; Marie, P.J.; Arnaud, C.D. Evolution of renal osteodystrophy: Correlation of bone histomorphometry and serum mineral and immunoreactive parathyroid hormone values before and after treatment with calcium carbonate or 25-hydroxycholecalciferol. Kidney Int. Suppl. 1975, 2, 102–112. [Google Scholar]
- Letteri, J.M.; Kleinman, L.M.; Ellis, K.N.; Caselnova, R.; Akhtar, M.; Cohn, S.H. Effects of 25-hydroxycholecalciferol on calcium metabolism in chronic renal failure. Adv. Exp. Med. Biol. 1977, 81, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Kovesdy, C.P. Clinical outcomes with active versus nutritional vitamin D compounds in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2009, 4, 1529–1539. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, R.; Georgianos, P.I. Con: Nutritional vitamin D replacement in chronic kidney disease and end-stage renal disease. Nephrol. Dial. Transpl. 2016, 31, 706–713. [Google Scholar] [CrossRef] [Green Version]
- Bover, J.; Gunnarsson, J.; Csomor, P.; Kaiser, E.; Cianciolo, G.; Lauppe, R. Impact of nutritional vitamin D supplementation on parathyroid hormone and 25-hydroxyvitamin D levels in non-dialysis chronic kidney disease: A meta-analysis. Clin. Kidney J. 2021, 14, 2177–2186. [Google Scholar] [CrossRef]
- Malluche, H.H.; Faugere, M.C. Renal osteodystrophy. N. Engl. J. Med. 1989, 321, 317–319. [Google Scholar] [CrossRef]
- Maxwell, D.R.; Benjamin, D.M.; Donahay, S.L.; Allen, M.K.; Hamburger, R.J.; Luft, F.C. Calcitriol in dialysis patients. Clin. Pharmacol. Ther. 1978, 23, 515–519. [Google Scholar] [CrossRef]
- Sperschneider, H.; Humbsch, K.; Abendroth, K. Orale Calcitriolstosstherapie bei Hämodialysepatienten. Auswirkungen auf die Histomorphometrie des Knochens bei renalem Hyperparathyreoidismus. Med. Klin. 1997, 92, 597–603. [Google Scholar] [CrossRef]
- Cozzolino, M.; Bernard, L.; Csomor, P.A. Active vitamin D increases the risk of hypercalcaemia in non-dialysis chronic kidney disease patients with secondary hyperparathyroidism: A systematic review and meta-analysis. Clin. Kidney J. 2021, 14, 2437–2443. [Google Scholar] [CrossRef] [PubMed]
- Sprague, S.M.; Strugnell, S.A.; Bishop, C.W. Extended-release calcifediol for secondary hyperparathyroidism in stage 3–4 chronic kidney disease. Expert Rev. Endocrinol. Metab. 2017, 12, 289–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lung, B.E.; Mowery, M.L.; Komatsu, D.E.E. StatPearls: Calcitriol; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022. [Google Scholar]
- Yee, J.; Rosenbaum, D.; Jacobs, J.W.; Sprague, S.M. Small Intestinal Phosphate Absorption: Novel Therapeutic Implications. Am. J. Nephrol. 2021, 52, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Goodman William, G.; Goldin, J.; Kuizon Beatriz, D.; Yoon, C.; Gales, B.; Sider, D.; Wang, Y.; Chung, J.; Emerick, A.; Greaser, L.; et al. Coronary-Artery Calcification in Young Adults with End-Stage Renal Disease Who Are Undergoing Dialysis. N. Engl. J. Med. 2020, 342, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, J.M.; Lowrie, E.G.; Hampers, C.L.; Merrill, J.P. Cardiovascular disease in uremic patients on hemodialysis. Kidney Int. Suppl. 1975, 2, 167–175. [Google Scholar]
- Llach, F.; Velasquez Forero, F. Secondary hyperparathyroidism in chronic renal failure: Pathogenic and clinical aspects. Am. J. Kidney Dis. 2001, 38, S20–S33. [Google Scholar] [CrossRef] [PubMed]
- Raggi, P.; Boulay, A.; Chasan-Taber, S.; Amin, N.; Dillon, M.; Burke, S.K.; Chertow, G.M. Cardiac calcification in adult hemodialysis patients. J. Am. Coll. Cardiol. 2002, 39, 695–701. [Google Scholar] [CrossRef] [Green Version]
- Morosetti, M.; Jankovic, L.; Cetani, F.; Cordisco, R.; Fortunato, L.; Friggi, A.; Frattarelli, D.; Palombo, G.; Pisani, G.; Rosa, M.; et al. High doses of intravenous calcitriol in the treatment of severe secondary hyperparathyroidism. J. Nephrol. 2004, 17, 95–100. [Google Scholar]
- Ketteler, M.; Rothe, H.; Krüger, T.; Biggar, P.H.; Schlieper, G. Mechanisms and treatment of extraosseous calcification in chronic kidney disease. Nat. Rev. Nephrol. 2011, 7, 509–516. [Google Scholar] [CrossRef]
- Schäfer, C.; Heiss, A.; Schwarz, A.; Westenfeld, R.; Ketteler, M.; Floege, J.; Müller-Esterl, W.; Schinke, T.; Jahnen-Dechent, W. The serum protein α2–Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J. Clin. Investig. 2003, 112, 357–366. [Google Scholar] [CrossRef]
- Lee, D. Vascular calcification: Inducers and inhibitors. Mater. Sci. Eng. B 2011, 176, 1133–1141. [Google Scholar] [CrossRef]
- Wolisi, G.O.; Moe, S.M. The role of vitamin D in vascular calcification in chronic kidney disease. Semin. Dial. 2005, 18, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, V.M.; Floege, J. Adynamic bone disease-bone and beyond. NDT Plus 2008, 1, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cozzolino, M.; Brandenburg, V. Paricalcitol and outcome: A manual on how a vitamin D receptor activator (VDRA) can help us to get down the “U”. Clin. Nephrol. 2009, 71, 593–601. [Google Scholar] [CrossRef]
- Wesseling-Perry, K.; Pereira, R.C.; Sahney, S.; Gales, B.; Wang, H.-J.; Elashoff, R.; Jüppner, H.; Salusky, I.B. Calcitriol and doxercalciferol are equivalent in controlling bone turnover, suppressing parathyroid hormone, and increasing fibroblast growth factor-23 in secondary hyperparathyroidism. Kidney Int. 2011, 79, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Isakova, T.; Xie, H.; Yang, W.; Xie, D.; Anderson, A.H.; Scialla, J.; Wahl, P.; Gutiérrez, O.M.; Steigerwalt, S.; He, J.; et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 2011, 305, 2432–2439. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, O.M.; Mannstadt, M.; Isakova, T.; Rauh-Hain, J.A.; Tamez, H.; Shah, A.; Smith, K.; Lee, H.; Thadhani, R.; Jüppner, H.; et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N. Engl. J. Med. 2008, 359, 584–592. [Google Scholar] [CrossRef] [Green Version]
- Suki, W.N.; Zabaneh, R.; Cangiano, J.L.; Reed, J.; Fischer, D.; Garrett, L.; Ling, B.N.; Chasan-Taber, S.; Dillon, M.A.; Blair, A.T.; et al. Effects of sevelamer and calcium-based phosphate binders on mortality in hemodialysis patients. Kidney Int. 2007, 72, 1130–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chertow, G.M.; Raggi, P.; McCarthy, J.T.; Schulman, G.; Silberzweig, J.; Kuhlik, A.; Goodman, W.G.; Boulay, A.; Burke, S.K.; Toto, R.D. The effects of sevelamer and calcium acetate on proxies of atherosclerotic and arteriosclerotic vascular disease in hemodialysis patients. Am. J. Nephrol. 2003, 23, 307–314. [Google Scholar] [CrossRef]
- Bikle, D.D. Clinical counterpoint: Vitamin D: New actions, new analogs, new therapeutic potential. Endocr. Rev. 1992, 13, 765–784. [Google Scholar] [CrossRef]
- Martin, K.J.; González, E.A.; Gellens, M.; Hamm, L.L.; Abboud, H.; Lindberg, J. 19-Nor-1-alpha-25-dihydroxyvitamin D2 (Paricalcitol) safely and effectively reduces the levels of intact parathyroid hormone in patients on hemodialysis. J. Am. Soc. Nephrol. 1998, 9, 1427–1432. [Google Scholar] [CrossRef] [PubMed]
- Llach, F.; Keshav, G.; Goldblat, M.V.; Lindberg, J.S.; Sadler, R.; Delmez, J.; Arruda, J.; Lau, A.; Slatopolsky, E. Suppression of parathyroid hormone secretion in hemodialysis patients by a novel vitamin D analogue: 19-nor-1,25-dihydroxyvitamin D2. Am. J. Kidney Dis. 1998, 32, S48–S54. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.; Wolf, M.; Lowrie, E.; Ofsthun, N.; Lazarus, J.M.; Thadhani, R. Survival of patients undergoing hemodialysis with paricalcitol or calcitriol therapy. N. Engl. J. Med. 2003, 349, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Thadhani, R.; Appelbaum, E.; Pritchett, Y.; Chang, Y.; Wenger, J.; Tamez, H.; Bhan, I.; Agarwal, R.; Zoccali, C.; Wanner, C.; et al. Vitamin D therapy and cardiac structure and function in patients with chronic kidney disease: The PRIMO randomized controlled trial. J. Am. Med. Assoc. 2012, 307, 674–684. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.Y.-M.; Fang, F.; Chan, J.; Wen, Y.-Y.; Qing, S.; Chan, I.H.-S.; Lo, G.; Lai, K.-N.; Lo, W.-K.; Lam, C.W.-K.; et al. Effect of paricalcitol on left ventricular mass and function in CKD—the OPERA trial. J. Am. Soc. Nephrol. 2014, 25, 175–186. [Google Scholar] [CrossRef]
- Petkovich, M.; Melnick, J.; White, J.; Tabash, S.; Strugnell, S.; Bishop, C.W. Modified-release oral calcifediol corrects vitamin D insufficiency with minimal CYP24A1 upregulation. J. Steroid Biochem. Mol. Biol. 2015, 148, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Sprague, S.M.; Crawford, P.W.; Melnick, J.Z.; Strugnell, S.A.; Ali, S.; Mangoo-Karim, R.; Lee, S.; Petkovich, P.M.; Bishop, C.W. Use of Extended-Release Calcifediol to Treat Secondary Hyperparathyroidism in Stages 3 and 4 Chronic Kidney Disease. Am. J. Nephrol. 2016, 44, 316–325. [Google Scholar] [CrossRef]
- Fadda, G.; Germain, M.J.; Broumand, V.; Nguyen, A.; McGarvey, N.; Gitlin, M.; Bishop, C.W.; Ashfaq, A. Real-World Assessment: Clinical Effectiveness and Safety of Extended-Release Calcifediol. Am. J. Nephrol. 2021, 52, 798–807. [Google Scholar] [CrossRef]
- Fernández-Martín, J.L.; Martínez-Camblor, P.; Dionisi, M.P.; Floege, J.; Ketteler, M.; London, G.; Locatelli, F.; Gorriz, J.L.; Rutkowski, B.; Ferreira, A.; et al. Improvement of mineral and bone metabolism markers is associated with better survival in haemodialysis patients: The COSMOS study. Nephrol. Dial. Transpl. 2015, 30, 1542–1551. [Google Scholar] [CrossRef] [Green Version]
- Pitt, B.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Bakris, G.L.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Schloemer, P.; et al. Cardiovascular Events with Finerenone in Kidney Disease and Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 2252–2263. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefánsson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.-F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- de Jong, M.A.; Petrykiv, S.I.; Laverman, G.D.; van Herwaarden, A.E.; de Zeeuw, D.; Bakker, S.J.L.; Heerspink, H.J.L.; de Borst, M.H. Effects of Dapagliflozin on Circulating Markers of Phosphate Homeostasis. Clin. J. Am. Soc. Nephrol. 2019, 14, 66–73. [Google Scholar] [CrossRef]
- Rau, M.; Thiele, K.; Hartmann, N.-U.K.; Möllmann, J.; Wied, S.; Hohl, M.; Marx, N.; Lehrke, M. Effects of empagliflozin on markers of calcium and phosphate homeostasis in patients with type 2 diabetes—Data from a randomized, placebo-controlled study. Bone Rep. 2022, 16, 101175. [Google Scholar] [CrossRef]
- Tomaschitz, A.; Verheyen, N.; Meinitzer, A.; Pieske, B.; Belyavskiy, E.; Brussee, H.; Haas, J.; März, W.; Pieske-Kraigher, E.; Verheyen, S.; et al. Effect of eplerenone on parathyroid hormone levels in patients with primary hyperparathyroidism: Results from the EPATH randomized, placebo-controlled trial. J. Hypertens. 2016, 34, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Ketteler, M.; Block, G.A.; Evenepoel, P.; Fukagawa, M.; Herzog, C.A.; McCann, L.; Moe, S.M.; Shroff, R.; Tonelli, M.A.; Toussaint, N.D.; et al. Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease-Mineral and Bone Disorder: Synopsis of the Kidney Disease: Improving Global Outcomes 2017 Clinical Practice Guideline Update. Ann. Intern. Med. 2018, 168, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Selamet, U.; Katz, R.; Ginsberg, C.; Rifkin, D.E.; Fried, L.F.; Kritchevsky, S.B.; Hoofnagle, A.N.; Bibbins-Domingo, K.; Drew, D.; Harris, T.; et al. Serum Calcitriol Concentrations and Kidney Function Decline, Heart Failure, and Mortality in Elderly Community-Living Adults: The Health, Aging, and Body Composition Study. Am. J. Kidney Dis. 2018, 72, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Isaka, Y.; Hamano, T.; Fujii, H.; Tsujimoto, Y.; Koiwa, F.; Sakaguchi, Y.; Tanaka, R.; Tomiyama, N.; Tatsugami, F.; Teramukai, S. Optimal Phosphate Control Related to Coronary Artery Calcification in Dialysis Patients. J. Am. Soc. Nephrol. 2021, 32, 723–735. [Google Scholar] [CrossRef]
- Dörr, K.; Kammer, M.; Reindl-Schwaighofer, R.; Lorenz, M.; Prikoszovich, T.; Marculescu, R.; Beitzke, D.; Wielandner, A.; Erben, R.G.; Oberbauer, R. Randomized trial of etelcalcetide for cardiac hypertrophy in hemodialysis. Circ. Res. 2021, 128, 1616–1625. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brandenburg, V.; Ketteler, M. Vitamin D and Secondary Hyperparathyroidism in Chronic Kidney Disease: A Critical Appraisal of the Past, Present, and the Future. Nutrients 2022, 14, 3009. https://doi.org/10.3390/nu14153009
Brandenburg V, Ketteler M. Vitamin D and Secondary Hyperparathyroidism in Chronic Kidney Disease: A Critical Appraisal of the Past, Present, and the Future. Nutrients. 2022; 14(15):3009. https://doi.org/10.3390/nu14153009
Chicago/Turabian StyleBrandenburg, Vincent, and Markus Ketteler. 2022. "Vitamin D and Secondary Hyperparathyroidism in Chronic Kidney Disease: A Critical Appraisal of the Past, Present, and the Future" Nutrients 14, no. 15: 3009. https://doi.org/10.3390/nu14153009
APA StyleBrandenburg, V., & Ketteler, M. (2022). Vitamin D and Secondary Hyperparathyroidism in Chronic Kidney Disease: A Critical Appraisal of the Past, Present, and the Future. Nutrients, 14(15), 3009. https://doi.org/10.3390/nu14153009