
Lipid Metabolism in Cartilage Development, Degeneration, and Regeneration


Abstract
:1. Introduction
2. Cartilage Development, Degeneration, and Regeneration
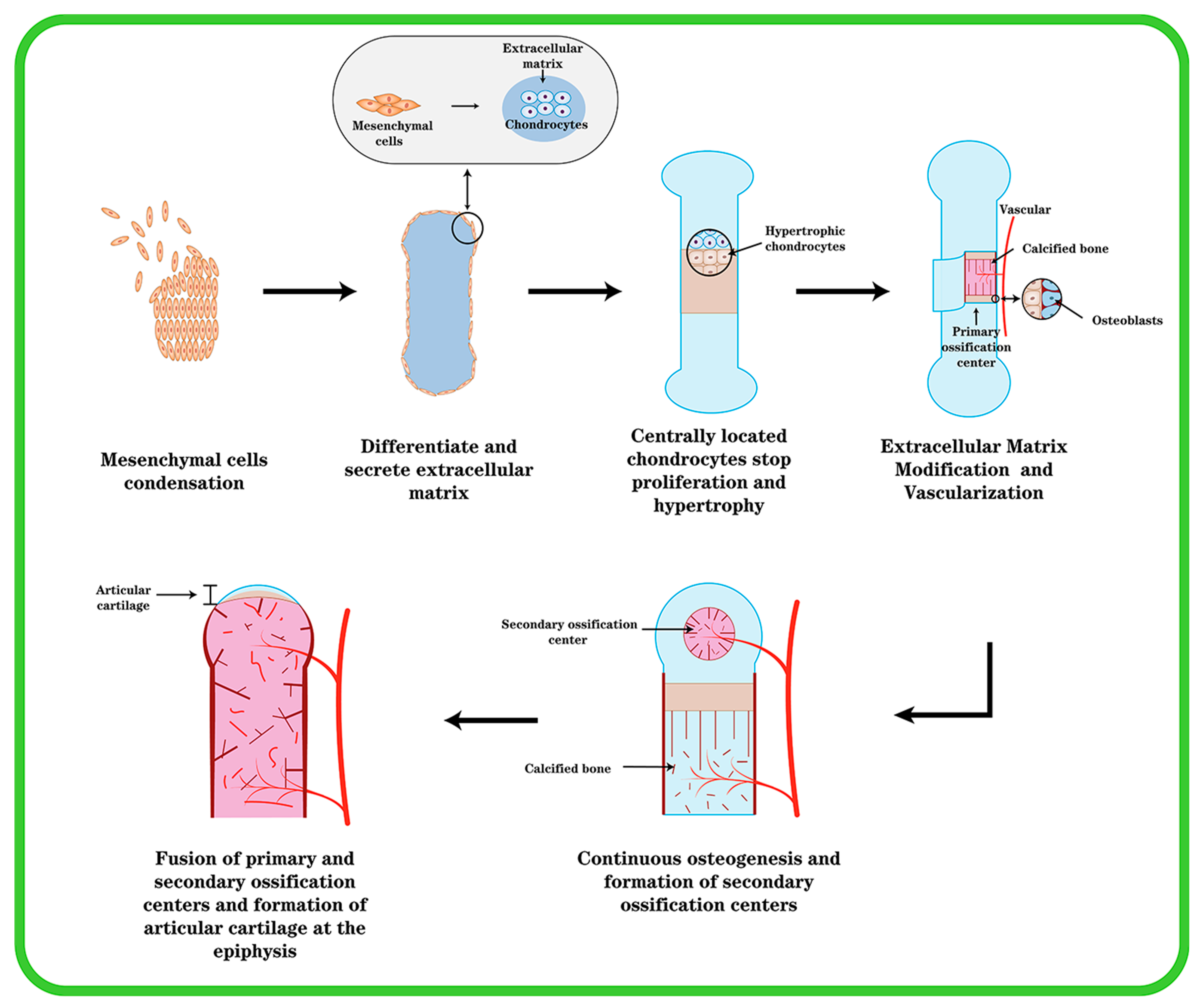
2.1. Chondrogenesis and Endochondral Ossification in Cartilage Development
2.2. Cartilage Homeostasis
2.3. Chondrocytes and Progenitor Cells in Cartilage Regeneration
2.4. ECMs and Chondrocytes in Cartilage Degeneration
3. Lipid Metabolism in Cartilage
3.1. Fatty Acid
3.2. Cholesterol Metabolism
3.3. Phospholipid Metabolism
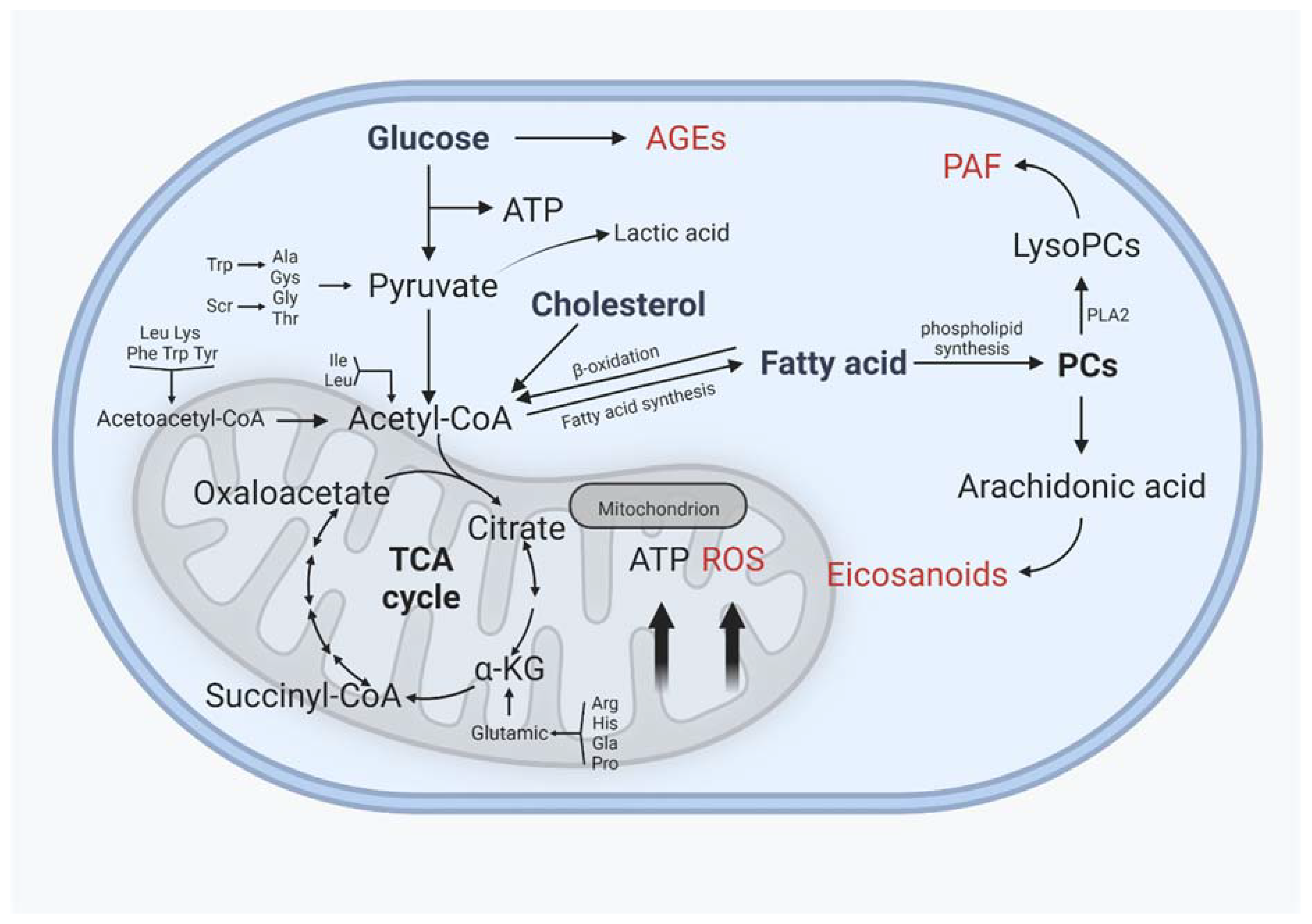
4. Interaction of Lipid Metabolism with Other Metabolisms
5. Lipid Metabolism and OA Treatment
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wachsmuth, L.; Söder, S.; Fan, Z.; Finger, F.; Aigner, T. Immunolocalization of matrix proteins in different human cartilage subtypes. Histol. Histopathol. 2006, 21, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the "omics" era. Matrix. Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Knudson, W.; Ishizuka, S.; Terabe, K.; Askew, E.B.; Knudson, C.B. The pericellular hyaluronan of articular chondrocytes. Matrix Biol. 2018, 78–79, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Tuan, R.S. Origin and function of cartilage stem/progenitor cells in osteoarthritis. Nat. Rev. Rheumatol. 2014, 11, 206–212. [Google Scholar] [CrossRef]
- Hunziker, E.B.; Quinn, T.M.; Häuselmann, H.-J. Quantitative structural organization of normal adult human articular cartilage. Osteoarthr. Cartil. 2002, 10, 564–572. [Google Scholar] [CrossRef]
- Archer, C.W.; Francis-West, P. The chondrocyte. Int. J. Biochem. Cell Biol. 2003, 35, 401–404. [Google Scholar] [CrossRef]
- Lin, Z.; Willers, C.; Xu, J.; Zheng, M.-H. The chondrocyte: Biology and clinical application. Tissue Eng. 2006, 12, 1971–1984. [Google Scholar] [CrossRef]
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; Seyama, Y.; Shaw, W.; et al. A comprehensive classification system for lipids. J. Lipid Res. 2005, 46, 839–861. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, K.; Yang, L.; Liu, R.; Chu, Y.; Qin, X.; Yang, P.; Yu, H. Lipid metabolism in inflammation-related diseases. Analyst 2018, 143, 4526–4536. [Google Scholar] [CrossRef]
- Schoeler, M.; Caesar, R. Dietary lipids, gut microbiota and lipid metabolism. Rev. Endocr. Metab. Disord. 2019, 20, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Horng, T. Lipid Metabolism in Regulation of Macrophage Functions. Trends Cell Biol. 2020, 30, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Erion, D.M.; Park, H.-J.; Lee, H.-Y. The role of lipids in the pathogenesis and treatment of type 2 diabetes and associated co-morbidities. BMB Rep. 2016, 49, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Villalvilla, A.; Gómez, R.; Largo, R.; Herrero-Beaumont, G. Lipid Transport and Metabolism in Healthy and Osteoarthritic Cartilage. Int. J. Mol. Sci. 2013, 14, 20793–20808. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Ornitz, D.M. Development of the Endochondral Skeleton. Cold Spring Harb. Perspect. Biol. 2013, 5, a008334. [Google Scholar] [CrossRef]
- DeLise, A.; Fischer, L.; Tuan, R. Cellular interactions and signaling in cartilage development. Osteoarthr. Cartil. 2000, 8, 309–334. [Google Scholar] [CrossRef]
- Karsenty, G.; Kronenberg, H.M.; Settembre, C. Genetic Control of Bone Formation. Annu. Rev. Cell Dev. Biol. 2009, 25, 629–648. [Google Scholar] [CrossRef]
- Mackie, E.J.; Ahmed, Y.A.; Tatarczuch, L.; Chen, K.-S.; Mirams, M. Endochondral ossification: How cartilage is converted into bone in the developing. Int. J. Biochem. Cell Biol. 2008, 40, 46–62. [Google Scholar] [CrossRef]
- Rim, Y.A.; Nam, Y.; Ju, J.H. The Role of Chondrocyte Hypertrophy and Senescence in Osteoarthritis Initiation and Progression. Int. J. Mol. Sci. 2020, 21, 2358. [Google Scholar] [CrossRef]
- Van Donkelaar, C.C.; Wilson, W. Mechanics of chondrocyte hypertrophy. Biomech. Model. Mechanobiol. 2011, 11, 655–664. [Google Scholar] [CrossRef]
- Van der Kraan, P.M.; van den Berg, W.B. Chondrocyte hypertrophy and osteoarthritis: Role in initiation and progression of cartilage degeneration? Osteoarthr. Cartil. 2012, 20, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, M.; Ohta, Y.; Larmour, C.; Enomoto-Iwamoto, M. Toward regeneration of articular cartilage. Birth Defects Res. Part C Embryo Today Rev. 2013, 99, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Heras, F.L.; Gahunia, H.K.; Pritzker, K.P. Articular Cartilage Development: A Molecular Perspective. Orthop. Clin. N. Am. 2012, 43, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Sun, H.; Bunpetch, V.; Koh, Y.; Wen, Y.; Wu, D.; Ouyang, H. The regulation of cartilage extracellular matrix homeostasis in joint cartilage degeneration and regeneration. Biomaterials 2020, 268, 120555. [Google Scholar] [CrossRef] [PubMed]
- Funaba, M.; Zimmerman, C.M.; Mathews, L.S. Modulation of Smad2-mediated signaling by extracellular signal-regulated kinase. J. Biol. Chem. 2002, 277, 41361–41368. [Google Scholar] [CrossRef] [PubMed]
- Ying, J.; Wang, P.; Zhang, S.; Xu, T.; Zhang, L.; Dong, R.; Xu, S.; Tong, P.; Wu, C.; Jin, H. Transforming growth factor-beta1 promotes articular cartilage repair through canonical Smad and Hippo pathways in bone mesenchymal stem cells. Life Sci. 2018, 192, 84–90. [Google Scholar] [CrossRef]
- ten Dijke, P.; Hill, C.S. New insights into TGF-beta-Smad signalling. Trends Biochem. Sci. 2004, 29, 265–273. [Google Scholar] [CrossRef]
- Davidson, E.N.B.; Remst, D.F.G.; Vitters, E.L.; van Beuningen, H.M.; Blom, A.B.; Goumans, M.J.; van den Berg, W.B.; van der Kraan, P.M. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in humans and mice. J. Immunol. 2009, 182, 7937–7945. [Google Scholar] [CrossRef]
- van der Kraan, P.; Davidson, E.B.; Berg, W.V.D. Bone Morphogenetic Proteins and articular cartilage: To serve and protect or a wolf in sheep clothing’s? Osteoarthr. Cartil. 2010, 18, 735–741. [Google Scholar] [CrossRef]
- Davidson, E.N.B.; Vitters, E.L.; Van Lent, P.L.; Van De Loo, F.A.; Berg, W.B.V.D.; Van Der Kraan, P.M. Elevated extracellular matrix production and degradation upon bone morphogenetic protein-2 (BMP-2) stimulation point toward a role for BMP-2 in cartilage repair and remodeling. Arthritis Res. Ther. 2007, 9, R102. [Google Scholar] [CrossRef]
- Elshaier, A.M.; Hakimiyan, A.A.; Rappoport, L.; Rueger, D.C.; Chubinskaya, S. Effect of interleukin-1beta on osteogenic protein 1-induced signaling in adult human articular chondrocytes. Arthritis Rheum. 2009, 60, 143–154. [Google Scholar] [CrossRef] [Green Version]
- Caron, M.; Emans, P.; Cremers, A.; Surtel, D.; Coolsen, M.; van Rhijn, L.; Welting, T. Hypertrophic differentiation during chondrogenic differentiation of progenitor cells is stimulated by BMP-2 but suppressed by BMP-7. Osteoarthr. Cartil. 2013, 21, 604–613. [Google Scholar] [CrossRef]
- Martin, J.A.; Buckwalter, J.A. Post-traumatic osteoarthritis: The role of stress induced chondrocyte damage. Biorheology 2006, 43, 517–521. [Google Scholar]
- Charlier, E.; Deroyer, C.; Ciregia, F.; Malaise, O.; Neuville, S.; Plener, Z.; Malaise, M.; de Seny, D. Chondrocyte dedifferentiation and osteoarthritis (OA). Biochem. Pharmacol. 2019, 165, 49–65. [Google Scholar] [CrossRef]
- Aigner, T.; Söder, S.; Gebhard, P.M.; McAlinden, A.; Haag, J. Mechanisms of disease: Role of chondrocytes in the pathogenesis of osteoarthritis—Structure, chaos and senescence. Nat. Clin. Pract. Rheumatol. 2007, 3, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Narcisi, R.; Cleary, M.A.; Sivasubramaniyan, K.; Brama, P.A.J.; van Osch, G.J.V.M. MSC Populations for Cartilage Regeneration. Cartilage 2017, 35–57. [Google Scholar] [CrossRef]
- Le, H.; Xu, W.; Zhuang, X.; Chang, F.; Wang, Y.; Ding, J. Mesenchymal stem cells for cartilage regeneration. J. Tissue Eng. 2020, 11. [Google Scholar] [CrossRef]
- Yu, Y.; Zheng, H.; Buckwalter, J.; Martin, J. Single cell sorting identifies progenitor cell population from full thickness bovine articular cartilage. Osteoarthr. Cartil. 2014, 22, 1318–1326. [Google Scholar] [CrossRef]
- Kurth, T.B.; Dell'Accio, F.; Crouch, V.; Augello, A.; Sharpe, P.T.; De Bari, C. Functional mesenchymal stem cell niches in adult mouse knee joint synovium in vivo. Arthritis Care Res. 2011, 63, 1289–1300. [Google Scholar] [CrossRef]
- Roelofs, A.J.; Zupan, J.; Riemen, A.H.K.; Kania, K.; Ansboro, S.; White, N.; Clark, S.M.; De Bari, C. Joint morphogenetic cells in the adult mammalian synovium. Nat. Commun. 2017, 8, 15040. [Google Scholar] [CrossRef]
- Koh, Y.-G.; Choi, Y.-J. Infrapatellar fat pad-derived mesenchymal stem cell therapy for knee osteoarthritis. Knee 2012, 19, 902–907. [Google Scholar] [CrossRef]
- Mantripragada, V.P.; Piuzzi, N.S.; Bova, W.A.; Boehm, C.; Obuchowski, N.A.; Lefebvre, V.; Midura, R.J.; Muschler, G.F. Donor-matched comparison of chondrogenic progenitors resident in human infrapatellar fat pad, synovium, and periosteum - implications for cartilage repair. Connect. Tissue Res. 2019, 60, 597–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGonagle, D.; Baboolal, T.; Jones, E. Native joint-resident mesenchymal stem cells for cartilage repair in osteoarthritis. Nat. Rev. Rheumatol. 2017, 13, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, I.; Ojima, M.; Suzuki, S.; Yamaga, M.; Horie, M.; Koga, H.; Tsuji, K.; Miyaguchi, K.; Ogishima, S.; Tanaka, H.; et al. Human mesenchymal stem cells in synovial fluid increase in the knee with degenerated cartilage and osteoarthritis. J. Orthop. Res. 2011, 30, 943–949. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.; Andreas, K.; Kalwitz, G.; Freymann, U.; Neumann, K.; Ringe, J.; Sittinger, M.; Häupl, T.; Kaps, C. Chemokine profile of synovial fluid from normal, osteoarthritis and rheumatoid arthritis patients: CCL25, CXCL10 and XCL1 recruit human subchondral mesenchymal progenitor cells. Osteoarthr. Cartil. 2010, 18, 1458–1466. [Google Scholar] [CrossRef]
- Kawai, H.; Sakamoto, F.; Taguchi, M.; Kitamura, M.; Sotomura, M.; Tsukamoto, G. 2-Oxo-1,3-dioxoles as specific substrates for measurement of arylesterase activity. Chem. Pharm. Bull 1991, 39, 1422–1425. [Google Scholar] [CrossRef]
- Marchan, J.; Wittig, O.; Diaz-Solano, D.; Gomez, M.; Cardier, J.E. Enhanced chondrogenesis from chondrocytes co-cultured on mesenchymal stromal cells: Implication for cartilage repair. Injury 2021, 53, 399–407. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, Y.; Wang, Q.; Fang, C.; Sun, Y.; Yuan, T.; Wang, Y.; Bao, R.; Zhao, N. Effect of bone marrow-derived stem cells on chondrocytes from patients with osteoarthritis. Mol. Med. Rep. 2015, 13, 1795–1800. [Google Scholar] [CrossRef]
- Xu, L.; Wu, Y.; Xiong, Z.; Zhou, Y.; Ye, Z.; Tan, W.-S. Mesenchymal Stem Cells Reshape and Provoke Proliferation of Articular Chondrocytes by Paracrine Secretion. Sci. Rep. 2016, 6, 32705. [Google Scholar] [CrossRef]
- Glyn-Jones, S.; Palmer, A.J.R.; Agricola, R.; Price, A.J.; Vincent, T.L.; Weinans, H.; Carr, A.J. Osteoarthritis. Lancet 2015, 386, 376–387. [Google Scholar] [CrossRef]
- O'Neill, T.W.; McCabe, P.S.; McBeth, J. Update on the epidemiology, risk factors and disease outcomes of osteoarthritis. Best Pr. Res. Clin. Rheumatol. 2018, 32, 312–326. [Google Scholar] [CrossRef]
- Huber-Lang, M.; Denk, S.; Fulda, S.; Erler, E.; Kalbitz, M.; Weckbach, S.; Schneider, E.M.; Weiss, M.; Kanse, S.M.; Perl, M. Cathepsin D is released after severe tissue trauma in vivo and is capable of generating C5a in vitro. Mol. Immunol. 2012, 50, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Burrage, P.S.; Mix, K.S.; Brinckerhoff, C.E. Matrix Metalloproteinases: Role In Arthritis. Front. Biosci. 2006, 11, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Liu, D.; Song, C.; Li, D.; Zhang, X.; Horecny, I.; Zhang, F.; Yan, Y.; Zhuang, L.; Li, J.; et al. Discovery of Isoindoline Amide Derivatives as Potent and Orally Bioavailable ADAMTS-4/5 Inhibitors for the Treatment of Osteoarthritis. ACS Pharmacol. Transl. Sci. 2022, 5, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Pratta, M.A.; Yao, W.; Decicco, C.; Tortorella, M.D.; Liu, R.-Q.; Copeland, R.A.; Magolda, R.; Newton, R.C.; Trzaskos, J.M.; Arner, E.C. Aggrecan Protects Cartilage Collagen from Proteolytic Cleavage. J. Biol. Chem. 2003, 278, 45539–45545. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Madsen, S.H.; Christiansen, C.; Henriksen, K.; Fosang, A.J.; Sondergaard, B.C. Cartilage degradation is fully reversible in the presence of aggrecanase but not matrix metalloproteinase activity. Arthritis Res. Ther. 2008, 10, R63. [Google Scholar] [CrossRef]
- Struglics, A.; Larsson, S.; Pratta, M.; Kumar, S.; Lark, M.; Lohmander, L. Human osteoarthritis synovial fluid and joint cartilage contain both aggrecanase- and matrix metalloproteinase-generated aggrecan fragments. Osteoarthr. Cartil. 2006, 14, 101–113. [Google Scholar] [CrossRef]
- Hu, Q.; Ecker, M. Overview of MMP-13 as a Promising Target for the Treatment of Osteoarthritis. Int. J. Mol. Sci. 2021, 22, 1742. [Google Scholar] [CrossRef]
- Kevorkian, L.; Young, D.A.; Darrah, C.; Donell, S.T.; Shepstone, L.; Porter, S.; Brockbank, S.M.V.; Edwards, D.R.; Parker, A.E.; Clark, I.M. Expression profiling of metalloproteinases and their inhibitors in cartilage. Arthritis Care Res. 2004, 50, 131–141. [Google Scholar] [CrossRef]
- Kozawa, E.; Cheng, X.W.; Urakawa, H.; Arai, E.; Yamada, Y.; Kitamura, S.; Sato, K.; Kuzuya, M.; Ishiguro, N.; Nishida, Y. Increased expression and activation of cathepsin K in human osteoarthritic cartilage and synovial tissues. J. Orthop. Res. 2015, 34, 127–134. [Google Scholar] [CrossRef]
- Aigner, T.; Kurz, B.; Fukui, N.; Sandell, L. Roles of chondrocytes in the pathogenesis of osteoarthritis. Curr. Opin. Rheumatol. 2002, 14, 578–584. [Google Scholar] [CrossRef]
- Goldring, S.R.; Goldring, M.B. Changes in the osteochondral unit during osteoarthritis: Structure, function and cartilage–bone crosstalk. Nat. Rev. Rheumatol. 2016, 12, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Valcourt, U.; Gouttenoire, J.; Aubert-Foucher, E.; Herbage, D.; Mallein-Gerin, F. Alternative splicing of type II procollagen pre-mRNA in chondrocytes is oppositely regulated by BMP-2 and TGF-β1. FEBS Lett. 2003, 545, 115–119. [Google Scholar] [CrossRef]
- Zheng, L.; Zhang, Z.; Sheng, P.; Mobasheri, A. The role of metabolism in chondrocyte dysfunction and the progression of osteoarthritis. Ageing Res. Rev. 2020, 66, 101249. [Google Scholar] [CrossRef]
- Santos, A.L.; Preta, G. Lipids in the cell: Organisation regulates function. Experientia 2018, 75, 1909–1927. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Eicosanoids. Essays Biochem. 2020, 64, 423–441. [Google Scholar] [PubMed]
- Maldonado-Valderrama, J.; Wilde, P.; Macierzanka, A.; Mackie, A. The role of bile salts in digestion. Adv. Colloid Interface Sci. 2011, 165, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, M.; Rolland, G.; Léonce, S.; Thomas, M.; Lesur, C.; Pérez, V.; de Nanteuil, G.; Bonnet, J. Effects of Ceramide on Apoptosis, Proteoglycan Degradation, and Matrix Metalloproteinase Expression in Rabbit Articular Cartilage. Biochem. Biophys. Res. Commun. 2000, 267, 438–444. [Google Scholar] [CrossRef]
- Sabatini, M.; Rolland, G.; Léonce, S.; Thomas, M.; Lesur, C.; Pérez, V.; de Nanteuil, G.; Bonnet, J. Isolated effects of external bath osmolality, solute concentration, and electrical charge on solute transport across articular cartilage. Med. Eng. Phys. 2016, 38, 1399–1407. [Google Scholar]
- Wang, Y.; Wei, L.; Zeng, L.; He, D.; Wei, X. Nutrition and degeneration of articular cartilage. Knee Surg. Sports Traumatol. Arthrosc. 2012, 21, 1751–1762. [Google Scholar] [CrossRef]
- Arkill, K.P.; Winlove, C.P. Fatty acid transport in articular cartilage. Arch. Biochem. Biophys. 2006, 456, 71–78. [Google Scholar] [CrossRef]
- Silva, M.O.; Gregory, J.L.; Ansari, N.; Stok, K.S. Molecular Signaling Interactions and Transport at the Osteochondral Interface: A Review. Front. Cell Dev. Biol. 2020, 8, 750. [Google Scholar] [CrossRef] [PubMed]
- Pouran, B.; Arbabi, V.; Bleys, R.L.; van Weeren, P.R.; Zadpoor, A.A.; Weinans, H. Solute transport at the interface of cartilage and subchondral bone plate: Effect of micro-architecture. J. Biomech. 2017, 52, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Sekar, S.; Shafie, S.R.; Prasadam, I.; Crawford, R.; Panchal, S.K.; Brown, L.; Xiao, Y. Saturated fatty acids induce development of both metabolic syndrome and osteoarthritis in rats. Sci. Rep. 2017, 7, 46457. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Baek, I.J.; Ryu, J.H.; Chun, C.H.; Jin, E.J. PPARalpha-ACOT12 axis is responsible for maintaining cartilage homeostasis through modulating de novo lipogenesis. Nat. Commun. 2022, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Jain, D.; McNeill, J.N.; Little, D.; Anderson, J.A.; Huebner, J.L.; Kraus, V.B.; Rodriguiz, R.M.; Wetsel, W.C.; Guilak, F. Dietary fatty acid content regulates wound repair and the pathogenesis of osteoarthritis following joint injury. Ann. Rheum Dis. 2015, 74, 2076–2083. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.F.; Infante, J.R. Molecular Pathways: Fatty Acid Synthase. Clin. Cancer Res. 2015, 21, 5434–5438. [Google Scholar] [CrossRef]
- Abu-Elheiga, L.; Matzuk, M.M.; Abo-Hashema, K.A.; Wakil, S.J. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science 2001, 291, 2613–2616. [Google Scholar] [CrossRef]
- Ichimura, A.; Hasegawa, S.; Kasubuchi, M.; Kimura, I. Free fatty acid receptors as therapeutic targets for the treatment of diabetes. Front. Pharmacol. 2014, 5, 236. [Google Scholar] [CrossRef]
- Miao, H.; Chen, L.; Hao, L.; Zhang, X.; Chen, Y.; Ruan, Z.; Liang, H. Stearic acid induces proinflammatory cytokine production partly through activation of lactate-HIF1alpha pathway in chondrocytes. Sci. Rep. 2015, 5, 13092. [Google Scholar] [CrossRef]
- Alvarez-Garcia, O.; Rogers, N.H.; Smith, R.G.; Lotz, M.K. Palmitate has proapoptotic and proinflammatory effects on articular cartilage and synergizes with interleukin-1. Arthritis Rheumatol. 2014, 66, 1779–1788. [Google Scholar] [CrossRef]
- Medina-Luna, D.; Santamaria-Olmedo, M.G.; Zamudio-Cuevas, Y.; Martinez-Flores, K.; Fernandez-Torres, J.; Martinez-Nava, G.A.; Clavijo-Cornejo, D.; Hernandez-Diaz, C.; Olivos-Meza, A.; Gomez-Quiroz, L.E.; et al. Hyperlipidemic microenvironment conditionates damage mechanisms in human chondrocytes by oxidative stress. Lipids Health Dis. 2017, 16, 114. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Mechanisms of Action of (n-3) Fatty Acids. J. Nutr. 2012, 142, 592S–599S. [Google Scholar] [CrossRef] [Green Version]
- Martindale, R.G.; Warren, M.M.; McClave, S.A. Does the use of specialized proresolving molecules in critical care offer a more focused approach to controlling inflammation than that of fish oils? Curr. Opin. Clin. Nutr. Metab Care 2016, 19, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Patterson, E.; Wall, R.; Fitzgerald, G.F.; Ross, R.; Stanton, C. Health Implications of High Dietary Omega-6 Polyunsaturated Fatty Acids. J. Nutr. Metab. 2012, 2012, 539426. [Google Scholar] [CrossRef] [PubMed]
- Adkisson, H.D., 4th; Risener Jr, F.S.; Zarrinkar, P.P.; Walla, M.D.; Christie, W.W.; Wuthier, R.E. Unique fatty acid composition of normal cartilage: Discovery of high levels of n-6 polyunsaturated fatty acids. FASEB J. 1991, 5, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Ioan-Facsinay, A.; Kloppenburg, M. Bioactive lipids in osteoarthritis: Risk or benefit? Curr. Opin. Rheumatol. 2018, 30, 108–113. [Google Scholar]
- Attur, M.; Krasnokutsky, S.; Statnikov, A.; Samuels, J.; Li, Z.; Friese, O.; Le Graverand-Gastineau, M.-P.H.; Rybak, L.; Kraus, V.B.; Jordan, J.M.; et al. Low-Grade Inflammation in Symptomatic Knee Osteoarthritis: Prognostic Value of Inflammatory Plasma Lipids and Peripheral Blood Leukocyte Biomarkers. Arthritis Rheumatol. 2015, 67, 2905–2915. [Google Scholar] [CrossRef]
- Miyamoto, M.; Ito, H.; Mukai, S.; Kobayashi, T.; Yamamoto, H.; Kobayashi, M.; Maruyama, T.; Akiyama, H.; Nakamura, T. Simultaneous stimulation of EP2 and EP4 is essential to the effect of prostaglandin E2in chondrocyte differentiation. Osteoarthr. Cartil. 2003, 11, 644–652. [Google Scholar] [CrossRef]
- Nishitani, K.; Ito, H.; Hiramitsu, T.; Tsutsumi, R.; Tanida, S.; Kitaori, T.; Yoshitomi, H.; Kobayashi, M.; Nakamura, T. PGE2 inhibits MMP expression by suppressing MKK4-JNK MAP kinase-c-JUN pathway via EP4 in human articular chondrocytes. J. Cell Biochem. 2010, 109, 425–433. [Google Scholar] [CrossRef]
- Alvarez-Soria, M.A.; Largo, R.; Sanchez-Pernaute, O.; Calvo, E.; Egido, J.; Herrero-Beaumont, G. Prostaglandin E2 receptors EP1 and EP4 are up-regulated in rabbit chondrocytes by IL-1beta, but not by TNFalpha. Rheumatol. Int. 2007, 27, 911–917. [Google Scholar] [CrossRef]
- Tuure, L.; Hämäläinen, M.; Nummenmaa, E.; Moilanen, T.; Moilanen, E. Downregulation of microsomal prostaglandin E synthase-1 (mPGES-1) expression in chondrocytes is regulated by MAP kinase phosphatase-1 (MKP-1). Int. Immunopharmacol. 2019, 71, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Soccio, R.E.; Breslow, J.L. Intracellular Cholesterol Transport. Arter. Thromb. Vasc. Biol. 2004, 24, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Cerqueira, N.M.F.S.A.; Oliveira, E.F.; Gesto, D.S.; Santos-Martins, D.; Moreira, C.; Moorthy, H.N.; Ramos, M.J.; Fernandes, P.A. Cholesterol Biosynthesis: A Mechanistic Overview. Biochemistry 2016, 55, 5483–5506. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhang, Z.; Shen, W.-J.; Azhar, S. Cellular cholesterol delivery, intracellular processing and utilization for biosynthesis of steroid hormones. Nutr. Metab. 2010, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Vallim, T.Q.D.A.; Tarling, E.J.; Edwards, P.A. Pleiotropic Roles of Bile Acids in Metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef]
- Schafer, W.R.; Trueblood, C.E.; Yang, C.-C.; Mayer, M.P.; Rosenberg, S.; Poulter, C.D.; Kim, S.-H.; Rine, J. Enzymatic Coupling of Cholesterol Intermediates to a Mating Pheromone Precursor and to the Ras Protein. Science 1990, 249, 1133–1139. [Google Scholar] [CrossRef]
- Incardona, J.P.; Eaton, S. Cholesterol in signal transduction. Curr. Opin. Cell Biol. 2000, 12, 193–203. [Google Scholar] [CrossRef]
- Girão, H.; Pereira, P.; Ramalho, J.; Quinlan, R.; Prescott, A. Cholesterol oxides mediated changes in cytoskeletal organisation involves Rho GTPases. Exp. Cell Res. 2003, 291, 502–513. [Google Scholar] [CrossRef]
- Wu, S.; De Luca, F. Role of cholesterol in the regulation of growth plate chondrogenesis and longitudinal bone growth. J. Biol. Chem. 2004, 279, 4642–4647. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler Thromb Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef]
- Parthasarathy, S.; Raghavamenon, A.; Garelnabi, M.O.; Santanam, N. Oxidized low-density lipoprotein. Methods Mol. Biol. 2010, 610, 403–417. [Google Scholar] [PubMed]
- Choi, M.-C.; Jo, J.; Park, J.; Kang, H.K.; Park, Y. NF-κB Signaling Pathways in Osteoarthritic Cartilage Destruction. Cells 2019, 8, 734. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Akagi, M.; Yoshida, K.; Hayakawa, S.; Sawamura, T.; Munakata, H.; Hamanishi, C. Oxidized low-density lipoprotein (ox-LDL) binding to lectin-like ox-LDL receptor-1 (LOX-1) in cultured bovine articular chondrocytes increases production of intracellular reactive oxygen species (ROS) resulting in the activation of NF-kappaB. Osteoarthr. Cartil. 2004, 12, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Kishimoto, H.; Akagi, M.; Zushi, S.; Teramura, T.; Onodera, Y.; Sawamura, T.; Hamanishi, C. Induction of hypertrophic chondrocyte-like phenotypes by oxidized LDL in cultured bovine articular chondrocytes through increase in oxidative stress. Osteoarthr. Cartil. 2010, 18, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Pearle, A.D.; Scanzello, C.R.; George, S.; Mandl, L.S.; DiCarlo, E.F.; Peterson, M.; Sculco, T.P.; Crow, M.K.C. Elevated high-sensitivity C-reactive protein levels are associated with local inflammatory findings in patients with osteoarthritis. Osteoarthr. Cartil. 2007, 15, 516–523. [Google Scholar] [CrossRef]
- Tsezou, A.; Iliopoulos, D.; Malizos, K.N.; Simopoulou, T. Impaired expression of genes regulating cholesterol efflux in human osteoarthritic chondrocytes. J. Orthop. Res. 2010, 28, 1033–1039. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Wang, N.; Tall, A.R. Role of HDL, ABCA1, and ABCG1 Transporters in Cholesterol Efflux and Immune Responses. Arter. Thromb. Vasc. Biol. 2010, 30, 139–143. [Google Scholar] [CrossRef]
- De Seny, D.; Cobraiville, G.; Charlier, E.; Neuville, S.; Esser, N.; Malaise, D.; Malaise, O.; Calvo, F.Q.; Relic, B.; Malaise, M.G. Acute-Phase Serum Amyloid A in Osteoarthritis: Regulatory Mechanism and Proinflammatory Properties. PLoS ONE 2013, 8, e66769. [Google Scholar] [CrossRef]
- Gentili, C.; Tutolo, G.; Pianezzi, A.; Cancedda, R.; Descalzi Cancedda, F. Cholesterol secretion and homeostasis in chondrocytes: A liver X receptor and retinoid X receptor heterodimer mediates apolipoprotein A1 expression. Matrix Biol. 2005, 24, 35–44. [Google Scholar] [CrossRef]
- de Seny, D.; Cobraiville, G.; Charlier, E.; Neuville, S.; Lutteri, L.; Le Goff, C.; Malaise, D.; Malaise, O.; Chapelle, J.P.; Relic, B.; et al. Apolipoprotein-A1 as a damage-associated molecular patterns protein in osteoarthritis: Ex vivo and in vitro pro-inflammatory properties. PLoS ONE 2015, 10, e0122904. [Google Scholar] [CrossRef]
- Farnaghi, S.; Crawford, R.; Xiao, Y.; Prasadam, I. Cholesterol metabolism in pathogenesis of osteoarthritis disease. Int. J. Rheum. Dis. 2017, 20, 131–140. [Google Scholar] [CrossRef] [PubMed]
- De Munter, W.; Blom, A.B.; Helsen, M.M.; Walgreen, B.; Van Der Kraan, P.M.; Ab Joosten, L.; Berg, W.B.V.D.; Van Lent, P.L. Cholesterol accumulation caused by low density lipoprotein receptor deficiency or a cholesterol-rich diet results in ectopic bone formation during experimental osteoarthritis. Arthritis Res. Ther. 2013, 15, R178. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.S.; Lee, G.; Song, W.H.; Koh, J.T.; Yang, J.; Kwak, J.S.; Kim, H.E.; Kim, S.K.; Son, Y.O.; Nam, H.; et al. The CH25H-CYP7B1-RORalpha axis of cholesterol metabolism regulates osteoarthritis. Nature 2019, 566, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.M.-G.; Beier, F. Liver X Receptor activation regulates genes involved in lipid homeostasis in developing chondrocytes. Osteoarthr. Cartil. Open 2020, 2, 100030. [Google Scholar] [CrossRef]
- Kostopoulou, F.; Gkretsi, V.; Malizos, K.N.; Iliopoulos, D.; Oikonomou, P.; Poultsides, L.; Tsezou, A. Central Role of SREBP-2 in the Pathogenesis of Osteoarthritis. PLoS ONE 2012, 7, e35753. [Google Scholar] [CrossRef]
- Kostopoulou, F.; Malizos, K.N.; Papathanasiou, I.; Tsezou, A. MicroRNA-33a regulates cholesterol synthesis and cholesterol efflux-related genes in osteoarthritic chondrocytes. Arthritis Res. Ther. 2015, 17, 42. [Google Scholar] [CrossRef]
- Smith, A.E.; Sigurbjörnsdóttir, E.S.; Steingrímsson, E.; Sigurbjörnsdóttir, S. Hedgehog signalling in bone and osteoarthritis: The role of Smoothened and cholesterol. FEBS J. 2022, 19, 16440. [Google Scholar] [CrossRef]
- Bernstein, P.; Sticht, C.; Jacobi, A.; Liebers, C.; Manthey, S.; Stiehler, M. Expression pattern differences between osteoarthritic chondrocytes and mesenchymal stem cells during chondrogenic differentiation. Osteoarthr. Cartil. 2010, 18, 1596–1607. [Google Scholar] [CrossRef]
- Fagone, P.; Jackowski, S. Membrane phospholipid synthesis and endoplasmic reticulum function. J. Lipid Res. 2009, 50, S311–S316. [Google Scholar] [CrossRef]
- Vance, J.E. Phospholipid Synthesis and Transport in Mammalian Cells. Traffic 2015, 16, 1–18. [Google Scholar] [CrossRef]
- Zhai, G.; Pelletier, J.-P.; Liu, M.; Aitken, D.; Randell, E.; Rahman, P.; Jones, G.; Martel-Pelletier, J. Activation of the Phosphatidylcholine to Lysophosphatidylcholine Pathway is Associated with Osteoarthritis Knee Cartilage Volume Loss Over Time. Sci. Rep. 2019, 9, 019–46185. [Google Scholar] [CrossRef] [PubMed]
- Tabe, S.; Hikiji, H.; Ariyoshi, W.; Hashidate-Yoshida, T.; Shindou, H.; Shimizu, T.; Okinaga, T.; Seta, Y.; Tominaga, K.; Nishihara, T. Lysophosphatidylcholine acyltransferase 4 is involved in chondrogenic differentiation of ATDC5 cells. Sci. Rep. 2017, 7, 16701. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Lee, H.Y.; Kwak, J.Y.; Park, J.I.; Yun, J.; Bae, Y.S. Sphingosine-1-phosphate stimulates rat primary chondrocyte proliferation. Biochem. Biophys. Res. Commun. 2006, 345, 67–73. [Google Scholar] [CrossRef]
- Garfinkel, R.J.; Dilisio, M.F.; Agrawal, D.K. Vitamin D and Its Effects on Articular Cartilage and Osteoarthritis. Orthop. J. Sports Med. 2017, 5. [Google Scholar] [CrossRef]
- Prabhu, A.V.; Luu, W.; Sharpe, L.J.; Brown, A.J. Cholesterol-mediated Degradation of 7-Dehydrocholesterol Reductase Switches the Balance from Cholesterol to Vitamin D Synthesis. J. Biol. Chem. 2016, 291, 8363–8373. [Google Scholar] [CrossRef]
- Asano, L.; Watanabe, M.; Ryoden, Y.; Usuda, K.; Yamaguchi, T.; Khambu, B.; Takashima, M.; Sato, S.-I.; Sakai, J.; Nagasawa, K.; et al. Vitamin D Metabolite, 25-Hydroxyvitamin D, Regulates Lipid Metabolism by Inducing Degradation of SREBP/SCAP. Cell Chem. Biol. 2017, 24, 207–217. [Google Scholar] [CrossRef]
- Martínez-Sena, T.; Soluyanova, P.; Guzmán, C.; Valdivielso, J.M.; Castell, J.V.; Jover, R. The Vitamin D Receptor Regulates Glycerolipid and Phospholipid Metabolism in Human Hepatocytes. Biomolecules 2020, 10, 493. [Google Scholar] [CrossRef]
- Rai, V.; Dietz, N.E.; Dilisio, M.F.; Radwan, M.M.; Agrawal, D.K. Vitamin D attenuates inflammation, fatty infiltration, and cartilage loss in the knee of hyperlipidemic microswine. Arthritis Res. Ther. 2016, 18, 203. [Google Scholar] [CrossRef]
- Pascual-Garrido, C.; Angeline, M.E.; Ma, R.; Chahla, J.; Voigt, C.; Deng, X.H.; Nguyen, J.; Warren, R.F.; Rodeo, S.A. Low Levels of Vitamin D have a Deleterious Effect on the Articular Cartilage in a Rat Model. HSS J. Musculoskelet. J. Hosp. Spéc. Surg. 2016, 12, 150–157. [Google Scholar] [CrossRef]
- Li, S.; Niu, G.; Dong, X.N.; Liu, Z.; Song, C.; Leng, H. Vitamin D Inhibits Activities of Metalloproteinase-9/-13 in Articular Cartilage. J. Nutr. Sci. Vitaminol. 2019, 65, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Jhun, J.; Woo, J.S.; Kwon, J.Y.; Na, H.S.; Cho, K.-H.; Kim, S.A.; Kim, S.J.; Moon, S.-J.; Park, S.-H.; Cho, M.-L. Vitamin D Attenuates Pain and Cartilage Destruction in OA Animals via Enhancing Autophagic Flux and Attenuating Inflammatory Cell Death. Immune Netw. 2022, 22, e34. [Google Scholar] [CrossRef] [PubMed]
- Rai, V.; Radwan, M.M.; Agrawal, D.K. IL-33, IL-37, and Vitamin D Interaction Mediate Immunomodulation of Inflammation in Degenerating Cartilage. Antibodies 2021, 10, 41. [Google Scholar] [CrossRef]
- Mobasheri, A. Glucose: An energy currency and structural precursor in articular cartilage and bone with emerging roles as an extracellular signaling molecule and metabolic regulator. Front. Endocrinol. 2012, 3, 00153. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Abel, E.D.; Long, F. Glucose metabolism induced by Bmp signaling is essential for murine skeletal. Nat. Commun. 2018, 9, 4831. [Google Scholar] [CrossRef]
- Rosa, S.C.; Gonçalves, J.; Judas, F.; Mobasheri, A.; Lopes, C.; Mendes, A.F. Impaired glucose transporter-1 degradation and increased glucose transport and oxidative stress in response to high glucose in chondrocytes from osteoarthritic versus normal human cartilage. Arthritis Res. Ther. 2009, 11, R80. [Google Scholar] [CrossRef]
- Rogatzki, M.J.; Ferguson, B.S.; Goodwin, M.L.; Gladden, L.B. Lactate is always the end product of glycolysis. Front. Neurosci. 2015, 9, 22. [Google Scholar] [CrossRef]
- Mookerjee, S.A.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 2017, 292, 7189–7207. [Google Scholar] [CrossRef]
- Martin, J.; Martini, A.; Molinari, A.; Morgan, W.; Ramalingam, W.; Buckwalter, J.; McKinley, T. Mitochondrial electron transport and glycolysis are coupled in articular cartilage. Osteoarthr. Cartil. 2012, 20, 323–329. [Google Scholar] [CrossRef]
- Otte, P. Basic cell metabolism of articular cartilage. Manometric studies. Z. Rheumatol. 1991, 50, 304–312. [Google Scholar]
- Alfano, B.; Barretta, L.; Del Giudice, A.; De Vito, S.; Di Francia, G.; Esposito, E.; Formisano, F.; Massera, E.; Miglietta, M.L.; Polichetti, T. A Review of Low-Cost Particulate Matter Sensors from the Depvelopers’ Perspectives. Sensors 2020, 20, 6819. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.B., Jr.; Setton, L.A.; Kensicki, E.; Bolognesi, M.P.; Toth, A.P.; Nettles, D.L. Global metabolic profiling of human osteoarthritic synovium. Osteoarthr. Cartil. 2012, 20, 64–67. [Google Scholar] [CrossRef]
- Leistad, L.; Feuerherm, A.; Faxvaag, A.; Johansen, B. Multiple phospholipase A2 enzymes participate in the inflammatory process in osteoarthritic cartilage. Scand. J. Rheumatol. 2011, 40, 308–316. [Google Scholar] [CrossRef]
- Tootsi, K.; Kals, J.; Zilmer, M.; Paapstel, K.; Ottas, A.; Märtson, A. Medium- and long-chain acylcarnitines are associated with osteoarthritis severity and arterial stiffness in end-stage osteoarthritis patients: A case-control study. Int. J. Rheum. Dis. 2018, 21, 1211–1218. [Google Scholar] [CrossRef]
- Yang, X.; Chen, W.; Zhao, X.; Chen, L.; Li, W.; Ran, J.; Wu, L. Pyruvate Kinase M2 Modulates the Glycolysis of Chondrocyte and Extracellular Matrix in Osteoarthritis. DNA Cell Biol. 2018, 37, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, T.; Sousa-Valente, J.; Malcangio, M. The Mon iodoacetate Model of Osteoarthritis Pain in the Mouse. J. Vis. Exp. 2016, e53746. [Google Scholar] [CrossRef]
- Das, R.; van Osch, G.; Kreukniet, M.; Oostra, J.; Weinans, H.; Jahr, H. Effects of individual control of pH and hypoxia in chondrocyte culture. J. Orthop. Res. 2009, 28, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.-S.; Yang, J.-I.; Kim, W.; Kim, H.-E.; Kim, S.-K.; Won, Y.; Son, Y.-O.; Chun, C.-H.; Chun, J.-S. Critical role for arginase II in osteoarthritis pathogenesis. Ann. Rheum. Dis. 2019, 78, 421–428. [Google Scholar] [CrossRef]
- Farnaghi, S.; Prasadam, I.; Cai, G.; Friis, T.; Du, Z.; Crawford, R.; Mao, X.; Xiao, Y. Protective effects of mitochondria-targeted antioxidants and statins on cholesterol-induced osteoarthritis. FASEB J. 2017, 31, 356–367. [Google Scholar] [CrossRef]
- Wang, Z.; Ni, S.; Zhang, H.; Fan, Y.; Xia, L.; Li, N. Silencing SGK1 alleviates osteoarthritis through epigenetic regulation of CREB1 and ABCA1 expression. Life Sci. 2020, 268, 118733. [Google Scholar] [CrossRef]
- He, H.; Lu, M.; Shi, H.; Yue, G.; Luo, H. Vaspin regulated cartilage cholesterol metabolism through miR155/LXRα and participated in the occurrence of osteoarthritis in rats. Life Sci. 2021, 269, 119096. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Yan, L.; Luo, L.; Gui, T.; Jang, B.; Amirshaghaghi, A.; You, T.; Tsourkas, A.; Qin, L.; Cheng, Z. Phospholipase A 2 inhibitor–loaded micellar nanoparticles attenuate inflammation and mitigate osteoarthritis progression. Sci. Adv. 2021, 7, eabe6374. [Google Scholar] [CrossRef] [PubMed]
- Stradner, M.H.; Gruber, G.; Angerer, H.; Huber, V.; Setznag, D.; Kremser, M.-L.; Moazedi-Fürst, F.C.; Windhager, R.; Graninger, W.B. Sphingosine 1-phosphate counteracts the effects of interleukin-1β in human chondrocytes. Arthritis Rheum. 2013, 65, 2113–2122. [Google Scholar] [CrossRef] [PubMed]
- Xiang, C.; Yang, K.; Liang, Z.; Wan, Y.; Cheng, Y.; Ma, D.; Zhang, H.; Hou, W.; Fu, P. Sphingosine-1-phosphate mediates the therapeutic effects of bone marrow mesenchymal stem cell-derived microvesicles on articular cartilage defect. Transl. Res. 2017, 193, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; Bierma-Zeinstra, S. Osteoarthritis. Lancet 2019, 393, 1745–1759. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author, Year | Study Population | Methods | Main Outcomes |
|---|---|---|---|
| Shufang Wu, 2003 [99] | Sprague Dawley rats | Treat with inhibitor of the cholesterol synthesis | Cholesterol production suppression inhibits chondrocyte proliferation, hypertrophy, and differentiation. Cholesterol controls growth plate chondrogenesis and longitudinal bone formation, potentially through enhancing IHH’s action in the growth plate. |
| H. Kishimoto, 2010 [104] | Bovine articular chondrocytes | Treat with ox-LDL | ox-LDL stimulates the hypertrophy of OA chondrocytes, in part, through binding to LOX-1. The binding of ox-LDL to LOX-1 promotes intracellular ROS generation and oxidative stress in chondrocytes. |
| Aspasia Tsezou, 2010 [106] | Human chondrocytes with OA | Treat with LXR agonist | In osteoarthritis patients, the expression of ABCA1 and ApoA1 transcriptional regulators LXRa and LXRb was dramatically reduced. LXR agonists enhance cholesterol efflux in chondrocytes with osteoarthritis. |
| C. Gentili, 2005 [109] | Chick chondrocytes | Treat with LXR/RXR agonists | Chondrocytes manufacture ApoA1 during development, and ligands that activate LXR or RXR may lead to a significant increase in ApoA1 expression and cholesterol efflux in thicker chondrocytes. Other LXR target genes are implicated in cholesterol transport and homeostasis, including ABCA1 and SREBP1. The expression of chondrocyte SAA is inhibited by ligands that activate LXR or RXR and induce ApoA1. |
| Dominique de Seny, 2015 [110] | Human chondrocytes with OA | Incubation with ApoA1, rhSAA, HDL, LDL | By boosting TLR4 activation, free ApoA1 particles in OA joints may directly or indirectly contribute to the local inflammatory process. ApoA1’s inflammatory characteristics on chondrocytes and fibroblast-like synoviocytes will be influenced by HDL and LDL concentrations that are tightly regulated. |
| Wouter de Munter, 2013 [112] | LDL receptor-deficient and wild-type control mice with OA | Cholesterol-rich diet | In OA, elevated LDL cholesterol levels stimulate the development of ectopic bone. The underlying process may include TGF -β activation and, to a lesser degree, BMP activation after ox-LDL absorption by synovial intimal macrophages. |
| W an-Su Choi, 2019 [113] | Mice with OA | High-cholesterol diet vs. regular diet | In osteoarthritic chondrocytes, increased cholesterol absorption, cholesterol hydroxylase levels, and hydroxysterol metabolite synthesis directly activate ROR, contributing to the pathogenesis of OA. Through LOX1-mediated augmentation of CH25H and CYP7B1 absorption and metabolism, increased cholesterol levels in chondrocytes induce experimental OA in mice. The CH25H-CYP7B1-ROR axis of cholesterol metabolism in chondrocytes is a crucial catabolic regulator in the etiology of osteoarthritis. |
| Margaret Man-Ger Sun, 2020 [114] | Mice primary chondrocytes | Treat with specific LXRagonist | Upon activation by its specific agonist, the LXR-Srebp1-Scd1 axis functions as a cholesterol sensor, lowering the buildup of free cholesterol in cells to mitigate cholesterol’s cytotoxic effects. LXR is activated by its particular agonist, and LXR’s protective impact on OA may be mediated, in part, by increased cholesterol excretion and suppression of chondrocyte hypertrophy. |
| Fotini Kostopoulou, 2015 [116] | Human cartilage with OA | Treat with TGF-β1 | miR-33a may influence the TGF-1/PI3K/Akt signaling pathway via the regulation of Smad7 expression. MiR-33 inhibition in OA chondrocytes raises the levels of ABCA1 and HDL, resulting in a reduction in MMP-13 expression. |
| Shirou Tabe, 2017 [122] | ATDC5 cells | Silence LPCA4 expression | Other than Col2, Sox9, and Runx2, knockdown of LPCAT4 decreased the mRNA expression of chondrogenic differentiation-related molecules. The expression of LPCAT4 rises during chondrogenic differentiation, and LPCAT4 is implicated in chondrocyte hypertrophy. |
| Mi-Kyoung Kim, 2006 [124] | Rat primary chondrocytes | Treat with S1P and PhS1P | Sphingosine kinase-1 is the key to the production of S1P from sphingosine. S1P and PhS1P lead to cellular proliferation of rat primary chondrocytes through a ptx-sensitive G-protein-mediated pathway, plc-mediated [Ca2+] increase and activation of MAPKs. |
| Hongming Miao, 2015 [79] | Male C57BL/6 mice | HFD | In a way reliant on LDH-a, circulating stearic acid elevates lactate levels in plasma and chondrocytes. Stearic acid increases the production of VEGF and cytokines that cause inflammation through a TLR4 pathway and a new lactate/HIF1 pathway. |
| Oscar lvarez-Garcia, 2014 [80] | Human chondrocytes and fibroblast-like synoviocytes | Human chondrocytes from normal and OA were treated with Palmitate or oleate in combination with IL-1 | Palmitate, but not oleate, stimulates IL-1 in normal chondrocytes, causing caspase activation and cell death, and upregulates IL-6 and COX2 in chondrocytes and fibroblast-like synoviocytes through Toll-like receptor-4 signaling. |
| Daniel edina-Luna, 2017 [81] | Human chondrocytes | Treatment with FFA blend consisting of palmitic and oleic acids vs. control | A high-fat milieu causes cartilage degradation by raising chondrocyte IL-1, ROS, and RNS, promoting autocrine production of IL-6 and IL-8. |
| Chia-Lung Wu, 2014 [75] | Mice with OA | HFD, rich in SFAs, n-3 PUFAs, n-6 PUFAs | n-3 PUFA supplementation may reduce the effects of obesity on OA and enhance healing. SFAs and omega-6 PUFAs are both harmful in OA following joint damage. SFAs may cause synovial macrophages to release IL-1 and TNF, both of which are implicated in cartilage degradation. |
| M. Miyamoto, 2003 [88] | Primary chondrocytes from rat rib cartilage | Treat with PGE2 | Simultaneous stimulation of EP2 and EP4 induces chondrocyte differentiation |
| H. Davis Adkisson, 1991 [85] | Cartilage from multiple species | Argentation high-performance liquid chromatographic(AHPLC) separationof fatty acid | Low levels of n-6 PUFAs and high levels of abnormal n-9 fatty acids found in normal cartilage |
| Kohei Nishitani, 2010 [89] | Human chondrocytes | Treat with PGE2 | PGE2 inhibits IL-1β-induced MMP-1 and MMP-13 synthesis through EP4 activation and suppression of MKK4, JNK MAP kinase, and c-JUN phosphorylation. |
| Sunderajhan Sekar, 2017 [73] | Rat | High-Carbohydrate Supplements with SFAs vs. cornstarch diet | High-fat diet can induce metabolic syndrome in rats. Palmitic acid and stearic acid combined with IL-1 mediate cartilage damage by increasing MMP-13, ADAMTS-4, and ADAMTS-5 gene expression and increase the severity of OA |
| Sujeong Park, 2022 [74] | ACOT12 knockout mice | Treatment with specific agonists and inhibitors of PPARα | ACOT12 is involved in maintaining cartilage homeostasis. Increased accumulation of acetyl-CoA due to PPARα deficiency leads to stimulation of cartilage-degrading enzymes and chondrocyte apoptosis through regulation of ACOT12. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, Z.; Zong, Z.; Deng, J.; Huang, J.; Liu, G.; Wei, B.; Cui, L.; Li, G.; Zhong, H.; Lin, S. Lipid Metabolism in Cartilage Development, Degeneration, and Regeneration. Nutrients 2022, 14, 3984. https://doi.org/10.3390/nu14193984
Su Z, Zong Z, Deng J, Huang J, Liu G, Wei B, Cui L, Li G, Zhong H, Lin S. Lipid Metabolism in Cartilage Development, Degeneration, and Regeneration. Nutrients. 2022; 14(19):3984. https://doi.org/10.3390/nu14193984
Chicago/Turabian StyleSu, Zhanpeng, Zhixian Zong, Jinxia Deng, Jianping Huang, Guihua Liu, Bo Wei, Liao Cui, Gang Li, Huan Zhong, and Sien Lin. 2022. "Lipid Metabolism in Cartilage Development, Degeneration, and Regeneration" Nutrients 14, no. 19: 3984. https://doi.org/10.3390/nu14193984
APA StyleSu, Z., Zong, Z., Deng, J., Huang, J., Liu, G., Wei, B., Cui, L., Li, G., Zhong, H., & Lin, S. (2022). Lipid Metabolism in Cartilage Development, Degeneration, and Regeneration. Nutrients, 14(19), 3984. https://doi.org/10.3390/nu14193984