
Emerging Evidence for the Widespread Role of Glutamatergic Dysfunction in Neuropsychiatric Diseases


{kind=link}
Abstract
:1. Introduction
1.1. Glutamine/Glutamate in the Peripheral Circulation
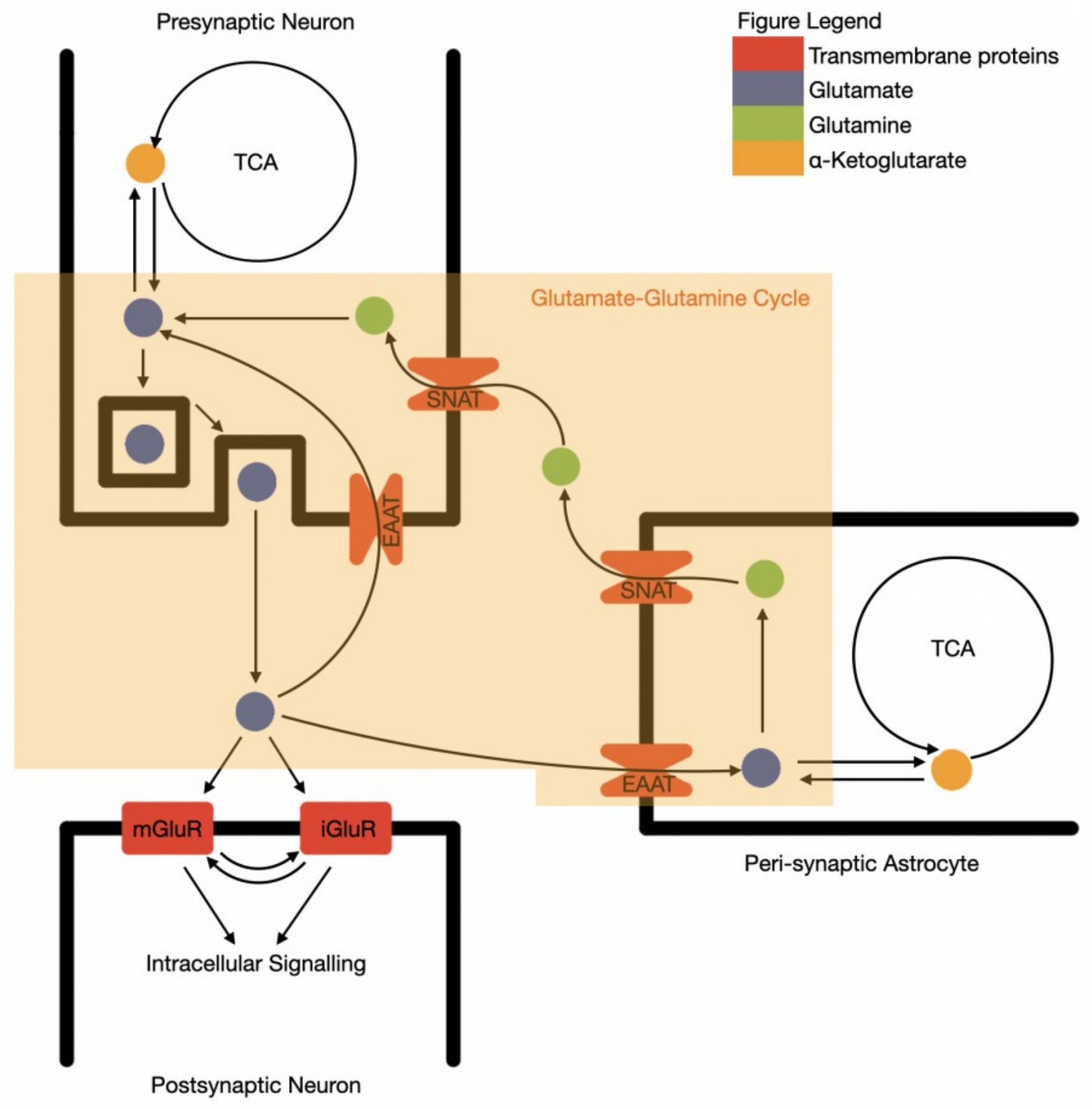
1.2. Glutamate Compartments in the Central Nervous System
1.3. Glutamatergic Receptors
1.4. Astrocyte Control of Glutamate Levels
2. Depression
2.1. The Glutamate Hypothesis of Depression
2.2. Glutamatergic Imaging Studies
2.3. Glutamate Mediates the Relationship of Stress with Depression
2.4. Glutamate in Inflammation Related Depression
2.5. The Kynurenine Pathway
2.6. Chronic Disease, Inflammation, and Depression
2.7. Chronic Disease-Induced Neurodegeneration
3. Glutamatergic Activity in Bipolar Disorder
4. The Glutamate Hypothesis of Schizophrenia
5. Neurodevelopmental Disorders
5.1. Obsessive Compulsive Disorder
5.2. Neurodevelopmental Genetic Associations
6. Neurodegenerative Conditions and Glutamate
7. Epilepsy
8. Brain Injury
9. Post-Traumatic Stress Disorder (PTSD)
10. Other Disorders
- (1)
- Binge eating disorder affects 3.5% of the population and, together with similar eating disorders, causes significant disability. Symptom overlaps occur with substance use disorders, and initial research indicates the involvement of GABA and glutamate modulation pathways and beneficial responses to glutamatergic modulators [189].
- (2)
- Anorexia nervosa is the most serious of the eating disorders, associated with high morbidity and mortality. However, the pathogenesis is poorly understood and there are no established pharmaceutical treatments. High field strength MRS (7T) shows reduced glutamate in the anterior cingulate cortex, occipital cortex, and putamen [190].
- (3)
- Personality disorders (PD) are a diverse group of mental disorders associated with dysfunction of social interactions and suicidal risk. Different subtypes have very different clinical presentations but all share difficulty with rigidity of thinking, social functioning, and behaviour, deriving from how the person perceives themselves and their relationships with others. Previously thought to be a “psychological” disorder with no organic basis, increasing evidence highlights abnormalities of prefrontal cortex glutamate activity. MRS studies show significant differences in NAA and Glu in the dorsolateral prefrontal cortex (DLPFC) between PD and healthy controls and across PD subtypes [191]. Interestingly, studies in PD have shown increased central pain suppression, pain adaptation, and dissociation from pain, especially in women and the borderline personality disorder subtype. Although the mechanisms are unclear, this hyposensitivity may underlie the high incidence of self-harming behaviour associated with this condition [192].
11. Effects of Gut Microbiota on CNS Disorders and Glutamate Dysfunction
12. Glutamatergic Function and Exercise
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef] [Green Version]
- Monteggia, L.M.; Malenka, R.C.; Deisseroth, K. Depression: The best way forward. Nature 2014, 515, 200–201. [Google Scholar] [CrossRef] [PubMed]
- Borbély, É.; Simon, M.; Fuchs, E.; Wiborg, O.; Czéh, B.; Helyes, Z. Novel drug developmental strategies for treatment-resistant depression. J. Cereb. Blood Flow Metab. 2021, 179, 1146–1186. [Google Scholar] [CrossRef] [PubMed]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef]
- Blaylock, R.L.; Faria, M. New concepts in the development of schizophrenia, autism spectrum disorders, and degenerative brain diseases based on chronic inflammation: A working hypothesis from continued advances in neuroscience research. Surg. Neurol. Int. 2021, 12, 556. [Google Scholar]
- Schousboe, A. Transport and metabolism of glutamate and GABA in neurons are glial cells. Int. Rev. Neurobiol. 1981, 22, 1–45. [Google Scholar]
- Fonnum, F. Glutamate: A Neurotransmitter in Mammalian Brain. J. Neurochem. 1984, 42, 1–11. [Google Scholar] [CrossRef]
- Franchini, L.; Stanic, J.; Ponzoni, L.; Mellone, M.; Carrano, N.; Musardo, S.; Zianni, E.; Olivero, G.; Marcello, E.; Pittaluga, A.; et al. Linking NMDA Receptor Synaptic Retention to Synaptic Plasticity and Cognition. iScience 2019, 19, 927–939. [Google Scholar] [CrossRef]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–62. [Google Scholar]
- Collingridge, G.L.; Lester, R.A. Excitatory amino acid receptors in the vertebrate central nervous system. Pharmacol. Rev. 1989, 41, 143–210. [Google Scholar]
- Kim, J.-H.; Marton, J.; Ametamey, S.M.; Cumming, P. A Review of Molecular Imaging of Glutamate Receptors. Molecules 2020, 25, 4749. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Mitchell, K.M.; Albahadily, F.N.; Michaelis, E.K.; Wilson, G.S. Direct measurement of glutamate release in the brain using a dual enzyme-based electrochemical sensor. Brain Res. 1994, 659, 117–125. [Google Scholar] [CrossRef]
- Rego, S.M.; Snyder, M.P. High Throughput Sequencing and Assessing Disease Risk. Cold Spring Harb. Perspect. Med. 2018, 9, a026849. [Google Scholar] [CrossRef]
- Gussew, A.; Rzanny, R.; Scholle, H.-C.; Kaiser, W.A.; Reichenbach, J.R. Quantitative Bestimmung von Glutamat im Hirn mithilfe der MR-Protonenspektroskopie bei 1.5 T und 3 T. RöFo-Fortschr. Geb. Röntgenstrahlen Bildgeb. Verfahr. 2008, 180, 722–732. [Google Scholar] [CrossRef]
- Majo, V.J.; Prabhakaran, J.; Mann, J.J.; Kumar, J.D. PET and SPECT tracers for glutamate receptors. Drug Discov. Today 2013, 18, 173–184. [Google Scholar] [CrossRef]
- Edition, F. Diagnostic and statistical manual of mental disorders (5th Ed.). Am. Psychiatr. Assoc. 2013, 21, 591–643. [Google Scholar]
- Cruzat, V.; Macedo Rogero, M.; Keane, K.N.; Curi, R.; Newsholme, P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018, 10, 1564. [Google Scholar] [CrossRef] [Green Version]
- Tapiero, H.; Mathé, G.; Couvreur, P.; Tew, K.D., II. Glutamine and glutamate. Biomed. Pharm. 2002, 56, 446–457. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- Castell, L.M. Glutamine Supplementation In Vitro and In Vivo, in Exercise and in Immunodepression. Sports Med. 2003, 33, 323–345. [Google Scholar] [CrossRef] [PubMed]
- Castell, L.M.; Newsholme, E.A. The relation between glutamine and the immunodepression observed in exercise. Amino Acids 2001, 20, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.L.; Hsu, C.S.; Yeh, S.L.; Chen, W.J. Dietary glutamine supplementation modulates Th1/Th2 cytokine and interleukin-6 expressions in septic mice. Cytokine 2005, 31, 329–334. [Google Scholar] [CrossRef]
- Chu, C.-C.; Hou, Y.-C.; Pai, M.-H.; Chao, C.-J.; Yeh, S.-L. Pretreatment with alanyl-glutamine suppresses T-helper-cell-associated cytokine expression and reduces inflammatory responses in mice with acute DSS-induced colitis. J. Nutr. Biochem. 2012, 23, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar]
- Foster, A.; Mena, E.; Fagg, G.; Cotman, C. Glutamate and aspartate binding sites are enriched in synaptic junctions isolated from rat brain. J. Neurosci. 1981, 1, 620–625. [Google Scholar] [CrossRef]
- Meldrum, B.S. Glutamate as a Neurotransmitter in the Brain: Review of Physiology and Pathology. J. Nutr. 2000, 130, 1007S–1015S. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, S.; Martin, S.; Martin, D. Regional distribution and relative amounts of glutamate decarboxylase isoforms in rat and mouse brain. Neurochem. Int. 1999, 35, 73–80. [Google Scholar] [CrossRef]
- Hawkins, R.A. The blood-brain barrier and glutamate. Am. J. Clin. Nutr. 2009, 90, 867S–874S. [Google Scholar] [CrossRef] [Green Version]
- Zlotnik, A.; Ohayon, S.; Gruenbaum, B.F.; Gruenbaum, S.E.; Mohar, B.; Boyko, M.; Klin, Y.; Sheiner, E.; Shaked, G.; Shapira, Y.; et al. Determination of Factors Affecting Glutamate Concentrations in the Whole Blood of Healthy Human Volunteers. J. Neurosurg. Anesthesiol. 2011, 23, 45–49. [Google Scholar] [CrossRef]
- Hawkins, R.A.; Viña, J.R. How Glutamate Is Managed by the Blood–Brain Barrier. Biology 2016, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Cichoż-Lach, H.; Michalak, A. Current pathogenetic aspects of hepatic encephalopathy and noncirrhotic hyperammonemic encephalopathy. World J. Gastroenterol. 2013, 19, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Attwell, D.; Laughlin, S.B. An Energy Budget for Signaling in the Grey Matter of the Brain. J. Cereb. Blood Flow Metab. 2001, 21, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. A Cellular Perspective on Brain Energy Metabolism and Functional Imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, E.; Rönnbäck, L. Altered neuronal-glial signaling in glutamatergic transmission as a unifying mechanism in chronic pain and mental fatigue. Neurochem. Res. 2004, 29, 989–996. [Google Scholar] [CrossRef]
- McKenna, M.C. Glutamate Pays Its Own Way in Astrocytes. Front. Endocrinol. 2013, 4, 191. [Google Scholar] [CrossRef] [Green Version]
- Hossmann, K.A. The hypoxic brain. Insights from ischemia research. Single Mol. Single Cell Seq. 1999, 474, 155–169. [Google Scholar]
- Reiner, A.; Levitz, J. Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef] [Green Version]
- Hollmann, M.; O’Shea-Greenfield, A.; Rogers, S.W.; Heinemann, S.F. Cloning by functional expression of a member of the glutamate receptor family. Nature 1989, 342, 643–648. [Google Scholar] [CrossRef]
- Hollmann, M.; Heinemann, S. Cloned glutamate receptors. Annu. Rev. Neurosci. 1994, 17, 31–108. [Google Scholar] [CrossRef]
- Hadzic, M.; Jack, A.; Wahle, P. Ionotropic glutamate receptors: Which ones, when, and where in the mammalian neocortex. J. Comp. Neurol. 2017, 525, 976–1033. [Google Scholar] [CrossRef] [PubMed]
- Levitz, J.; Habrian, C.; Bharill, S.; Fu, Z.; Vafabakhsh, R.; Isacoff, E.Y. Mechanism of Assembly and Cooperativity of Homomeric and Heteromeric Metabotropic Glutamate Receptors. Neuron 2016, 92, 143–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The glutamate/GABA-glutamine cycle: Aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.V.; Markussen, K.H.; Jakobsen, E.; Schousboe, A.; Waagepetersen, H.S.; Rosenberg, P.A.; Aldana, B.I. Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacology 2021, 196, 108719. [Google Scholar] [CrossRef]
- Vos, T.; Flaxman, A.D.; Naghavi, M.; Lozano, R.; Michaud, C.; Ezzati, M.; Shibuya, K.; Salomon, J.A.; Abdalla, S.; Aboyans, V.; et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2163–2196. [Google Scholar] [CrossRef]
- Tolentino, J.C.; Schmidt, S.L. DSM-5 Criteria and Depression Severity: Implications for Clinical Practice. Front. Psychiatry 2018, 9, 450. [Google Scholar] [CrossRef] [Green Version]
- Mulinari, S. Monoamine Theories of Depression: Historical Impact on Biomedical Research. J. Hist. Neurosci. 2012, 21, 366–392. [Google Scholar] [CrossRef]
- Kalkman, H.O. Novel Treatment Targets Based on Insights in the Etiology of Depression: Role of IL-6 Trans-Signaling and Stress-Induced Elevation of Glutamate and ATP. Pharmaceuticals 2019, 12, 113. [Google Scholar] [CrossRef] [Green Version]
- Chourbaji, S.; Vogt, M.A.; Fumagalli, F.; Sohr, R.; Frasca, A.; Brandwein, C.; Hörtnagl, H.; Riva, M.A.; Sprengel, R.; Gass, P. AMPA receptor subunit 1 (GluR-A) knockout mice model the glutamate hypothesis of depression. FASEB J. 2008, 22, 3129–3134. [Google Scholar] [CrossRef]
- Alexander, L.; Jelen, L.A.; Mehta, M.A.; Young, A.H. The anterior cingulate cortex as a key locus of ketamine’s antidepressant action. Neurosci. Biobehav. Rev. 2021, 127, 531–554. [Google Scholar] [CrossRef]
- Zanos, P.; Gould, T.D. Mechanisms of ketamine action as an antidepressant. Mol. Psychiatry 2018, 23, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Fullana, M.N.; Ruiz-Bronchal, E.; Ferrés-Coy, A.; Juarez-Escoto, E.; Artigas, F.; Bortolozzi, A. Regionally selective knockdown of astroglial glutamate transporters in infralimbic cortex induces a depressive phenotype in mice. Glia 2019, 67, 1122–1137. [Google Scholar] [CrossRef] [PubMed]
- Luykx, J.; Laban, K.; Heuvel, M.V.D.; Boks, M.; Mandl, R.; Kahn, R.; Bakker, S. Region and state specific glutamate downregulation in major depressive disorder: A meta-analysis of 1H-MRS findings. Neurosci. Biobehav. Rev. 2012, 36, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, S.; Takamiya, A.; Noda, Y.; Horita, N.; Wada, M.; Tsugawa, S.; Plitman, E.; Sano, Y.; Tarumi, R.; ElSalhy, M.; et al. Glutamatergic neurometabolite levels in major depressive disorder: A systematic review and meta-analysis of proton magnetic resonance spectroscopy studies. Mol. Psychiatry 2019, 24, 952–964. [Google Scholar] [CrossRef] [Green Version]
- Godlewska, B.R.; Masaki, C.; Sharpley, A.L.; Cowen, P.J.; Emir, U.E. Brain glutamate in medication-free depressed patients: A proton MRS study at 7 Tesla. Psychol. Med. 2017, 48, 1731–1737. [Google Scholar] [CrossRef] [Green Version]
- Kantrowitz, J.T.; Dong, Z.; Milak, M.S.; Rashid, R.; Kegeles, L.S.; Javitt, D.C.; Lieberman, J.A.; Mann, J.J. Ventromedial prefrontal cortex/anterior cingulate cortex Glx, glutamate, and GABA levels in medication-free major depressive disorder. Transl. Psychiatry 2021, 11, 1–6. [Google Scholar] [CrossRef]
- Persson, J.; Wall, A.; Weis, J.; Gingnell, M.; Antoni, G.; Lubberink, M.; Bodén, R. Inhibitory and excitatory neurotransmitter systems in depressed and healthy: A positron emission tomography and magnetic resonance spectroscopy study. Psychiatry Res. Neuroimaging 2021, 315, 111327. [Google Scholar] [CrossRef]
- Charney, D.S.; Manji, H.K. Life Stress, Genes, and Depression: Multiple Pathways Lead to Increased Risk and New Opportunities for Intervention. Sci. STKE 2004, 2004, re5. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, L.L.; Gawuga, C.E.; Tyrka, A.R.; Lee, J.K.; Anderson, G.M.; Price, L.H. Association between Plasma IL-6 Response to Acute Stress and Early-Life Adversity in Healthy Adults. Neuropsychopharmacology 2010, 35, 2617–2623. [Google Scholar] [CrossRef] [Green Version]
- Dickerson, S.S.; Gable, S.L.; Irwin, M.R.; Aziz, N.; Kemeny, M.E. Social-evaluative threat and proinflammatory cytokine regulation: An experimental laboratory investigation. Psychol. Sci. 2009, 20, 1237–1244. [Google Scholar] [CrossRef] [Green Version]
- Bierhaus, A.; Wolf, J.; Andrassy, M.; Rohleder, N.; Humpert, P.M.; Petrov, D.; Ferstl, R.; von Eynatten, M.; Wendt, T.; Rudofsky, G.; et al. A mechanism converting psychosocial stress into mononuclear cell activation. Proc. Natl. Acad. Sci. USA 2003, 100, 1920–1925. [Google Scholar] [CrossRef] [Green Version]
- Powell, N.D.; Sloan, E.K.; Bailey, M.T.; Arevalo, J.M.; Miller, G.E.; Chen, E.; Kobor, M.S.; Reader, B.F.; Sheridan, J.F.; Cole, S.W. Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via β-adrenergic induction of myelopoiesis. Proc. Natl. Acad. Sci. USA 2013, 110, 16574–16579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pace, T.W.; Mletzko, T.C.; Alagbe, O.; Musselman, D.L.; Nemeroff, C.B.; Miller, A.H.; Heim, C.M. Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am. J. Psychiatry 2006, 163, 1630–1633. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.W.; Anda, R.F.; Tiemeier, H.; Felitti, V.J.; Edwards, V.J.; Croft, J.B.; Giles, W.H. Adverse Childhood Experiences and the Risk of Premature Mortality. Am. J. Prev. Med. 2009, 37, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Rajamanickam, S.; Justice, N.J. Local Corticotropin-Releasing Factor Signaling in the Hypothalamic Paraventricular Nucleus. J. Neurosci. 2018, 38, 1874–1890. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, N.C. How GABA generates depolarization. J. Physiol. 2010, 588, 757–758. [Google Scholar] [CrossRef]
- De Laurentiis, A.; Pisera, D.; Lasaga, M.; del Carmen Díaz, M.; Theas, S.; Duvilanski, B.; Seilicovich, A. Effect of interleukin-6 and tumor necrosis factor-alpha on GABA release from mediobasal hypothalamus and posterior pituitary. Neuroimmunomodulation 2000, 7, 77–83. [Google Scholar] [CrossRef]
- Lüscher, B.; Möhler, H. Brexanolone, a neurosteroid antidepressant, vindicates the GABAergic deficit hypothesis of depression and may foster resilience. F1000Research 2019, 8, 751. [Google Scholar] [CrossRef] [Green Version]
- Oquendo, M.A.; Echavarria, G.; Galfalvy, H.; Grunebaum, M.F.; Burke, A.; Barrera, A.; Cooper, T.B.; Malone, K.M.; Mann, J.J. Lower Cortisol Levels in Depressed Patients with Comorbid Post-Traumatic Stress Disorder. Neuropsychopharmacology 2002, 28, 591–598. [Google Scholar] [CrossRef]
- Raber, J. Detrimental effects of chronic hypothalamic—Pituitary—Adrenal axis activation. Mol. Neurobiol. 1998, 18, 1–22. [Google Scholar] [CrossRef]
- Ferrini, F.; De Koninck, Y. Microglia Control Neuronal Network Excitability via BDNF Signalling. Neural Plast. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tata, D.A.; Anderson, B.J. The effects of chronic glucocorticoid exposure on dendritic length, synapse numbers and glial volume in animal models: Implications for hippocampal volume reductions in depression. Physiol. Behav. 2010, 99, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Liston, C.; Miller, M.M.; Goldwater, D.S.; Radley, J.J.; Rocher, A.B.; Hof, P.R.; Morrison, J.H.; McEwen, B.S. Stress-induced alterations in prefrontal cortical dendritic morphology predict selective impairments in perceptual attentional set-shifting. J. Neurosci. 2006, 26, 7870–7874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harden, L.M.; Kent, S.; Pittman, Q.J.; Roth, J. Fever and sickness behavior: Friend or foe? Brain Behav. Immun. 2015, 50, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Raison, C.L.; Miller, A.H. The evolutionary significance of depression in Pathogen Host Defense (PATHOS-D). Mol. Psychiatry 2012, 18, 15–37. [Google Scholar] [CrossRef]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and Its Discontents: The Role of Cytokines in the Pathophysiology of Major Depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef] [Green Version]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A Meta-Analysis of Cytokines in Major Depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Garner, K.M.; Amin, R.; Johnson, R.W.; Scarlett, E.J.; Burton, M.D. Microglia priming by interleukin-6 signaling is enhanced in aged mice. J. Neuroimmunol. 2018, 324, 90–99. [Google Scholar] [CrossRef]
- Campbell, I.L.; Erta, M.; Lim, S.L.; Frausto, R.; May, U.; Rose-John, S.; Scheller, J.; Hidalgo, J. Trans-Signaling Is a Dominant Mechanism for the Pathogenic Actions of Interleukin-6 in the Brain. J. Neurosci. 2014, 34, 2503–2513. [Google Scholar] [CrossRef] [Green Version]
- Finsterwald, C.; Alberini, C.M. Stress and glucocorticoid receptor-dependent mechanisms in long-term memory: From adaptive responses to psychopathologies. Neurobiol. Learn. Mem. 2013, 112, 17–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savitz, J. Role of Kynurenine Metabolism Pathway Activation in Major Depressive Disorders. Curr. Top Behav. Neurosci. 2016, 31, 249–267. [Google Scholar] [CrossRef]
- Fujigaki, H.; Yamamoto, Y.; Saito, K. L-Tryptophan-kynurenine pathway enzymes are therapeutic target for neuropsychiatric diseases: Focus on cell type differences. Neuropharmacology 2016, 112, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R. Kynurenines and glutamate: Multiple links and therapeutic implications. Adv. Pharmacol. 2016, 76, 13–37. [Google Scholar] [PubMed] [Green Version]
- Martin, K.S.; Azzolini, M.; Ruas, J. The kynurenine connection: How exercise shifts muscle tryptophan metabolism and affects energy homeostasis, the immune system, and the brain. Am. J. Physiol. Physiol. 2020, 318, C818–C830. [Google Scholar] [CrossRef] [PubMed]
- Gaynes, B.N.; Lloyd, S.W.; Lux, L.; Gartlehner, G.; Hansen, R.A.; Brode, S.; Jonas, D.E.; Evans, T.S.; Viswanathan, M.; Lohr, K.N. Repetitive transcranial magnetic stimulation for treatment-resistant depression: A systematic review and meta-analysis. J. Clin. Psychiatry 2014, 75, 477–489. [Google Scholar] [CrossRef]
- Erchinger, V.J.; Ersland, L.; Aukland, S.M.; Abbott, C.C.; Oltedal, L. Magnetic Resonance Spectroscopy in Depressed Subjects Treated with Electroconvulsive Therapy—A Systematic Review of Literature. Front. Psychiatry 2021, 12. [Google Scholar] [CrossRef]
- Dandekar, M.P.; Fenoy, A.J.; Carvalho, A.F.; Soares, J.C.; Quevedo, J. Deep brain stimulation for treatment-resistant depression: An integrative review of preclinical and clinical findings and translational implications. Mol. Psychiatry 2018, 23, 1094–1112. [Google Scholar] [CrossRef]
- Brahim, L.O.; Lambert, S.D.; Feeley, N.; Coumoundouros, C.; Schaffler, J.; McCusker, J.; Moodie, E.E.M.; Kayser, J.; Kolne, K.; Belzile, E.; et al. The effects of self-management interventions on depressive symptoms in adults with chronic physical disease(s) experiencing depressive symptomatology: A systematic review and meta-analysis. BMC Psychiatry 2021, 21, 584. [Google Scholar]
- Roy, T.; Lloyd, C.E. Epidemiology of depression and diabetes: A systematic review. J. Affect. Disord. 2012, 142, S8–S21. [Google Scholar] [CrossRef]
- Rosenblat, J.D.; Kurdyak, P.; Cosci, F.; Berk, M.; Maes, M.; Brunoni, A.R.; Li, M.; Rodin, G.; McIntyre, R.S.; Carvalho, A.F. Depression in the medically ill. Aust. N. Z. J. Psychiatry 2020, 54, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Martin-Subero, M.; Anderson, G.; Kanchanatawan, B.; Berk, M.; Maes, M. Comorbidity between depression and inflammatory bowel disease explained by immune-inflammatory, oxidative, and nitrosative stress; tryptophan catabolite; and gut–brain pathways. CNS Spectrums 2015, 21, 184–198. [Google Scholar] [CrossRef] [Green Version]
- Nerurkar, L.; Siebert, S.; McInnes, I.B.; Cavanagh, J. Rheumatoid arthritis and depression: An inflammatory perspective. Lancet Psychiatry 2018, 6, 164–173. [Google Scholar] [CrossRef]
- Halaris, A. Inflammation-Associated Co-morbidity Between Depression and Cardiovascular. Disease 2016, 31, 45–70. [Google Scholar] [CrossRef]
- Du, Y.-J.; Yang, C.-J.; Li, B.; Wu, X.; Lv, Y.-B.; Jin, H.-L.; Cao, Y.-X.; Sun, J.; Luo, Q.-L.; Gong, W.-Y.; et al. Association of pro-inflammatory cytokines, cortisol and depression in patients with chronic obstructive pulmonary disease. Psychoneuroendocrinology 2014, 46, 141–152. [Google Scholar] [CrossRef]
- Patel, N.; Nadkarni, A.; Cardwell, L.A.; Vera, N.; Frey, C.; Patel, N.; Feldman, S.R. Psoriasis, Depression, and Inflammatory Overlap: A Review. Am. J. Clin. Dermatol. 2017, 18, 613–620. [Google Scholar] [CrossRef]
- Sasaki-Hamada, S.; Sanai, E.; Kanemaru, M.; Kamanaka, G.; Oka, J.I. Long-term exposure to high glucose induces changes in the expression of AMPA receptor subunits and glutamate transmission in primary cultured cortical neurons. Biochem. Biophys. Res. Commun. 2021, 589, 48–54. [Google Scholar] [CrossRef]
- Siebenhüner, A.R.; Rossel, J.B.; Schreiner, P.; Butter, M.; Greuter, T.; Krupka, N.; Jordi, S.B.U.; Biedermann, L.; Rogler, G.; Misselwitz, B.; et al. Effects of anti-TNF therapy and immunomodulators on anxiety and depressive symptoms in patients with inflammatory bowel disease: A 5-year analysis. Therap. Adv. Gastroenterol. 2021, 14, 17562848211033763. [Google Scholar] [CrossRef]
- Li, Z.R.; Han, Y.S.; Liu, Z.; Zhao, H.Q.; Liu, J.; Yang, H.; Wang, Y.H. GR/NF-κB signaling pathway regulates hippocampal inflammatory responses in diabetic rats with chronic unpredictable mild stress. Eur. J. Pharmacol. 2021, 895, 173861. [Google Scholar] [CrossRef]
- Simon, G.E. Treating depression in patients with chronic disease: Recognition and treatment are crucial; depression worsens the course of a chronic illness. West J. Med. 2001, 175, 292–293. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-S.; Lobbestael, E.; Vermeire, S.; Sabino, J.; Cleynen, I. Inflammatory bowel disease and Parkinson’s disease: Common pathophysiological links. Gut 2021, 70, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Spilling, C.A.; Bajaj, M.-P.K.; Burrage, D.R.; Ruickbie, S.; Thai, N.J.; Baker, E.H.; Jones, P.W.; Barrick, T.R.; Dodd, J.W. Contributions of cardiovascular risk and smoking to chronic obstructive pulmonary disease (COPD)-related changes in brain structure and function. Int. J. Chronic Obstr. Pulm. Dis. 2019, 14, 1855–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, S.C.; Harre, U.; Purohit, P.; Dietel, K.; Kienhöfer, D.; Hahn, J.; Baum, W.; Herrmann, M.; Schett, G.; Mielenz, D. Neurodegeneration Enhances the Development of Arthritis. J. Immunol. 2017, 198, 2394–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheong, J.L.; de Pablo-Fernandez, E.; Foltynie, T.; Noyce, A.J. The Association Between Type 2 Diabetes Mellitus and Parkinson’s Disease. J. Park. Dis. 2020, 10, 775–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sima, A.A.F. Encephalopathies: The emerging diabetic complications. Geol. Rundsch. 2010, 47, 279–293. [Google Scholar] [CrossRef]
- Mahalakshmi, B.; Maurya, N.; Lee, S.-D.; Kumar, V.B. Possible Neuroprotective Mechanisms of Physical Exercise in Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 5895. [Google Scholar] [CrossRef]
- Lda, S.C.; Alencar, Á.P.; Neto, P.J.N.; dos Santos, M.D.S.V.; da Silva, C.G.L.; Pinheiro, S.D.F.L.; Silveira, R.T.; Bianco, B.A.V.; Júnior, R.F.F.; Lima, M.A.P. Risk factors for suicide in bipolar disorder: A systematic review. J. Affect. Disord. 2015, 170, 237–254. [Google Scholar]
- Chitty, K.M.; Lagopoulos, J.; Hickie, I.B.; Hermens, D.F. Hippocampal glutamatergic/NMDA receptor functioning in bipolar disorder: A study combining mismatch negativity and proton magnetic resonance spectroscopy. Psychiatry Res. Neuroimaging 2015, 233, 88–94. [Google Scholar] [CrossRef]
- Chitty, K.M.; Lagopoulos, J.; Lee, R.S.; Hickie, I.; Hermens, D. A systematic review and meta-analysis of proton magnetic resonance spectroscopy and mismatch negativity in bipolar disorder. Eur. Neuropsychopharmacol. 2013, 23, 1348–1363. [Google Scholar] [CrossRef]
- Reddy-Thootkur, M.; Kraguljac, N.V.; Lahti, A.C. The role of glutamate and GABA in cognitive dysfunction in schizophrenia and mood disorders—A systematic review of magnetic resonance spectroscopy studies. Schizophr. Res. 2020. [Google Scholar] [CrossRef]
- Merikangas, K.R.; Jin, R.; He, J.-P.; Kessler, R.C.; Lee, S.; Sampson, N.A.; Viana, M.C.; Andrade, L.H.; Hu, C.; Karam, E.G.; et al. Prevalence and Correlates of Bipolar Spectrum Disorder in the World Mental Health Survey Initiative. Arch. Gen. Psychiatry 2011, 68, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.L.; Marquardt, T.; Hurducas, C.; Spyridi, S.; Barnes, A.; Smith, R.; Cowen, P.J.; McShane, R.; Hawton, K.; Malhi, G.S.; et al. Ketamine and other glutamate receptor modulators for depression in adults with bipolar disorder. Cochrane Database Syst. Rev. 2021, 2021. [Google Scholar] [CrossRef]
- de Bartolomeis, A.; Buonaguro, E.F.; Iasevoli, F.; Tomasetti, C. The emerging role of dopamine-glutamate interaction and of the postsynaptic density in bipolar disorder pathophysiology: Implications for treatment. J. Psychopharmacol. 2014, 28, 505–526. [Google Scholar] [CrossRef] [PubMed]
- Wurfel, B.E.; Drevets, W.C.; Bliss, S.A.; McMillin, J.R.; Suzuki, H.; Ford, B.N.; Morris, H.M.; Teague, T.K.; Dantzer, R.; Savitz, J.B. Serum kynurenic acid is reduced in affective psychosis. Transl. Psychiatry 2017, 7, e1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erhardt, S.; Schwieler, L.; Imbeault, S.; Engberg, G. The kynurenine pathway in schizophrenia and bipolar disorder. Neuropharmacology 2017, 112, 297–306. [Google Scholar] [CrossRef] [PubMed]
- The ICD-10 Classification of Mental and Behavioural Disorders; World Health Organization (WHO): Geneva, Switzerland, 1992.
- Brisch, R.; Saniotis, A.; Wolf, R.; Bielau, H.; Bernstein, H.-G.; Steiner, J.; Bogerts, B.; Braun, A.K.; Jankowski, Z.; Kumaritlake, J.; et al. The role of dopamine in schizophrenia from a neurobiological and evolutionary perspective: Old fashioned, but still in vogue. Front Psychiatry 2014, 5, 47. [Google Scholar] [CrossRef]
- Jentsch, J.D.; Roth, R.H. The neuropsychopharmacology of phencyclidine: From NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 1999, 20, 201–225. [Google Scholar] [CrossRef] [Green Version]
- Howes, O.; McCutcheon, R.; Stone, J. Glutamate and dopamine in schizophrenia: An update for the 21st century. J. Psychopharmacol. 2015, 29, 97–115. [Google Scholar] [CrossRef] [Green Version]
- Smucny, J.; Carter, C.S.; Maddock, R.J. Magnetic resonance spectroscopic evidence of increased choline in the dorsolateral prefrontal and visual cortices in recent onset schizophrenia. Neurosci. Lett. 2021, 770, 136410. [Google Scholar] [CrossRef]
- Marsman, A.; Heuvel, M.V.D.; Klomp, D.W.J.; Kahn, R.S.; Luijten, P.R.; Pol, H.H. Glutamate in Schizophrenia: A Focused Review and Meta-Analysis of 1H-MRS Studies. Schizophr. Bull. 2011, 39, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Merritt, K.; Egerton, A.; Kempton, M.J.; Taylor, M.J.; McGuire, P.K. Nature of glutamate alterations in schizophrenia: A meta-analysis of proton magnetic resonance spectroscopy studies. JAMA Psychiatry 2016, 73, 665–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahara, T.; Tsugawa, S.; Noda, Y.; Ueno, F.; Honda, S.; Kinjo, M.; Segawa, H.; Hondo, N.; Mori, Y.; Watanabe, H.; et al. Glutamatergic and GABAergic metabolite levels in schizophrenia-spectrum disorders: A meta-analysis of 1H-magnetic resonance spectroscopy studies. Mol. Psychiatry 2021, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Tomitaka, S.; Tomitaka, M.; Tolliver, B.K.; Sharp, F.R. Bilateral blockade of NMDA receptors in anterior thalamus by dizocilpine (MK-801) injures pyramidal neurons in rat retrosplenial cortex. Eur. J. Neurosci. 2000, 12, 1420–1430. [Google Scholar] [CrossRef] [PubMed]
- Sneeboer, M.A.; van Mierlo, H.C.; Stotijn, E.; MacIntyre, D.J.; Smith, C.; Kahn, R.S.; Hol, E.M.; de Witte, L.D. Increased number of T-lymphocytes in post-mortem brain tissue of patients with schizophrenia. Schizophr. Res. 2019, 216, 526–528. [Google Scholar] [CrossRef]
- Dinesh, A.A.; Islam, J.; Khan, J.; Turkheimer, F.; Vernon, A.C. Effects of Antipsychotic Drugs: Cross Talk Between the Nervous and Innate Immune System. CNS Drugs 2020, 34, 1229–1251. [Google Scholar] [CrossRef] [PubMed]
- Khandaker, G.M.; Zimbron, J.; Lewis, G.; Jones, P. Prenatal maternal infection, neurodevelopment and adult schizophrenia: A systematic review of population-based studies. Psychol. Med. 2012, 43, 239–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.S.; Derkits, E.J. Prenatal Infection and Schizophrenia: A Review of Epidemiologic and Translational Studies. Am. J. Psychiatry 2010, 167, 261–280. [Google Scholar] [CrossRef] [Green Version]
- Prata, J.; Santos, S.G.; Almeida, M.I.; Coelho, R.; Barbosa, M.A. Bridging Autism Spectrum Disorders and Schizophrenia through inflammation and biomarkers—Pre-clinical and clinical investigations. J. Neuroinflammation 2017, 14, 1–33. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Geng, H.; Liu, W.; Zhang, G. Prenatal, perinatal, and postnatal factors associated with autism: A meta-analysis. Medicine (Baltimore) 2017, 96, e6696. [Google Scholar] [CrossRef]
- Smith, S.E.P.; Li, J.; Garbett, K.; Mirnics, K.; Patterson, P.H. Maternal Immune Activation Alters Fetal Brain Development through Interleukin-6. J. Neurosci. 2007, 27, 10695–10702. [Google Scholar] [CrossRef] [Green Version]
- Gober, R.; Ardalan, M.; Shiadeh, S.M.J.; Duque, L.; Garamszegi, S.P.; Ascona, M.; Barreda, A.; Sun, X.; Mallard, C.; Vontell, R.T. Microglia activation in postmortem brains with schizophrenia demonstrates distinct morphological changes between brain regions. Brain Pathol. 2021, 32, e13003. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Deng, W.; Gong, Q.; Huang, C.; Jiang, L.; Li, M.; He, Z.; Wang, Q.; Ma, X.; Wang, Y.; et al. Extensive brain structural network abnormality in first-episode treatment-naive patients with schizophrenia: Morphometrical and covariation study. Psychol. Med. 2014, 44, 2489–2501. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, D.R.; Haroon, E.; Miller, A.H.; Strauss, G.P.; Buckley, P.F.; Miller, B.J. TNF-α and IL-6 are associated with the deficit syndrome and negative symptoms in patients with chronic schizophrenia. Schizophr. Res. 2018, 199, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Koolschijn, R.S.; Shpektor, A.; Clarke, W.T.; Ip, I.B.; Dupret, D.; Emir, U.E.; Barron, H.C. Author response: Memory recall involves a transient break in excitatory-inhibitory balance. Elife 2021, 10, e70071. [Google Scholar] [CrossRef]
- Coustals, N.; Martelli, C.; Brunet-Lecomte, M.; Petillion, A.; Romeo, B.; Benyamina, A. Chronic smoking and cognition in patients with schizophrenia: A meta-analysis. Schizophr. Res. 2020, 222, 113–121. [Google Scholar] [CrossRef]
- Vancampfort, D.; Firth, J.; Schuch, F.; Rosenbaum, S.; Mugisha, J.; Hallgren, M.; Probst, M.; Ward, P.; Gaughran, F.; De Hert, M.; et al. Sedentary behavior and physical activity levels in people with schizophrenia, bipolar disorder and major depressive disorder: A global systematic review and meta-analysis. World Psychiatry 2017, 16, 308–315. [Google Scholar] [CrossRef]
- Pandurangi, A.K.; Buckley, P.F. Inflammation, Antipsychotic Drugs, and Evidence for Effectiveness of Anti-inflammatory Agents in Schizophrenia. Curr. Top. Behav. Neurosci. 2019, 44, 227–244. [Google Scholar] [CrossRef]
- Pardo-De-Santayana, G.; Juncal-Ruiz, M.; Vázquez-Bourgon, J.; Riesco-Dávila, L.; de la Foz, V.O.-G.; Pelayo-Terán, J.M.; López-Hoyos, M.; Crespo-Facorro, B. Active psychosis and pro-inflammatory cytokines in first-episode of psychosis. J. Psychiatr. Res. 2020, 134, 150–157. [Google Scholar] [CrossRef]
- Jeppesen, R.; Christensen, R.H.B.; Pedersen, E.M.J.; Nordentoft, M.; Hjorthøj, C.; Köhler-Forsberg, O.; Benros, M.E. Efficacy and safety of anti-inflammatory agents in treatment of psychotic disorders–A comprehensive systematic review and meta-analysis. Brain Behav. Immun. 2020, 90, 364–380. [Google Scholar] [CrossRef]
- Tomasik, J.; Yolken, R.H.; Bahn, S.; Dickerson, F.B. Immunomodulatory Effects of Probiotic Supplementation in Schizophrenia Patients: A Randomized, Placebo-Controlled Trial. Biomark. Insights 2015, 10, BMI.S22007–54. [Google Scholar] [CrossRef]
- Thapar, A.; Cooper, M.; Rutter, M. Neurodevelopmental disorders. Lancet Psychiatry 2017, 4, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Gzielo, K.; Nikiforuk, A. Astroglia in Autism Spectrum Disorder. Int. J. Mol. Sci. 2021, 22, 11544. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E.M.; Lindemann, L.; Jønch, A.E.; Apostol, G.; Bear, M.F.; Carpenter, R.L.; Crawley, J.N.; Curie, A.; Des Portes, V.; Hossain, F.; et al. Drug development for neurodevelopmental disorders: Lessons learned from fragile X syndrome. Nat. Rev. Drug Discov. 2018, 17, 280–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, S.K.; Pradhan, S.; du Plessis, A.J.; Ben-Ari, Y.; Limperopoulos, C. GABA and glutamate in the preterm neonatal brain: In-vivo measurement by magnetic resonance spectroscopy. NeuroImage 2021, 238, 118215. [Google Scholar] [CrossRef]
- Kang, Q.Q.; Li, X.; Tong, G.L.; Fan, Y.L.; Shi, L. Magnetic resonance spectroscopy features of the thalamus and the cerebellum and their association with clinical features in children with autism spectrum disorder: A prospective study. Zhongguo Dang Dai Er Ke Za Zhi 2021, 23, 1250–1255. [Google Scholar]
- Xie, M.-J.; Iwata, K.; Ishikawa, Y.; Nomura, Y.; Tani, T.; Murata, K.; Fukazawa, Y.; Matsuzaki, H. Autistic-Like Behavior and Impairment of Serotonin Transporter and AMPA Receptor Trafficking in N-Ethylmaleimide Sensitive Factor Gene-Deficient Mice. Front. Genet. 2021, 14, 1855–1866. [Google Scholar] [CrossRef]
- Eltokhi, A.; Santuy, A.; Merchan-Perez, A.; Sprengel, R. Glutamatergic Dysfunction and Synaptic Ultrastructural Alterations in Schizophrenia and Autism Spectrum Disorder: Evidence from Human and Rodent Studies. Int. J. Mol. Sci. 2020, 22, 59. [Google Scholar] [CrossRef]
- Chisholm, K.; Lin, A.; Abu-Akel, A.; Wood, S. The association between autism and schizophrenia spectrum disorders: A review of eight alternate models of co-occurrence. Neurosci. Biobehav. Rev. 2015, 55, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Bejjani, A.; O’Neill, J.; Kim, J.A.; Frew, A.J.; Yee, V.W.; Ly, R.; Kitchen, C.; Salamon, N.; McCracken, J.T.; Toga, A.W.; et al. Elevated Glutamatergic Compounds in Pregenual Anterior Cingulate in Pediatric Autism Spectrum Disorder Demonstrated by 1H MRS and 1H MRSI. PLoS ONE 2012, 7, e38786. [Google Scholar] [CrossRef]
- Bernardi, S.; Anagnostou, E.; Shen, J.; Kolevzon, A.; Buxbaum, J.D.; Hollander, E.; Hof, P.R.; Fan, J. In vivo 1H-magnetic resonance spectroscopy study of the attentional networks in autism. Brain Res. 2011, 1380, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Purkayastha, P.; Malapati, A.; Yogeeswari, P.; Sriram, D. A Review on GABA/Glutamate Pathway for Therapeutic Intervention of ASD and ADHD. Curr. Med. Chem. 2015, 22, 1850–1859. [Google Scholar] [CrossRef] [PubMed]
- Hollestein, V.; Buitelaar, J.K.; Brandeis, D.; Banaschewski, T.; Kaiser, A.; Hohmann, S.; Oranje, B.; Gooskens, B.; Durston, S.; Williams, S.C.; et al. Developmental changes in fronto-striatal glutamate and their association with functioning during inhibitory control in autism spectrum disorder and obsessive compulsive disorder. NeuroImage: Clin. 2021, 30, 102622. [Google Scholar] [CrossRef] [PubMed]
- He, J.L.; Oeltzschner, G.; Mikkelsen, M.; Deronda, A.; Harris, A.D.; Crocetti, D.; Wodka, E.L.; Mostofsky, S.H.; Edden, R.A.E.; Puts, N.A.J. Region-specific elevations of glutamate + glutamine correlate with the sensory symptoms of autism spectrum disorders. Transl. Psychiatry 2021, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, E.J.; Power, H.; Fawcett, J.M. Women are at greater risk of OCD than men: A meta-analytic review of OCD prevalence worldwide. J. Clin. Psychiatry 2020, 81, 19r13085. [Google Scholar] [CrossRef]
- Tanaka, K. Astroglia and Obsessive Compulsive Disorder. Adv. Neurobiol. 2021, 26, 139–149. [Google Scholar] [CrossRef]
- Batistuzzo, M.C.; Sottili, B.A.; Shavitt, R.G.; Lopes, A.C.; Cappi, C.; de Mathis, M.A.; Pastorello, B.; Diniz, J.B.; Silva, R.M.F.; Miguel, E.C.; et al. Lower Ventromedial Prefrontal Cortex Glutamate Levels in Patients with Obsessive–Compulsive Disorder. Front. Psychiatry 2021, 12, 668304. [Google Scholar] [CrossRef]
- Aida, T.; Yoshida, J.; Nomura, M.; Tanimura, A.; Iino, Y.; Soma, M.; Bai, N.; Ito, Y.; Cui, W.; Aizawa, H.; et al. Astroglial Glutamate Transporter Deficiency Increases Synaptic Excitability and Leads to Pathological Repetitive Behaviors in Mice. Neuropsychopharmacology 2015, 40, 1569–1579. [Google Scholar] [CrossRef]
- Wu, K.; Hanna, G.L.; Easter, P.; Kennedy, J.L.; Rosenberg, D.R.; Arnold, P.D. Glutamate system genes and brain volume alterations in pediatric obsessive-compulsive disorder: A preliminary study. Psychiatry Res. Neuroimaging 2013, 211, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Lesch, K.; Merker, S.; Reif, A.; Novak, M. Dances with black widow spiders: Dysregulation of glutamate signalling enters centre stage in ADHD. Eur. Neuropsychopharmacol. 2012, 23, 479–491. [Google Scholar] [CrossRef]
- Soto, D.; Altafaj, X.; Sindreu, C.; Bayés, A. Glutamate receptor mutations in psychiatric and neurodevelopmental disorders. Commun. Integr. Biol. 2014, 7, e27887. [Google Scholar] [CrossRef] [Green Version]
- Durães, F.; Pinto, M.; Sousa, M.E. Old Drugs as New Treatments for Neurodegenerative Diseases. Pharmaceuticals 2018, 11, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamorro, Á.; Dirnagl, U.; Urra, X.; Planas, A.M. Neuroprotection in acute stroke: Targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016, 15, 869–881. [Google Scholar] [CrossRef]
- Cougnoux, A.; Yerger, J.C.; Fellmeth, M.; Serra-Vinardell, J.; Navid, F.; Wassif, C.A.; Cawley, N.X.; Porter, F.D. Reduction of glutamate neurotoxicity: A novel therapeutic approach for Niemann-Pick disease, type C1. Mol. Genet. Metab. 2021, 134, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2004, 45, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Iovino, L.; Tremblay, M.E.; Civiero, L. Glutamate-induced excitotoxicity in Parkinson’s disease: The role of glial cells. J. Pharmacol. Sci. 2020, 144, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Rebec, G.V. Corticostriatal network dysfunction in Huntington’s disease: Deficits in neural processing, glutamate transport, and ascorbate release. CNS Neurosci. Ther. 2018, 24, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Van Laar, V.S.; Roy, N.; Liu, A.; Rajprohat, S.; Arnold, B.; Dukes, A.; Holbein, C.D.; Berman, S.B. Glutamate excitotoxicity in neurons triggers mitochondrial and endoplasmic reticulum accumulation of Parkin, and, in the presence of N-acetyl cysteine, mitophagy. Neurobiol. Dis. 2015, 74, 180–193. [Google Scholar] [CrossRef] [Green Version]
- Vongthip, W.; Sillapachaiyaporn, C.; Kim, K.-W.; Sukprasansap, M.; Tencomnao, T. Thunbergia laurifolia Leaf Extract Inhibits Glutamate-Induced Neurotoxicity and Cell Death through Mitophagy Signaling. Antioxidants 2021, 10, 1678. [Google Scholar] [CrossRef]
- de Lima, I.B.Q.; Ribeiro, F.M. The Implication of Glial Metabotropic Glutamate Receptors in Alzheimer’s Disease. Curr. Neuropharmacol. 2021, 20, 1. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P. Chronic glutamate toxicity in neurodegenerative diseases-what is the evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef]
- Limbad, C.; Oron, T.R.; Alimirah, F.; Davalos, A.R.; Tracy, T.E.; Gan, L.; Desprez, P.-Y.; Campisi, J. Astrocyte senescence promotes glutamate toxicity in cortical neurons. PLoS ONE 2020, 15, e0227887. [Google Scholar] [CrossRef] [PubMed]
- Olsen, I.; Singhrao, S.K.; Potempa, J. Citrullination as a plausible link to periodontitis, rheumatoid arthritis, atherosclerosis and Alzheimer’s disease. J. Oral Microbiol. 2018, 10, 1487742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Nasrallah, H.A. The use of memantine in neuropsychiatric disorders: An overview. Ann. Clin. Psychiatry 2018, 30, 234–248. [Google Scholar] [PubMed]
- Landi, D.; Vollaro, S.; Pellegrino, G.; Mulas, D.; Ghazaryan, A.; Falato, E.; Pasqualetti, P.; Rossini, P.; Filippi, M. Oral fingolimod reduces glutamate-mediated intracortical excitability in relapsing–remitting multiple sclerosis. Clin. Neurophysiol. 2015, 126, 165–169. [Google Scholar] [CrossRef]
- de la Rubia-Ortí, J.E.; Fernández, D.; Platero, F.; García-Pardo, M.P. Can ketogenic diet improve Alzheimer’s disease? Association with anxiety, depression, and glutamate system. Front. Nutr. 2021, 8, 744398. [Google Scholar] [CrossRef]
- Manford, M. Recent advances in epilepsy. J. Neurol. 2017, 264, 1811–1824. [Google Scholar] [CrossRef] [Green Version]
- Hanada, T. Ionotropic Glutamate Receptors in Epilepsy: A Review Focusing on AMPA and NMDA Receptors. Biomolecules 2020, 10, 464. [Google Scholar] [CrossRef] [Green Version]
- Yılmaz, C.; Karali, K.; Fodelianaki, G.; Gravanis, A.; Chavakis, T.; Charalampopoulos, I.; Alexaki, V.I. Neurosteroids as regulators of neuroinflammation. Front. Neuroendocrinol. 2019, 55, 100788. [Google Scholar] [CrossRef]
- Armstrong, R.A. Risk factors for Alzheimer’s disease. Folia Neuropathol. 2019, 57, 87–105. [Google Scholar] [CrossRef] [Green Version]
- Yan, E.B.; Frugier, T.; Lim, E.; Heng, R.B.; Sundaram, G.; Tan, M.; Rosenfeld, J.V.; Walker, D.W.; Guillemin, G.; Morganti-Kossmann, C. Activation of the kynurenine pathway and increased production of the excitotoxin quinolinic acid following traumatic brain injury in humans. J. Neuroinflammation 2015, 12, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Eisele, A.; Hill-Strathy, M.; Michels, L.; Rauen, K. Magnetic Resonance Spectroscopy following Mild Traumatic Brain Injury: A Systematic Review and Meta-Analysis on the Potential to Detect Posttraumatic Neurodegeneration. Neurodegener. Dis. 2020, 20, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Faye, C.; McGowan, J.C.; Denny, C.A.; David, D.J. Neurobiological Mechanisms of Stress Resilience and Implications for the Aged Population. Curr. Neuropharmacol. 2018, 16, 234–270. [Google Scholar] [CrossRef] [PubMed]
- Rosso, I.M.; Crowley, D.J.; Silveri, M.M.; Rauch, S.L.; Jensen, J.E. Hippocampus Glutamate and N-Acetyl Aspartate Markers of Excitotoxic Neuronal Compromise in Posttraumatic Stress Disorder. Neuropsychopharmacology 2017, 42, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Averill, L.A.; Purohit, P.; Averill, C.L.; Boesl, M.A.; Krystal, J.H.; Abdallah, C.G. Glutamate dysregulation and glutamatergic therapeutics for PTSD: Evidence from human studies. Neurosci. Lett. 2016, 649, 147–155. [Google Scholar] [CrossRef] [Green Version]
- Feder, A.; Rutter, S.B.; Schiller, D.; Charney, D.S. The emergence of ketamine as a novel treatment for posttraumatic stress disorder. Adv. Pharmacol. 2020, 89, 261–286. [Google Scholar] [CrossRef]
- Zohar, J.; Amital, D.; Miodownik, C.; Kotler, M.; Bleich, A.; Lane, R.M.; Austin, C. Double-Blind Placebo-Controlled Pilot Study of Sertraline in Military Veterans with Posttraumatic Stress Disorder. J. Clin. Psychopharmacol. 2002, 22, 190–195. [Google Scholar] [CrossRef]
- Perez-Garcia, G.; Sosa, M.A.G.; De Gasperi, R.; Tschiffely, A.E.; McCarron, R.M.; Hof, P.R.; Gandy, S.; Ahlers, S.T.; Elder, G.A. Blast-induced "PTSD": Evidence from an animal model. Neuropharmacology 2018, 145, 220–229. [Google Scholar] [CrossRef]
- Guardia, D.; Rolland, B.; Karila, L.; Cottencin, O. GABAergic and Glutamatergic Modulation in Binge Eating: Therapeutic Approach. Curr. Pharm. Des. 2011, 17, 1396–1409. [Google Scholar] [CrossRef]
- Godlewska, B.R.; Pike, A.; Sharpley, A.L.; Ayton, A.; Park, R.J.; Cowen, P.J.; Emir, U.E. Brain glutamate in anorexia nervosa: A magnetic resonance spectroscopy case control study at 7 Tesla. Psychopharmacology 2016, 234, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Smesny, S.; Große, J.; Gussew, A.; Langbein, K.; Schönfeld, N.; Wagner, G.; Valente, M.; Reichenbach, J.R. Prefrontal glutamatergic emotion regulation is disturbed in cluster B and C personality disorders—A combined 1H/31P-MR spectroscopic study. J. Affect. Disord. 2018, 227, 688–697. [Google Scholar] [CrossRef]
- Defrin, R.; Sagy, N.C.; Biran, I.; Goor-Aryeh, I.; Shai, R.; Ginzburg, K. Enhanced pain modulation capacity among individuals with borderline personality disorder: A possible mechanism underlying their hypoalgesia. Eur. J. Pain 2019, 24, 544–554. [Google Scholar] [CrossRef] [PubMed]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.-N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, K.M.; Kang, N.; Bienenstock, J.; Foster, J.A. Reduced anxiety-like behavior and central neurochemical change in germ-free mice. Neurogastroenterol. Motil. 2010, 23, 255-e119. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, L.; Wang, X.; Wang, F.; Zhang, J.; Jiang, R.; Wang, X.; Wang, K.; Liu, Z.; Xia, Z.; et al. Similar Fecal Microbiota Signatures in Patients with Diarrhea-Predominant Irritable Bowel Syndrome and Patients with Depression. Clin. Gastroenterol. Hepatol. 2016, 14, 1602–1611.e5. [Google Scholar] [CrossRef]
- Wang, H.; Liu, L.; Rao, X.; Zeng, B.; Yu, Y.; Zhou, C.; Zeng, L.; Zheng, P.; Pu, J.; Xu, S.; et al. Integrated phosphoproteomic and metabolomic profiling reveals perturbed pathways in the hippocampus of gut microbiota dysbiosis mice. Transl. Psychiatry 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Maqsood, R.; Stone, T.W. The Gut-Brain Axis, BDNF, NMDA and CNS Disorders. Neurochem. Res. 2016, 41, 2819–2835. [Google Scholar] [CrossRef]
- Barrio, C.; Arias-Sánchez, S.; Martín-Monzón, I. The gut microbiota-brain axis, psychobiotics and its influence on brain and behaviour: A systematic review. Psychoneuroendocrinology 2021, 137, 105640. [Google Scholar] [CrossRef]
- Jangi, S.; Gandhi, R.; Cox, L.; Li, N.; Von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L.; et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 12015. [Google Scholar] [CrossRef] [Green Version]
- Quigley, E.M.M. Microbiota-Brain-Gut Axis and Neurodegenerative Diseases. Curr. Neurol. Neurosci. Rep. 2017, 17, 94. [Google Scholar] [CrossRef]
- Zhu, F.; Ju, Y.; Wang, W.; Wang, Q.; Guo, R.; Ma, Q.; Sun, Q.; Fan, Y.; Xie, Y.; Yang, Z.; et al. Metagenome-wide association of gut microbiome features for schizophrenia. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Z.; Yang, R.; Wang, W.; Qi, L.; Huang, T. Associations between gut microbiota and Alzheimer’s disease, major depressive disorder, and schizophrenia. J. Neuroinflammation 2020, 17, 288. [Google Scholar] [CrossRef] [PubMed]
- Sanada, K.; Nakajima, S.; Kurokawa, S.; Barceló-Soler, A.; Ikuse, D.; Hirata, A.; Yoshizawa, A.; Tomizawa, Y.; Salas-Valero, M.; Noda, Y.; et al. Gut microbiota and major depressive disorder: A systematic review and meta-analysis. J. Affect. Disord. 2020, 266, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Wang, K.; Hu, J. Effect of Probiotics on Depression: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Nutrients 2016, 8, 483. [Google Scholar] [CrossRef] [PubMed]
- Stripling, J.; Rodriguez, M. Current Evidence in Delivery and Therapeutic Uses of Fecal Microbiota Transplantation in Human Diseases—Clostridium difficile Disease and Beyond. Am. J. Med. Sci. 2018, 356, 424–432. [Google Scholar] [CrossRef]
- Meyyappan, A.C.; Forth, E.; Wallace, C.J.K.; Milev, R. Effect of fecal microbiota transplant on symptoms of psychiatric disorders: A systematic review. BMC Psychiatry 2020, 20, 299. [Google Scholar]
- Zhu, F.; Guo, R.; Wang, W.; Ju, Y.; Wang, Q.; Ma, Q.; Sun, Q.; Fan, Y.; Xie, Y.; Yang, Z.; et al. Transplantation of microbiota from drug-free patients with schizophrenia causes schizophrenia-like abnormal behaviors and dysregulated kynurenine metabolism in mice. Mol. Psychiatry 2019, 25, 2905–2918. [Google Scholar] [CrossRef]
- Kang, D.-W.; Adams, J.B.; Gregory, A.C.; Borody, T.; Chittick, L.; Fasano, A.; Khoruts, A.; Geis, E.; Maldonado, J.; McDonough-Means, S.; et al. Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: An open-label study. Microbiome 2017, 5, 1–16. [Google Scholar] [CrossRef]
- Makkawi, S.; Camara-Lemarroy, C.; Metz, L. Fecal microbiota transplantation associated with 10 years of stability in a patient with SPMS. Neurol.-Neuroimmunol. Neuroinflammation 2018, 5, e459. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Xu, H.; Luo, Q.; He, J.; Li, M.; Chen, H.; Chen, H.; Tang, W.; Zhou, Y. Fecal microbiota transplantation to treat Parkinson’s disease with constipation: A case report. Medicine (Baltimore) 2019, 98, e16163. [Google Scholar] [CrossRef]
- Meyyappan, A.C.; Milev, R. The safety, efficacy, and tolerability of microbial ecosystem therapeutic-2 in people with major depression and/or generalized anxiety disorder: Protocol for a phase 1, open-label study. JMIR Res. Protoc. 2020, 9, e17223. [Google Scholar]
- Vivar, C.; van Praag, H. Running changes the brain: The long and the short of it. Physiology (Bethesda) 2017, 32, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Glutamate and Neurotrophic Factors in Neuronal Plasticity and Disease. Ann. N. Y. Acad. Sci. 2008, 1144, 97–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Wu, Y.; Jia, J. Exercise preconditioning and brain ischemic tolerance. Neuroscience 2011, 177, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Schuch, F.B.; Vancampfort, D.; Richards, J.; Rosenbaum, S.; Ward, P.; Stubbs, B. Exercise as a treatment for depression: A meta-analysis adjusting for publication bias. J. Psychiatr. Res. 2016, 77, 42–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firth, J.; Stubbs, B.; Rosenbaum, S.; Vancampfort, D.; Malchow, B.; Schuch, F.; Elliott, R.; Nuechterlein, K.H.; Yung, A.R. Aerobic Exercise Improves Cognitive Functioning in People with Schizophrenia: A Systematic Review and Meta-Analysis. Schizophr. Bull. 2016, 43, 546–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bremer, E.; Crozier, M.; Lloyd, M. A systematic review of the behavioural outcomes following exercise interventions for children and youth with autism spectrum disorder. Autism 2016, 20, 899–915. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.Á.A.; Ma, M.Á.B.; García-Casares, N. Efecto del ejercicio físico en la enfermedad de Alzheimer. Una revisión sistemática [Effect of physical exercise on Alzheimer’s disease. A systematic review]. Aten. Primaria 2020, 52, 307–318. [Google Scholar] [CrossRef]
- Rao, A.K.; Chou, A.; Bursley, B.; Smulofsky, J.; Jezequel, J. Systematic Review of the Effects of Exercise on Activities of Daily Living in People with Alzheimer’s Disease. Am. J. Occup. Ther. 2014, 68, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.W.; Plassman, B.L.; Burke, J.; Benjamin, S. Preventing Alzheimer’s disease and cognitive decline. Evid. Rep. Technol. Assess (Full Rep) 2010, 193, 1–727. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGrath, T.; Baskerville, R.; Rogero, M.; Castell, L. Emerging Evidence for the Widespread Role of Glutamatergic Dysfunction in Neuropsychiatric Diseases. Nutrients 2022, 14, 917. https://doi.org/10.3390/nu14050917
McGrath T, Baskerville R, Rogero M, Castell L. Emerging Evidence for the Widespread Role of Glutamatergic Dysfunction in Neuropsychiatric Diseases. Nutrients. 2022; 14(5):917. https://doi.org/10.3390/nu14050917
Chicago/Turabian StyleMcGrath, Thomas, Richard Baskerville, Marcelo Rogero, and Linda Castell. 2022. "Emerging Evidence for the Widespread Role of Glutamatergic Dysfunction in Neuropsychiatric Diseases" Nutrients 14, no. 5: 917. https://doi.org/10.3390/nu14050917
APA StyleMcGrath, T., Baskerville, R., Rogero, M., & Castell, L. (2022). Emerging Evidence for the Widespread Role of Glutamatergic Dysfunction in Neuropsychiatric Diseases. Nutrients, 14(5), 917. https://doi.org/10.3390/nu14050917