
The Therapeutic Role of Ketogenic Diet in Neurological Disorders


Abstract
:1. Ketogenic Diet
1.1. History of the Ketogenic Diet
1.2. Types and Characteristics of Ketogenic Diets
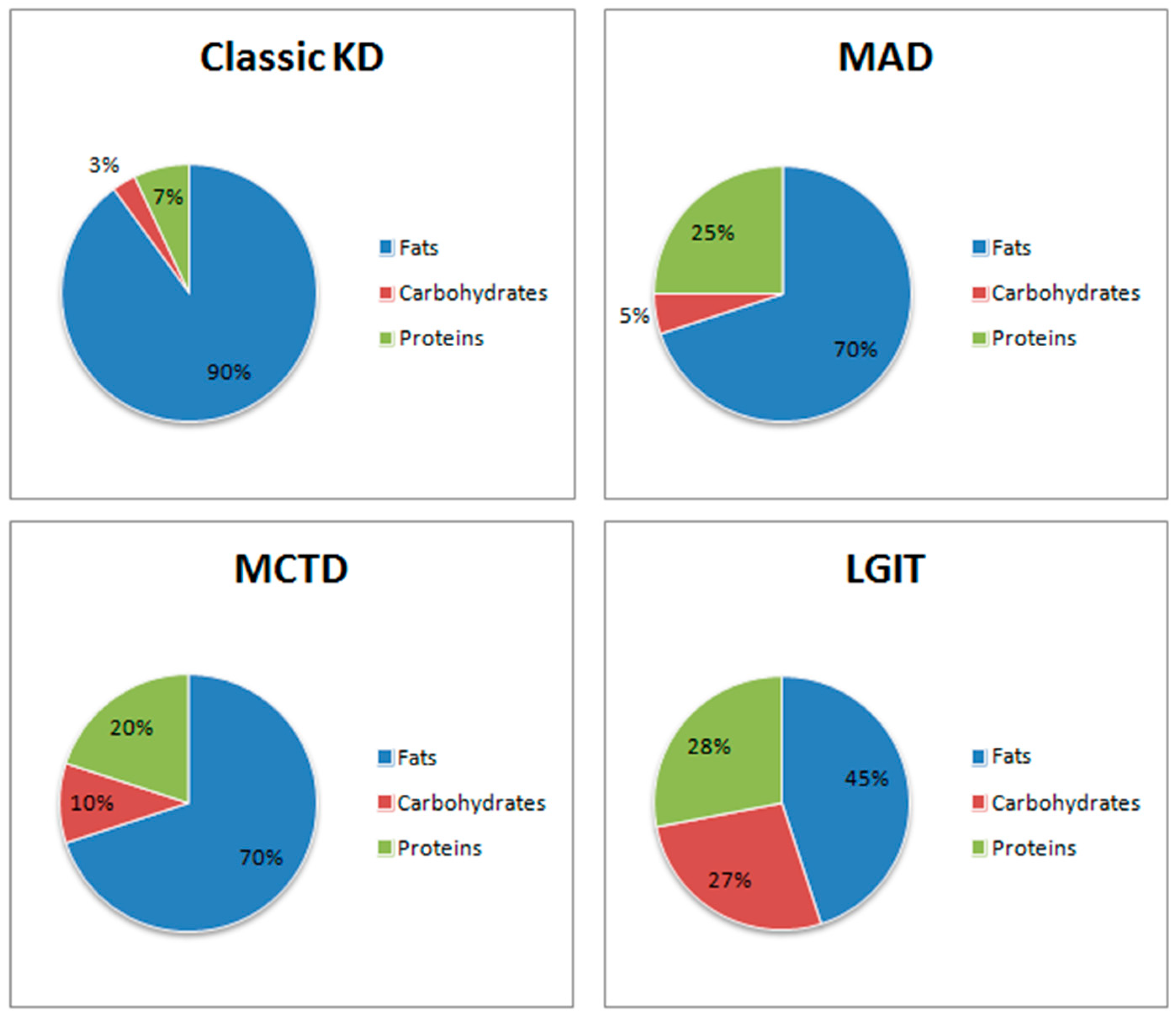
1.2.1. Classic Ketogenic Diet (CKD)
1.2.2. Modifications of Classic Ketogenic Diet
High-Protein Ketogenic Diet (MAD)
Medium-Chain Triglycerides Diet (MCTD)
Very Low Calorie Ketogenic Diet (VLCKD)
Low Glycaemic Index Treatment (LGIT)
Cyclical Ketogenic Diet (CKD)
Targeted Ketogenic Diet (TKD)
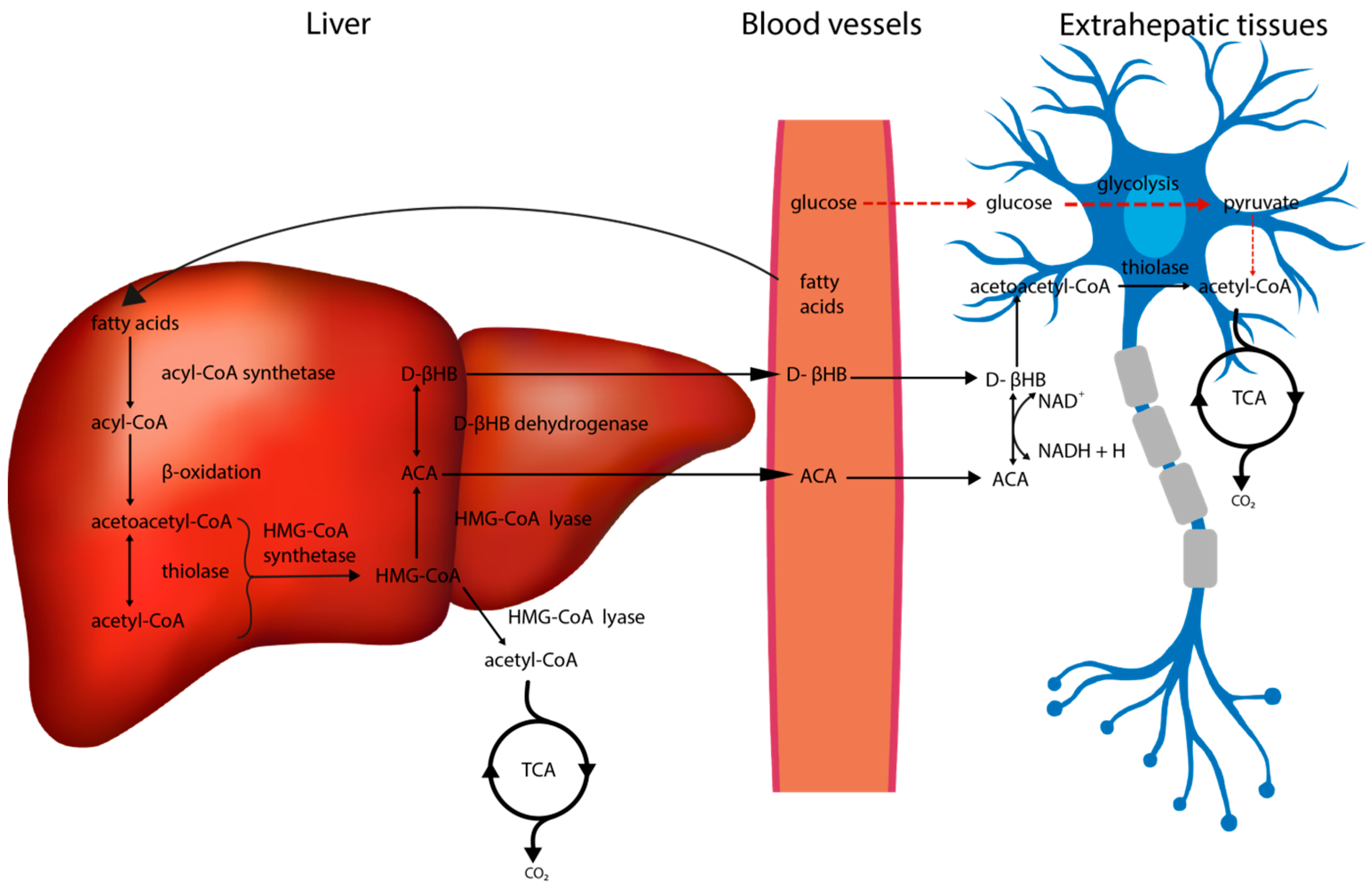
2. Metabolic Alterations in the Brain Associated with the Ketogenic Diet
2.1. The Impact of the Ketogenic Diet on Glucose Metabolism
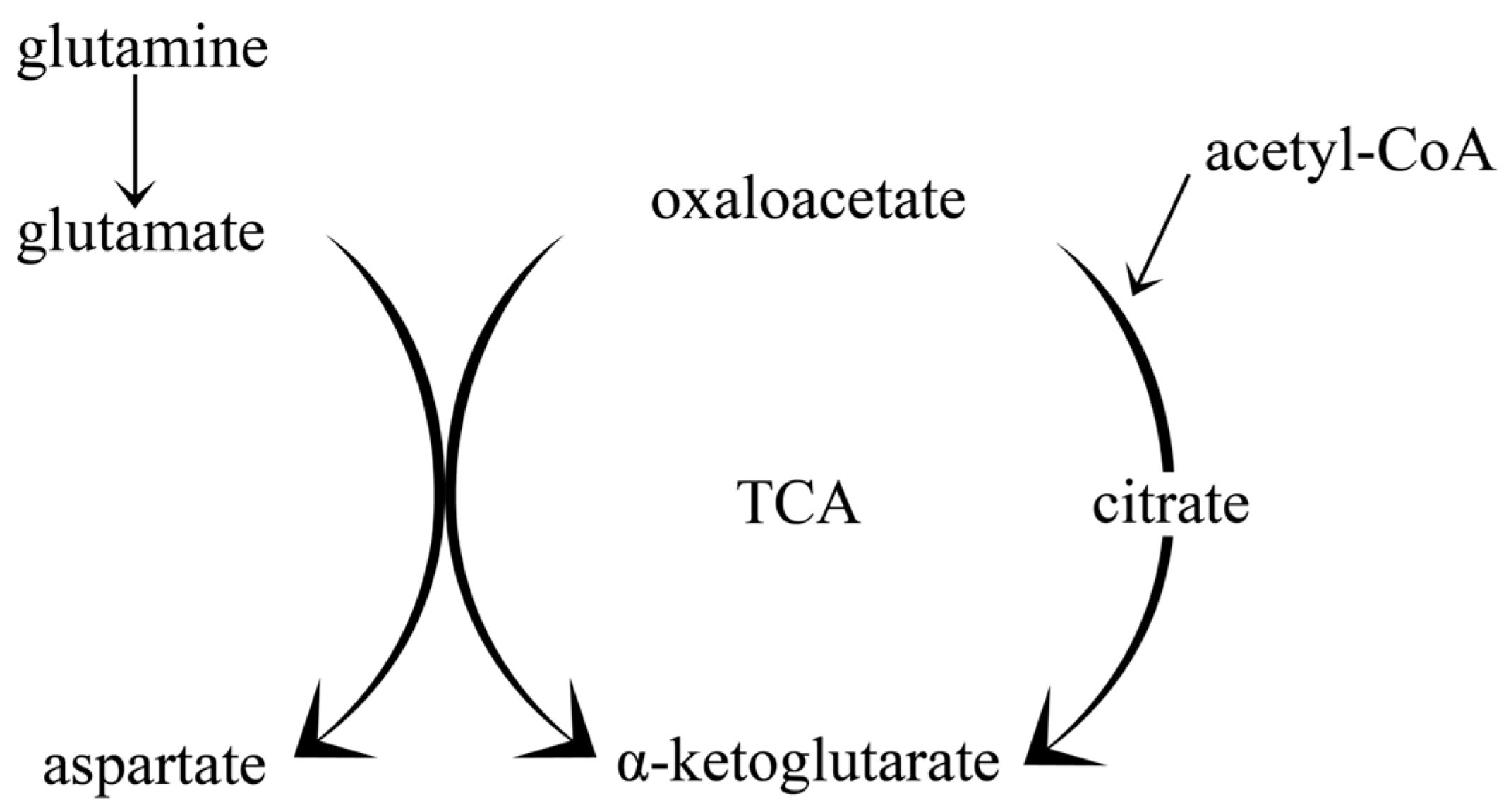
2.2. The Impact of the Ketogenic Diet on Amino Acid Metabolism and Neurotransmitter Synthesis; Glutamate-Glutamine Cycle
2.3. The Impact of the Ketogenic Diet on Insulin Signalling
2.4. The Impact of the Ketogenic Diet on Oxidative Stress
2.5. The Impact of the Ketogenic Diet on Neuroinflammation
2.6. The Impact of the Ketogenic Diet on Brain-Derived Neurotrophic Factor (BDNF)
2.7. The Impact of the Ketogenic Diet on Activity of ATP-sensitive Potassium Channels
2.8. The Impact of the Ketogenic Diet on Beta Amyloid and Tau Protein Synthesis
3. The Impact of the Ketogenic Diet on Gut Microbiota
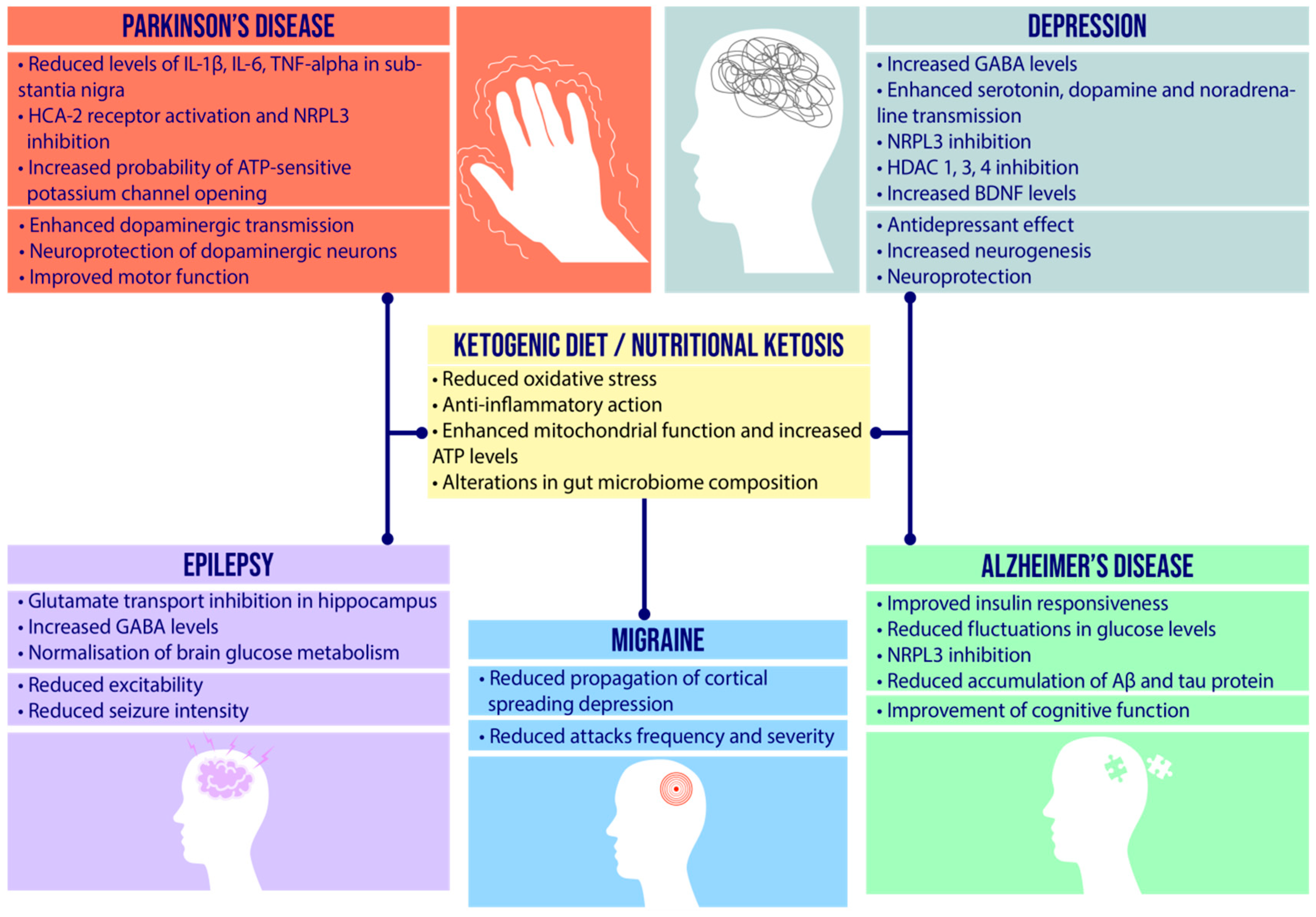
4. Etiopathogenesis of Neurological Diseases and Therapeutic Role of Ketogenic Diet
4.1. Epilepsy
4.1.1. Etiopathogenesis and Potential Role of Ketogenic Diet
4.1.2. Indications for a Ketogenic Diet
4.1.3. Clinical Data
4.2. Depression
4.2.1. Etiopathogenesis and Potential Role of Ketogenic Diet
4.2.2. Clinical Data
4.3. Migraine
4.3.1. Etiopathogenesis and Potential Role of Ketogenic Diet
4.3.2. Clinical Data
4.4. Alzheimer’s Disease
4.4.1. Etiopathogenesis and Potential Role of Ketogenic Diet
4.4.2. Medical Foods
4.4.3. Clinical Data
4.5. Parkinson’s Disease
4.5.1. Etiopathogenesis and Potential Role of Ketogenic Diet
4.5.2. Clinical Data
5. Adverse Effects of the Ketogenic Diet
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kwon, H.E.; Kim, H.D. Recent aspects of ketogenic diet in neurological disorders. Acta Epileptol. 2021, 3, 21. [Google Scholar] [CrossRef]
- Zilberter, Y.; Zilberter, T. Glucose-Sparing Action of Ketones Boosts Functions Exclusive to Glucose in the Brain. Eneuro 2020, 7, ENEURO.0303-20.2020. [Google Scholar] [CrossRef] [PubMed]
- Niepoetter, P.; Gopalan, C. The Effects of Ketogenic Diets on Psychiatric Disorders Involving Mitochondrial Dysfunction: A Literature Review of the Influence of Dieting on Autism, Depression, Anxiety, and Schizophrenia. HAPS Educ. 2019, 23, 426–431. [Google Scholar] [CrossRef]
- Tillery, E.E.; Ellis, K.D.; Threatt, T.B.; Reyes, H.A.; Plummer, C.S.; Barney, L.R. The use of the ketogenic diet in the treatment of psychiatric disorders. Mental Health Clin. 2021, 11, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412–426. [Google Scholar] [CrossRef]
- Fu, S.; Wang, J.; Xue, W.; Liu, H.; Liu, B.; Zeng, Y.; Li, S.; Huang, B.; Lv, Q.; Wang, W.; et al. Anti-inflammatory effects of BHBA in both in vivo and in vitro Parkinson’s disease models are mediated by GPR109A-dependent mechanisms. J. Neuroinflamm. 2015, 12, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimazu, T.; Hirschey, M.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of Oxidative Stress by β-Hydroxybutyrate, an Endogenous Histone Deacetylase Inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, P.; Xu, X.; Zhang, Y.; Gong, Y.; Hu, W.; Gao, M.; Wu, Y.; Ling, Y.; Zhao, X.; et al. The ketone body metabolite β-hydroxybutyrate induces an antidepression-associated ramification of microglia via HDACs inhibition-triggered Akt-small RhoGTPase activation. Glia 2018, 66, 256–278. [Google Scholar] [CrossRef]
- Qiao, G.; Lv, T.; Zhang, M.; Chen, P.; Sun, Q.; Zhang, J.; Li, Q. β-hydroxybutyrate (β-HB) exerts anti-inflammatory and antioxidant effects in lipopolysaccharide (LPS)-stimulated macrophages in Liza haematocheila. Fish Shellfish Immunol. 2020, 107, 444–451. [Google Scholar] [CrossRef]
- Noh, H.S.; Hah, Y.S.; Nilufar, R.; Han, J.; Bong, J.H.; Kang, S.S.; Cho, G.J.; Choi, W.S. Acetoacetate protects neuronal cells from oxidative glutamate toxicity. J. Neurosci. Res. 2006, 83, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Maalouf, M.; Sullivan, P.G.; Davis, L.; Kim, D.Y.; Rho, J.M. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 2007, 145, 256–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, P.G.; Rippy, N.A.; Dorenbos, K.; Concepcion, R.C.; Agarwal, A.K.; Rho, J.M. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann. Neurol. 2004, 55, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Hasan-Olive, M.M.; Lauritzen, K.H.; Ali, M.; Rasmussen, L.J.; Storm-Mathisen, J.; Bergersen, L.H. A Ketogenic Diet Improves Mitochondrial Biogenesis and Bioenergetics via the PGC1α-SIRT3-UCP2 Axis. Neurochem Res. 2019, 44, 22–37. [Google Scholar] [CrossRef]
- Zou, X.H.; Sun, L.H.; Yang, W.; Li, B.J.; Cui, R.J. Potential role of insulin on the pathogenesis of depression. Cell Prolif. 2020, 53. [Google Scholar] [CrossRef] [Green Version]
- Wheless, J.W. History of the ketogenic diet. Epilepsia 2008, 49, 3–5. [Google Scholar] [CrossRef]
- Vining, E.P.G. A Multicenter Study of the Efficacy of the Ketogenic Diet. Arch Neurol. 1998, 55, 1433. [Google Scholar] [CrossRef] [Green Version]
- Freeman, J.M.; Vining, E.P.G.; Pillas, D.J.; Pyzik, P.L.; Casey, J.C.; Kelly, L.M. The Efficacy of the Ketogenic Diet—1998: A Prospective Evaluation of Intervention in 150 Children. Pediatrics 1998, 102, 1358–1363. [Google Scholar] [CrossRef]
- Neal, E.G.; Chaffe, H.; Schwartz, R.H.; Lawson, M.S.; Edwards, N.; Fitzsimmons, G.; Whitney, A.; Cross, J.H. The ketogenic diet for the treatment of childhood epilepsy: A randomised controlled trial. Lancet Neurol. 2008, 7, 500–506. [Google Scholar] [CrossRef]
- Ułamek-Kozioł, M.; Pluta, R. To treat or not to treat Alzheimer’s disease by the ketogenic diet? That is the question. Neural. Regen. Res. 2020, 15, 857–858. [Google Scholar] [CrossRef]
- Pavón, S.; Lázaro, E.; Martínez, O.; Amayra, I.; López-Paz, J.F.; Caballero, P.; Al-Rashaida, M.; Luna, P.M.; García, M.; Pérez, M.; et al. Ketogenic diet and cognition in neurological diseases: A systematic review. Nutr. Rev. 2021, 79, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.; Hallett, M.; Ehrlich, D. Nutritional Ketosis in Parkinson’s Disease—A Review of Remaining Questions and Insights. Neurotherapeutics 2021, 18, 1637–1649. [Google Scholar] [CrossRef] [PubMed]
- Pondel, N.; Liśkiewicz, D.; Liśkiewicz, A. Dieta ketogeniczna-mechanizm działania i perspektywy zastosowania w terapii: Dane z badań klinicznych [Ketogenic diet—Mechanism of action and perspectives for the use in the therapy: Data from clinical studies]. Postepy Biochem. 2020, 66, 270–286. [Google Scholar] [CrossRef] [PubMed]
- Kossoff, E.H.; Dorward, J.L. The Modified Atkins Diet. Epilepsia 2008, 49, 37–41. [Google Scholar] [CrossRef]
- Kossoff, E.H.; McGrogan, J.R.; Bluml, R.M.; Pillas, D.J.; Rubenstein, J.E.; Vining, E.P. A Modified Atkins Diet Is Effective for the Treatment of Intractable Pediatric Epilepsy. Epilepsia 2006, 47, 421–424. [Google Scholar] [CrossRef]
- Rusek, M.; Pluta, R.; Ułamek-Kozioł, M.; Czuczwar, S.J. Ketogenic diet in alzheimer’s disease. Int. J. Mol. Sci. 2019, 20, 3892. [Google Scholar] [CrossRef] [Green Version]
- Huttenlocher, P.R.; Wilbourn, A.J.; Signore, J.M. Medium-Chain Triglycerides as a Therapy for Intractable Childhood Epilepsy. Neurology 1971, 21, 1097. [Google Scholar] [CrossRef]
- Zhou, W.; Mukherjee, P.; Kiebish, M.A.; Markis, W.T.; Mantis, J.G.; Seyfried, T.N. The Calorically Restricted Ketogenic Diet, an Effective Alternative Therapy for Malignant Brain Cancer. Nutr. Metab. 2007, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Licha, D.; Vidali, S.; Aminzadeh-Gohari, S.; Alka, O.; Breitkreuz, L.; Kohlbacher, O.; Reischl, R.J.; Feichtinger, R.G.; Kofler, B.; Huber, C.G. Untargeted Metabolomics Reveals Molecular Effects of Ketogenic Diet on Healthy and Tumor Xenograft Mouse Models. Int. J. Mol. Sci. 2019, 20, 3873. [Google Scholar] [CrossRef] [Green Version]
- Hughes, S.D.; Kanabus, M.; Anderson, G.; Hargreaves, I.P.; Rutherford, T.; Donnell, M.O.; Cross, J.H.; Rahman, S.; Eaton, S.; Heales, S.J.R. The Ketogenic Diet Component Decanoic Acid Increases Mitochondrial Citrate Synthase and Complex I Activity in Neuronal Cells. J. Neurochem. 2014, 129, 426–433. [Google Scholar] [CrossRef]
- Feinman, R.D.; Pogozelski, W.K.; Astrup, A.; Bernstein, R.K.; Fine, E.J.; Westman, E.C.; Accurso, A.; Frassetto, L.; Gower, B.A.; McFarlane, S.I.; et al. Dietary Carbohydrate Restriction as the First Approach in Diabetes Management: Critical Review and Evidence Base. Nutrition 2015, 31, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega-López, S.; Venn, B.; Slavin, J. Relevance of the Glycemic Index and Glycemic Load for Body Weight, Diabetes, and Cardiovascular Disease. Nutrients 2018, 10, 1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kysel, P.; Haluzíková, D.; Doležalová, R.P.; Laňková, I.; Lacinová, Z.; Kasperová, B.J.; Trnovská, J.; Hrádková, V.; Mráz, M.; Vilikus, Z.; et al. The Influence of Cyclical Ketogenic Reduction Diet vs. Nutritionally Balanced Reduction Diet on Body Composition, Strength, and Endurance Performance in Healthy Young Males: A Randomized Controlled Trial. Nutrients 2020, 12, 2832. [Google Scholar] [CrossRef] [PubMed]
- Owen, O.E.; Morgan, A.P.; Kemp, H.G.; Sullivan, J.M.; Herrera, M.G.; Cahill, G.F. Brain Metabolism during Fasting. J. Clin. Invest. 1967, 46, 1589–1595. [Google Scholar] [CrossRef] [PubMed]
- Masino, S.A.; Rho, J.M. Mechanisms of Ketogenic Diet Action. In Jasper’s Basic Mechanisms of the Epilepsies; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; National Center for Biotechnology Information: Bethesda, MD, USA, 2012; pp. 1157–1182. [Google Scholar]
- Leino, R.L.; Gerhart, D.Z.; Duelli, R.; Enerson, B.E.; Drewes, L.R. Diet-Induced Ketosis Increases Monocarboxylate Transporter (MCT1) Levels in Rat Brain. Neurochem. Int. 2001, 38, 519–527. [Google Scholar] [CrossRef]
- Bentourkia, M.; Tremblay, S.; Pifferi, F.; Rousseau, J.; Lecomte, R.; Cunnane, S. PET Study of 11 C-Acetoacetate Kinetics in Rat Brain during Dietary Treatments Affecting Ketosis. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E796–E801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelking, L.R. Ketone Body Formation and Utilization. In Textb of Veterinary Physiological Chemistry; Elsevier: City of Jackson, MS, USA, 2015; pp. 450–457. [Google Scholar] [CrossRef]
- Mujica-Parodi, L.R.; Amgalan, A.; Sultan, S.F.; Antal, B.; Sun, X.; Skiena, S.; Lithen, A.; Adra, N.; Ratai, E.-M.; Weistuch, C.; et al. Diet Modulates Brain Network Stability, a Biomarker for Brain Aging, in Young Adults. Proc. Natl. Acad. Sci. USA 2020, 117, 6170–6177. [Google Scholar] [CrossRef] [Green Version]
- Taggart, A.K.P.; Kero, J.; Gan, X.; Cai, T.-Q.; Cheng, K.; Ippolito, M.; Ren, N.; Kaplan, R.; Wu, K.; Wu, T.-J.; et al. (D)-β-Hydroxybutyrate Inhibits Adipocyte Lipolysis via the Nicotinic Acid Receptor PUMA-G. J. Biol. Chem. 2005, 280, 26649–26652. [Google Scholar] [CrossRef] [Green Version]
- Elamin, M.; Ruskin, D.N.; Masino, S.A.; Sacchetti, P. Ketone-Based Metabolic Therapy: Is Increased NAD+ a Primary Mechanism? Front Mol. Neurosci. 2017, 10, 377. [Google Scholar] [CrossRef]
- Ma, S.; Suzuki, K. Keto-Adaptation and Endurance Exercise Capacity, Fatigue Recovery, and Exercise-Induced Muscle and Organ Damage Prevention: A Narrative Review. Sports 2019, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Hyde, P.N.; Lustberg, M.B.; Miller, V.J.; LaFountain, R.A.; Volek, J.S. Pleiotropic Effects of Nutritional Ketosis: Conceptual Framework for Keto-Adaptation as a Breast Cancer Therapy. Cancer Treat. Res. Commun. 2017, 12, 32–39. [Google Scholar] [CrossRef]
- Cotter, D.G.; Schugar, R.C.; Wentz, A.E.; André d’Avignon, D.; Crawford, P.A. Successful Adaptation to Ketosis by Mice with Tissue-Specific Deficiency of Ketone Body Oxidation. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E363–E374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokoloff, L. Measurement of Local Cerebral Glucose Utilization and Its Relation to Local Functional Activity in the Brain. Adv. Exp. Med. Biol. 1991, 291, 21–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kuang, Y.; Xu, K.; Harris, D.; Lee, Z.; LaManna, J.; Puchowicz, M.A. Ketosis Proportionately Spares Glucose Utilization in Brain. J. Cereb. Blood Flow Metab. 2013, 33, 1307–1311. [Google Scholar] [CrossRef] [Green Version]
- Hertz, L.; Peng, L.; Dienel, G.A. Energy Metabolism in Astrocytes: High Rate of Oxidative Metabolism and Spatiotemporal Dependence on Glycolysis/Glycogenolysis. J. Cereb. Blood Flow Metab. 2007, 27, 219–249. [Google Scholar] [CrossRef]
- Hertz, L.; Rothman, D.L. Glucose, Lactate, β-Hydroxybutyrate, Acetate, GABA, and Succinate as Substrates for Synthesis of Glutamate and GABA in the Glutamine–Glutamate/GABA Cycle. Adv. Neurobiol. 2016, 13, 9–42. [Google Scholar] [CrossRef]
- Duelli, R.; Kuschinsky, W. Brain Glucose Transporters: Relationship to Local Energy Demand. Physiology 2001, 16, 71–76. [Google Scholar] [CrossRef]
- Cheng, C.M.; Kelley, B.; Wang, J.; Strauss, D.; Eagles, D.A.; Bondy, C.A. A Ketogenic Diet Increases Brain Insulin-Like Growth Factor Receptor and Glucose Transporter Gene Expression. Endocrinology 2003, 144, 2676–2682. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, A.M.; Hernandez-Garzón, E.; Perez-Domper, P.; Perez-Alvarez, A.; Mederos, S.; Matsui, T.; Santi, A.; Trueba-Saiz, A.; García-Guerra, L.; Pose-Utrilla, J.; et al. Insulin Regulates Astrocytic Glucose Handling Through Cooperation With IGF-I. Diabetes 2017, 66, 64–74. [Google Scholar] [CrossRef] [Green Version]
- Melø, T.M.; Nehlig, A.; Sonnewald, U. Neuronal–Glial Interactions in Rats Fed a Ketogenic Diet. Neurochem. Int. 2006, 48, 498–507. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate Uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Juge, N.; Yoshida, Y.; Yatsushiro, S.; Omote, H.; Moriyama, Y. Vesicular Glutamate Transporter Contains Two Independent Transport Machineries. J. Biol. Chem. 2006, 281, 39499–39506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juge, N.; Gray, J.A.; Omote, H.; Miyaji, T.; Inoue, T.; Hara, C.; Uneyama, H.; Edwards, R.H.; Nicoll, R.A.; Moriyama, Y. Metabolic Control of Vesicular Glutamate Transport and Release. Neuron 2010, 68, 99–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yudkoff, M.; Daikhin, Y.; Nissim, I.; Lazarow, A.; Nissim, I. Brain Amino Acid Metabolism and Ketosis. J. Neurosci. Res. 2001, 66, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Norenberg, M.D.; Martinez-Hernandez, A. Fine Structural Localization of Glutamine Synthetase in Astrocytes of Rat Brain. Brain Res. 1979, 161, 303–310. [Google Scholar] [CrossRef]
- Sakai, R.; Cohen, D.M.; Henry, J.F.; Burrin, D.G.; Reeds, P.J. Leucine-Nitrogen Metabolism in the Brain of Conscious Rats: Its Role as a Nitrogen Carrier in Glutamate Synthesis in Glial and Neuronal Metabolic Compartments. J. Neurochem. 2004, 88, 612–622. [Google Scholar] [CrossRef]
- Erecińska, M.; Nelson, D.; Daikhin, Y.; Yudkoff, M. Regulation of GABA Level in Rat Brain Synaptosomes: Fluxes Through Enzymes of the GABA Shunt and Effects of Glutamate, Calcium, and Ketone Bodies. J. Neurochem. 2002, 67, 2325–2334. [Google Scholar] [CrossRef]
- Tokarz, V.L.; MacDonald, P.E.; Klip, A. The Cell Biology of Systemic Insulin Function. J. Cell Biol. 2018, 217, 2273–2289. [Google Scholar] [CrossRef] [Green Version]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global Estimates of Diabetes Prevalence for 2017 and Projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef]
- Ginter, E.; Simko, V. Type 2 Diabetes Mellitus, Pandemic in 21st Century. Adv. Exp. Med. Biol. 2013, 771, 42–50. [Google Scholar] [CrossRef]
- Unnikrishnan, R.; Pradeepa, R.; Joshi, S.R.; Mohan, V. Type 2 Diabetes: Demystifying the Global Epidemic. Diabetes 2017, 66, 1432–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plum, L.; Schubert, M.; Brüning, J.C. The Role of Insulin Receptor Signaling in the Brain. Trends Endocrinol. Metab. 2005, 16, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Lauritzen, H.P.M.M.; Ussar, S.; Christensen, J.H.; Mori, M.A.; Bross, P.; Kahn, C.R. Leptin Regulation of Hsp60 Impacts Hypothalamic Insulin Signaling. J. Clin. Invest. 2013, 123, 4667–4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, A.M.; Torres-Alemán, I. The Many Faces of Insulin-like Peptide Signalling in the Brain. Nat. Rev. Neurosci. 2012, 13, 225–239. [Google Scholar] [CrossRef]
- Salcedo-Tello, P.; Ortiz-Matamoros, A.; Arias, C. GSK3 Function in the Brain during Development, Neuronal Plasticity, and Neurodegeneration. Int. J. Alzheimers Dis. 2011, 2011, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gupta, D.; Kurhe, Y.; Radhakrishnan, M. Antidepressant Effects of Insulin in Streptozotocin Induced Diabetic Mice: Modulation of Brain Serotonin System. Physiol. Behav. 2014, 129, 73–78. [Google Scholar] [CrossRef]
- Verdile, G.; Keane, K.N.; Cruzat, V.F.; Medic, S.; Sabale, M.; Rowles, J.; Wijesekara, N.; Martins, R.N.; Fraser, P.E.; Newsholme, P. Inflammation and Oxidative Stress: The Molecular Connectivity between Insulin Resistance, Obesity, and Alzheimer’s Disease. Mediators Inflamm. 2015, 2015, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guénette, S. Insulin-Degrading Enzyme Regulates the Levels of Insulin, Amyloid β-Protein, and the β-Amyloid Precursor Protein Intracellular Domain in Vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-K.; Kumar, P.; Fu, Q.; Rosen, K.M.; Querfurth, H.W. The Insulin/Akt Signaling Pathway Is Targeted by Intracellular β-Amyloid. Mol. Biol. Cell 2009, 20, 1533–1544. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the Brain: There and Back Again. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Kellar, D.; Craft, S. Brain Insulin Resistance in Alzheimer’s Disease and Related Disorders: Mechanisms and Therapeutic Approaches. Lancet Neurol. 2020, 19, 758–766. [Google Scholar] [CrossRef]
- Xu, W.; Hu, X.; Zhang, X.; Ling, C.; Wang, C.; Gao, L. Cognitive Impairment and Related Factors Among Middle-Aged and Elderly Patients with Type 2 Diabetes from a Bio-Psycho-Social Perspective. Diabetes Metab. Syndr. Obes. 2021, 14, 4361–4369. [Google Scholar] [CrossRef] [PubMed]
- Stoykovich, S.; Gibas, K. APOE Ε4, the Door to Insulin-resistant Dyslipidemia and Brain Fog? A Case Study. Alzheimers Dement. 2019, 11, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Morrill, S.J.; Gibas, K.J. Ketogenic Diet Rescues Cognition in ApoE4+ Patient with Mild Alzheimer’s Disease: A Case Study. Diabetes Metab Syndr. 2019, 13, 1187–1191. [Google Scholar] [CrossRef]
- Fortier, M.; Castellano, C.; St-Pierre, V.; Myette-Côté, É.; Langlois, F.; Roy, M.; Morin, M.; Bocti, C.; Fulop, T.; Godin, J.; et al. A Ketogenic Drink Improves Cognition in Mild Cognitive Impairment: Results of a 6-month RCT. Alzheimer’s Dement. 2021, 17, 543–552. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA Damage: Mechanisms, Mutation, and Disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [Green Version]
- Włodarek, D. Role of Ketogenic Diets in Neurodegenerative Diseases (Alzheimer’s Disease and Parkinson’s Disease). Nutrients 2019, 11, 169. [Google Scholar] [CrossRef] [Green Version]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The Role of Reactive Oxygen Species in the Pathogenesis of Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease: A Mini Review. Oxid. Med. Cell Longev. 2016, 2016, 1–15. [Google Scholar] [CrossRef]
- Navab, M.; Gharavi, N.; Watson, A.D. Inflammation and Metabolic Disorders. Curr. Opin. Clin. Nutr. Metab. Care. 2008, 11, 459–464. [Google Scholar] [CrossRef]
- Delerive, P.; de Bosscher, K.; Besnard, S.; vanden Berghe, W.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.-C.; Tedgui, A.; Haegeman, G.; Staels, B. Peroxisome Proliferator-Activated Receptor α Negatively Regulates the Vascular Inflammatory Gene Response by Negative Cross-Talk with Transcription Factors NF-ΚB and AP-1. J. Biol. Chem. 1999, 274, 32048–32054. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Cheng, B. Neuroprotective and Anti-Inflammatory Activities of Ketogenic Diet on MPTP-Induced Neurotoxicity. J Mol. Neurosci. 2010, 42, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Milder, J.B.; Liang, L.-P.; Patel, M. Acute Oxidative Stress and Systemic Nrf2 Activation by the Ketogenic Diet. Neurobiol. Dis. 2010, 40, 238–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zandi-Nejad, K.; Takakura, A.; Jurewicz, M.; Chandraker, A.K.; Offermanns, S.; Mount, D.; Abdi, R. The Role of HCA2 (GPR109A) in Regulating Macrophage Function. FASEB J. 2013, 27, 4366–4374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullingford, T.E. The Ketogenic Diet; Fatty Acids, Fatty Acid-Activated Receptors and Neurological Disorders. Prostaglandins Leukot Essent Fatty Acids. 2004, 70, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Monsalve, M.; Borniquel, S.; Valle, I.; Lamas, S. Mitochondrial dysfunction in human pathologies. Front Biosci. 2007, 12, 1131–1153. [Google Scholar] [CrossRef] [Green Version]
- Jeong, E.A.; Jeon, B.T.; Shin, H.J.; Kim, N.; Lee, D.H.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Roh, G.S. Ketogenic Diet-Induced Peroxisome Proliferator-Activated Receptor-γ Activation Decreases Neuroinflammation in the Mouse Hippocampus after Kainic Acid-Induced Seizures. Exp. Neurol. 2011, 232, 195–202. [Google Scholar] [CrossRef]
- Lee, G.-S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The Calcium-Sensing Receptor Regulates the NLRP3 Inflammasome through Ca2+ and CAMP. Nature 2012, 492, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Tschopp, J.; Schroder, K. NLRP3 Inflammasome Activation: The Convergence of Multiple Signalling Pathways on ROS Production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Shao, B.-Z.; Cao, Q.; Liu, C. Targeting NLRP3 Inflammasome in the Treatment of CNS Diseases. Front Mol. Neurosci. 2018, 11, 320. [Google Scholar] [CrossRef]
- Conner, J.M.; Lauterborn, J.C.; Yan, Q.; Gall, C.M.; Varon, S. Distribution of Brain-Derived Neurotrophic Factor (BDNF) Protein and MRNA in the Normal Adult Rat CNS: Evidence for Anterograde Axonal Transport. J. Neurosci. 1997, 17, 2295–2313. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Nagappan, G.; Lu, Y. BDNF and Synaptic Plasticity, Cognitive Function, and Dysfunction. Handb. Exp. Pharmacol. 2014, 220, 223–250. [Google Scholar] [CrossRef] [PubMed]
- Koppel, S.J.; Swerdlow, R.H. Neuroketotherapeutics: A Modern Review of a Century-Old Therapy. Neurochem. Int. 2018, 117, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Elesawy, B.H.; Raafat, B.M.; al Muqbali, A.; Abbas, A.M.; Sakr, H.F. The Impact of Intermittent Fasting on Brain-Derived Neurotrophic Factor, Neurotrophin 3, and Rat Behavior in a Rat Model of Type 2 Diabetes Mellitus. Brain Sci. 2021, 11, 242. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Zhu, R.; Zhu, L.; Qiu, T.; Cao, Z.; Kang, T. Potassium Channels: Structures, Diseases, and Modulators. Chem. Biol. Drug Des. 2014, 83, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Inagaki, N. Neuroprotection by K Channels. J. Mol. Cell Cardiol. 2005, 38, 945–949. [Google Scholar] [CrossRef]
- Xie, J.; Duan, L.; Qian, X.; Huang, X.; Ding, J.; Hu, G. KATP Channel Openers Protect Mesencephalic Neurons against MPP+-Induced Cytotoxicity via Inhibition of ROS Production. J. Neurosci. Res. 2010, 88, 428–437. [Google Scholar] [CrossRef]
- Karschin, C.; Ecke, C.; Ashcroft, F.M.; Karschin, A. Overlapping Distribution of K ATP Channel-Forming Kir6.2 Subunit and the Sulfonylurea Receptor SUR1 in Rodent Brain. FEBS Lett. 1997, 401, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.-M.; Lee, C.R. Intrinsic and Integrative Properties of Substantia Nigra Pars Reticulata Neurons. Neuroscience 2011, 198, 69–94. [Google Scholar] [CrossRef] [Green Version]
- Bröer, S. Not Part of the Temporal Lobe, but Still of Importance? Substantia Nigra and Subthalamic Nucleus in Epilepsy. Front Syst. Neurosci. 2020, 14, 581826. [Google Scholar] [CrossRef]
- Hu, B.; Wang, D.; Xia, Z.; Yang, A.; Zhang, J.; Shi, Q.; Dai, H. Regulation and Control Roles of the Basal Ganglia in the Development of Absence Epileptiform Activities. Cogn. Neurodyn. 2020, 14, 137–154. [Google Scholar] [CrossRef]
- Deransart, C.; Hellwig, B.; Heupel-Reuter, M.; Leger, J.-F.; Heck, D.; Lucking, C.H. Single-Unit Analysis of Substantia Nigra Pars Reticulata Neurons in Freely Behaving Rats with Genetic Absence Epilepsy. Epilepsia 2003, 44, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Berg, J.; Yellen, G. Ketogenic Diet Metabolites Reduce Firing in Central Neurons by Opening KATP Channels. J. Neurosci. 2007, 27, 3618–3625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutas, A.; Birnbaumer, L.; Yellen, G. Metabolism Regulates the Spontaneous Firing of Substantia Nigra Pars Reticulata Neurons via K ATP and Nonselective Cation Channels. J. Neurosci. 2014, 34, 16336–16347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, G.R.; Lutas, A.; Martinez-Francois, J.R.; Yellen, G. Single KATP Channel Opening in Response to Action Potential Firing in Mouse Dentate Granule Neurons. J. Neurosci. 2011, 31, 8689–8696. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.Y.; Abdelwahab, M.G.; Lee, S.H.; O’Neill, D.; Thompson, R.J.; Duff, H.J.; Sullivan, P.G.; Rho, J.M. Ketones Prevent Oxidative Impairment of Hippocampal Synaptic Integrity through KATP Channels. PLoS ONE 2015, 10, e0119316. [Google Scholar] [CrossRef] [Green Version]
- Brownlow, M.L.; Benner, L.; D’Agostino, D.; Gordon, M.N.; Morgan, D. Ketogenic Diet Improves Motor Performance but Not Cognition in Two Mouse Models of Alzheimer’s Pathology. PLoS ONE 2013, 8, e75713. [Google Scholar] [CrossRef] [Green Version]
- Gano, L.B.; Patel, M.; Rho, J.M. Ketogenic Diets, Mitochondria, and Neurological Diseases. J. Lipid Res. 2014, 55, 2211–2228. [Google Scholar] [CrossRef] [Green Version]
- Cryan, J.F.; O’Riordan, K.J.; Sandhu, K.; Peterson, V.; Dinan, T.G. The Gut Microbiome in Neurological Disorders. Lancet Neurol. 2020, 19, 179–194. [Google Scholar] [CrossRef]
- Quigley, E.M.M. Microbiota-Brain-Gut Axis and Neurodegenerative Diseases. Curr. Neurol. Neurosci. Rep. 2017, 17, 94. [Google Scholar] [CrossRef]
- Winter, G.; Hart, R.A.; Charlesworth, R.P.G.; Sharpley, C.F. Gut Microbiome and Depression: What We Know and What We Need to Know. Rev. Neurosci. 2018, 29, 629–643. [Google Scholar] [CrossRef]
- Alfonsetti, M.; Castelli, V.; d’Angelo, M. Are We What We Eat? Impact of Diet on the Gut–Brain Axis in Parkinson’s Disease. Nutrients 2022, 14, 380. [Google Scholar] [CrossRef] [PubMed]
- Osadchiy, V.; Martin, C.R.; Mayer, E.A. The Gut-Brain Axis and the Microbiome: Mechanisms and Clinical Implications. Clin. Gastroenterol. Hepatol. 2019, 17, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Jašarević, E.; Morrison, K.E.; Bale, T.L. Sex Differences in the Gut Microbiome–Brain Axis across the Lifespan. Philos. Trans. R Soc. Lond B Biol. Sci. 2016, 371, 1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBlanc, J.G.; Milani, C.; de Giori, G.S.; Sesma, F.; van Sinderen, D.; Ventura, M. Bacteria as Vitamin Suppliers to Their Host: A Gut Microbiota Perspective. Curr. Opin. Biotechnol. 2013, 24, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Barrett, E.; Ross, R.P.; O’Toole, P.W.; Fitzgerald, G.F.; Stanton, C. γ-Aminobutyric Acid Production by Culturable Bacteria from the Human Intestine. J. Appl. Microbiol. 2012, 113, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.L.; Wolin, M.J. Pathways of Acetate, Propionate, and Butyrate Formation by the Human Fecal Microbial Flora. Appl. Environ. Microbiol. 1996, 62, 1589–1592. [Google Scholar] [CrossRef] [Green Version]
- Andoh, A. Physiological Role of Gut Microbiota for Maintaining Human Health. Digestion 2016, 93, 176–181. [Google Scholar] [CrossRef]
- Duncan, S.H.; Louis, P.; Thomson, J.M.; Flint, H.J. The Role of PH in Determining the Species Composition of the Human Colonic Microbiota. Environ. Microbiol. 2009, 11, 2112–2122. [Google Scholar] [CrossRef]
- Caputi, V.; Giron, M. Microbiome-Gut-Brain Axis and Toll-Like Receptors in Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 1689. [Google Scholar] [CrossRef] [Green Version]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.-J. Review Article: The Role of Butyrate on Colonic Function. Aliment Pharmacol. Ther. 2007, 27, 104–119. [Google Scholar] [CrossRef]
- Huang, R.; Wang, K.; Hu, J. Effect of Probiotics on Depression: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Nutrients 2016, 8, 483. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.T.; Walsh, R.F.L.; Sheehan, A.E. Prebiotics and Probiotics for Depression and Anxiety: A Systematic Review and Meta-Analysis of Controlled Clinical Trials. Neurosci. Biobehav. Rev. 2019, 102, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Ansari, F.; Pourjafar, H.; Tabrizi, A.; Homayouni, A. The Effects of Probiotics and Prebiotics on Mental Disorders: A Review on Depression, Anxiety, Alzheimer, and Autism Spectrum Disorders. Curr. Pharm. Biotechnol. 2020, 21, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Zhou, Q.; Qiu, C.-Z.; Dai, W.-K.; Wang, H.-P.; Li, Y.-H.; Liao, J.-X.; Lu, X.-G.; Lin, S.-F.; Ye, J.-H.; et al. Ketogenic Diet Poses a Significant Effect on Imbalanced Gut Microbiota in Infants with Refractory Epilepsy. World J. Gastroenterol. 2017, 23, 6164–6171. [Google Scholar] [CrossRef] [PubMed]
- Tagliabue, A.; Ferraris, C.; Uggeri, F.; Trentani, C.; Bertoli, S.; de Giorgis, V.; Veggiotti, P.; Elli, M. Short-Term Impact of a Classical Ketogenic Diet on Gut Microbiota in GLUT1 Deficiency Syndrome: A 3-Month Prospective Observational Study. Clin. Nutr. ESPEN 2017, 17, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, S.; Zhou, Y.; Yu, L.; Zhang, L.; Wang, Y. Altered Gut Microbiome Composition in Children with Refractory Epilepsy after Ketogenic Diet. Epilepsy Res. 2018, 145, 163–168. [Google Scholar] [CrossRef]
- Dahlin, M.; Prast-Nielsen, S. The Gut Microbiome and Epilepsy. EBioMedicine 2019, 44, 741–746. [Google Scholar] [CrossRef] [Green Version]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Januszewski, S.; Pluta, R. Ketogenic Diet and Epilepsy. Nutrients 2019, 11, 2510. [Google Scholar] [CrossRef] [Green Version]
- Paoli, A.; Mancin, L.; Bianco, A.; Thomas, E.; Mota, J.F.; Piccini, F. Ketogenic Diet and Microbiota: Friends or Enemies? Genes 2019, 10, 534. [Google Scholar] [CrossRef] [Green Version]
- Basciani, S.; Camajani, E.; Contini, S.; Persichetti, A.; Risi, R.; Bertoldi, L.; Strigari, L.; Prossomariti, G.; Watanabe, M.; Mariani, S.; et al. Very-Low-Calorie Ketogenic Diets With Whey, Vegetable, or Animal Protein in Patients With Obesity: A Randomized Pilot Study. J. Clin. Endocrinol. Metab. 2020, 105, 2939–2949. [Google Scholar] [CrossRef]
- Ang, Q.Y.; Alexander, M.; Newman, J.C.; Tian, Y.; Cai, J.; Upadhyay, V.; Turnbaugh, J.A.; Verdin, E.; Hall, K.D.; Leibel, R.L.; et al. Ketogenic Diets Alter the Gut Microbiome Resulting in Decreased Intestinal Th17 Cells. Cell 2020, 181, 1263–1275.e16. [Google Scholar] [CrossRef] [PubMed]
- Gourbeyre, P.; Denery, S.; Bodinier, M. Probiotics, Prebiotics, and Synbiotics: Impact on the Gut Immune System and Allergic Reactions. J. Leukoc Biol. 2011, 89, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Wang, A.C.; Parikh, I.; Green, S.J.; Hoffman, J.D.; Chlipala, G.; Murphy, M.P.; Sokola, B.S.; Bauer, B.; Hartz, A.M.S.; et al. Ketogenic Diet Enhances Neurovascular Function with Altered Gut Microbiome in Young Healthy Mice. Sci. Rep. 2018, 8, 6670. [Google Scholar] [CrossRef] [Green Version]
- Olson, C.A.; Vuong, H.E.; Yano, J.M.; Liang, Q.Y.; Nusbaum, D.J.; Hsiao, E.Y. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell 2018, 173, 1728–1741.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Lin, S.; Vanhoutte, P.M.; Woo, C.W.; Xu, A. Akkermansia Muciniphila Protects Against Atherosclerosis by Preventing Metabolic Endotoxemia-Induced Inflammation in Apoe−/− Mice. Circulation 2016, 133, 2434–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eor, J.Y.; Son, Y.J.; Kim, J.Y.; Kang, H.C.; Youn, S.E.; Kim, J.H.; Kim, S.H. Neuroprotective Effect of Both Synbiotics and Ketogenic Diet in a Pentylenetetrazol-Induced Acute Seizure Murine Model. Epilepsy Res. 2021, 174, 106668. [Google Scholar] [CrossRef] [PubMed]
- Mu, C.; Nikpoor, N.; Tompkins, T.A.; Rho, J.M.; Scantlebury, M.H.; Shearer, J. Probiotics Counteract Hepatic Steatosis Caused by Ketogenic Diet and Upregulate AMPK Signaling in a Model of Infantile Epilepsy. eBioMedicine 2022, 76, 103838. [Google Scholar] [CrossRef]
- Patel, M. Targeting Oxidative Stress in Central Nervous System Disorders. Trends Pharmacol. Sci. 2016, 37, 768–778. [Google Scholar] [CrossRef] [Green Version]
- Floyd, R.A. Antioxidants, Oxidative Stress, and Degenerative Neurological Disorders. Proc. Soc. Exp. Biol. Med. 1999, 222, 236–245. [Google Scholar] [CrossRef]
- Ma, Q.; Xing, C.; Long, W.; Wang, H.Y.; Liu, Q.; Wang, R.-F. Impact of Microbiota on Central Nervous System and Neurological Diseases: The Gut-Brain Axis. J. Neuroinflamm. 2019, 16, 53. [Google Scholar] [CrossRef] [Green Version]
- Gross, E.C.; Klement, R.J.; Schoenen, J.; D’Agostino, D.P.; Fischer, D. Potential Protective Mechanisms of Ketone Bodies in Migraine Prevention. Nutrients 2019, 11, 811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matin, N.; Tabatabaie, O.; Falsaperla, R.; Lubrano, R.; Pavone, P.; Mahmood, F.; Gullotta, M.; Serra, A.; di Mauro, P.; Cocuzza, S.; et al. Epilepsy and Innate Immune System: A Possible Immunogenic Predisposition and Related Therapeutic Implications. Hum. Vaccin. Immunother. 2015, 11, 2021–2029. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Smith, J.; Patel, M. Metabolic Dysfunction and Oxidative Stress in Epilepsy. Int. J. Mol. Sci. 2017, 18, 2365. [Google Scholar] [CrossRef] [Green Version]
- Yuen, A.W.C.; Keezer, M.R.; Sander, J.W. Epilepsy Is a Neurological and a Systemic Disorder. Epilepsy Behav. 2018, 78, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Terrone, G.; Balosso, S.; Pauletti, A.; Ravizza, T.; Vezzani, A. Inflammation and Reactive Oxygen Species as Disease Modifiers in Epilepsy. Neuropharmacology 2020, 167, 107742. [Google Scholar] [CrossRef] [PubMed]
- Menezes, L.F.S.; Sabiá Júnior, E.F.; Tibery, D.V.; dos Anjos Carneiro, L.; Schwartz, E.F. Epilepsy-Related Voltage-Gated Sodium Channelopathies: A Review. Front Pharmacol. 2020, 11, 1276. [Google Scholar] [CrossRef]
- Bartolini, E.; Campostrini, R.; Kiferle, L.; Pradella, S.; Rosati, E.; Chinthapalli, K.; Palumbo, P. Epilepsy and Brain Channelopathies from Infancy to Adulthood. Neurol. Sci. 2020, 41, 749–761. [Google Scholar] [CrossRef]
- Hildebrand, M.S.; Damiano, J.A.; Mullen, S.A.; Bellows, S.T.; Oliver, K.L.; Dahl, H.-H.M.; Scheffer, I.E.; Berkovic, S.F. Glucose Metabolism Transporters and Epilepsy: Only GLUT1 Has an Established Role. Epilepsia 2014, 55, e18–e21. [Google Scholar] [CrossRef] [Green Version]
- Koch, H.; Weber, Y.G. The Glucose Transporter Type 1 (Glut1) Syndromes. Epilepsy Behav. 2019, 91, 90–93. [Google Scholar] [CrossRef]
- Stafstrom, C.E. Hyperglycemia Lowers Seizure Threshold. Epilepsy Curr. 2003, 3, 148–149. [Google Scholar] [CrossRef]
- Martínez-François, J.R.; Fernández-Agüera, M.C.; Nathwani, N.; Lahmann, C.; Burnham, V.L.; Danial, N.N.; Yellen, G. BAD and KATP Channels Regulate Neuron Excitability and Epileptiform Activity. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kossoff, E.H. International Consensus Statement on Clinical Implementation of the Ketogenic Diet: Agreement, Flexibility, and Controversy. Epilepsia 2008, 49, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Kossoff, E.H.; Zupec-Kania, B.A.; Auvin, S.; Ballaban-Gil, K.R.; Christina Bergqvist, A.G.; Blackford, R.; Buchhalter, J.R.; Caraballo, R.H.; Cross, J.H.; Dahlin, M.G.; et al. Optimal Clinical Management of Children Receiving Dietary Therapies for Epilepsy: Updated Recommendations of the International Ketogenic Diet Study Group. Epilepsia Open 2018, 3, 175–192. [Google Scholar] [CrossRef] [PubMed]
- Kossoff, E.; Cervenka, M. Ketogenic Dietary Therapy Controversies for Its Second Century. Epilepsy Curr. 2020, 20, 125–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louw, E.; Aldaz, V.; Harvey, J.; Roan, M.; Hurk, D.; Cross, J.H.; Auvin, S.; Forbes, E.; Bor, B.; Olieman, J.; et al. Optimal Clinical Management of Children Receiving Ketogenic Parenteral Nutrition: A Clinical Practice Guide. Dev. Med. Child Neurol. 2020, 62, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kverneland, M.; Selmer, K.K.; Nakken, K.O.; Iversen, P.O.; Taubøll, E. A Prospective Study of the Modified Atkins Diet for Adults with Idiopathic Generalized Epilepsy. Epilepsy Behav. 2015, 53, 197–201. [Google Scholar] [CrossRef]
- IJff, D.M.; Postulart, D.; Lambrechts, D.A.J.E.; Majoie, M.H.J.M.; de Kinderen, R.J.A.; Hendriksen, J.G.M.; Evers, S.M.A.A.; Aldenkamp, A.P. Cognitive and Behavioral Impact of the Ketogenic Diet in Children and Adolescents with Refractory Epilepsy: A Randomized Controlled Trial. Epilepsy Behav. 2016, 60, 153–157. [Google Scholar] [CrossRef]
- Kim, J.A.; Yoon, J.-R.; Lee, E.J.; Lee, J.S.; Kim, J.T.; Kim, H.D.; Kang, H.-C. Efficacy of the Classic Ketogenic and the Modified Atkins Diets in Refractory Childhood Epilepsy. Epilepsia 2016, 57, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Goel, S.; Jain, P.; Agarwala, A.; Aneja, S. Evaluation of a Simplified Modified Atkins Diet for Use by Parents with Low Levels of Literacy in Children with Refractory Epilepsy: A Randomized Controlled Trial. Epilepsy Res. 2016, 127, 152–159. [Google Scholar] [CrossRef]
- Ashrafi, M.R.; Hosseini, S.A.; Zamani, G.R.; Mohammadi, M.; Tavassoli, A.; Badv, R.S.; Heidari, M.; Karimi, P.; Malamiri, R.A. The Efficacy of the Ketogenic Diet in Infants and Young Children with Refractory Epilepsies Using a Formula-Based Powder. Acta Neurol. Belg. 2017, 117, 175–182. [Google Scholar] [CrossRef]
- Lambrechts, D.A.J.E.; de Kinderen, R.J.A.; Vles, J.S.H.; de Louw, A.J.A.; Aldenkamp, A.P.; Majoie, H.J.M. A Randomized Controlled Trial of the Ketogenic Diet in Refractory Childhood Epilepsy. Acta Neurol. Scand. 2017, 135, 231–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baby, N.; Vinayan, K.P.; Pavithran, N.; Grace Roy, A. A Pragmatic Study on Efficacy, Tolerability and Long Term Acceptance of Ketogenic Diet Therapy in 74 South Indian Children with Pharmacoresistant Epilepsy. Seizure 2018, 58, 41–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kverneland, M.; Molteberg, E.; Iversen, P.O.; Veierød, M.B.; Taubøll, E.; Selmer, K.K.; Nakken, K.O. Effect of Modified Atkins Diet in Adults with Drug-Resistant Focal Epilepsy: A Randomized Clinical Trial. Epilepsia 2018, 59, 1567–1576. [Google Scholar] [CrossRef] [PubMed]
- Guzel, O.; Uysal, U.; Arslan, N. Efficacy and Tolerability of Olive Oil-Based Ketogenic Diet in Children with Drug-Resistant Epilepsy: A Single Center Experience from Turkey. Eur. J. Paediatr. Neurol. 2019, 23, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjurulf, B.; Magnus, P.; Hallböök, T.; Strømme, P. Potassium Citrate and Metabolic Acidosis in Children with Epilepsy on the Ketogenic Diet: A Prospective Controlled Study. Dev. Med. Child Neurol. 2020, 62, 57–61. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Dabla, S.; Kaushik, J.S. Modified Atkins Diet vs. Low Glycemic Index Treatment for Drug-Resistant Epilepsy in Children: An Open Label, Randomized Controlled Trial. Indian Pediatr. 2021, 58, 815–819. [Google Scholar] [CrossRef]
- Lakshminarayanan, K.; Agarawal, A.; Panda, P.K.; Sinha, R.; Tripathi, M.; Pandey, R.M.; Gulati, S. Efficacy of Low Glycemic Index Diet Therapy (LGIT) in Children Aged 2–8 Years with Drug-Resistant Epilepsy: A Randomized Controlled Trial. Epilepsy Res. 2021, 171, 106574. [Google Scholar] [CrossRef]
- Poorshiri, B.; Barzegar, M.; Tahmasebi, S.; Shiva, S.; Raeisi, S.; Ebadi, Z. The Efficacy Comparison of Classic Ketogenic Diet and Modified Atkins Diet in Children with Refractory Epilepsy: A Clinical Trial. Acta Neurol. Belg. 2021, 121, 483–487. [Google Scholar] [CrossRef]
- Saveanu, R.V.; Nemeroff, C.B. Etiology of Depression: Genetic and Environmental Factors. Psychiatr. Clin. N. Am. 2012, 35, 51–71. [Google Scholar] [CrossRef]
- Malhi, G.S.; Mann, J.J. Depression. Lancet 2018, 392, 2299–2312. [Google Scholar] [CrossRef]
- Evans-Lacko, S.; Aguilar-Gaxiola, S.; Al-Hamzawi, A.; Alonso, J.; Benjet, C.; Bruffaerts, R.; Chiu, W.T.; Florescu, S.; de Girolamo, G.; Gureje, O.; et al. Socio-Economic Variations in the Mental Health Treatment Gap for People with Anxiety, Mood, and Substance Use Disorders: Results from the WHO World Mental Health (WMH) Surveys. Psychol. Med. 2018, 48, 1560–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammen, C. Risk Factors for Depression: An Autobiographical Review. Annu. Rev. Clin. Psychol. 2018, 14, 1–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Włodarczyk, A.; Cubała, W.J.; Stawicki, M. Ketogenic Diet for Depression: A Potential Dietary Regimen to Maintain Euthymia? Prog Neuropsychopharmacol. Biol. Psychiatry 2021, 109, 110257. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Yun, M.; Oh, Y.J.; Choi, H.J. Mind-Altering with the Gut: Modulation of the Gut-Brain Axis with Probiotics. J. Microbiol. 2018, 56, 172–182. [Google Scholar] [CrossRef]
- Reigstad, C.S.; Salmonson, C.E.; Rainey, J.F.; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut Microbes Promote Colonic Serotonin Production through an Effect of Short-Chain Fatty Acids on Enterochromaffin Cells. FASEB J. 2015, 29, 1395–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwahara, A.; Matsuda, K.; Kuwahara, Y.; Asano, S.; Inui, T.; Marunaka, Y. Microbiota-Gut-Brain Axis: Enteroendocrine Cells and the Enteric Nervous System Form an Interface between the Microbiota and the Central Nervous System. Biomed. Res. 2020, 41, 199–216. [Google Scholar] [CrossRef]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus Strain Regulates Emotional Behavior and Central GABA Receptor Expression in a Mouse via the Vagus Nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [Green Version]
- de la Rubia Ortí, J.E.; Fernández, D.; Platero, F.; García-Pardo, M.P. Can Ketogenic Diet Improve Alzheimer’s Disease? Association With Anxiety, Depression, and Glutamate System. Front. Nutr. 2021, 8, 744398. [Google Scholar] [CrossRef]
- Ari, C.; Kovács, Z.; Juhasz, G.; Murdun, C.; Goldhagen, C.R.; Koutnik, A.M.; Poff, A.M.; Kesl, S.L.; D’Agostino, D.P. Exogenous Ketone Supplements Reduce Anxiety-Related Behavior in Sprague-Dawley and Wistar Albino Glaxo/Rijswijk Rats. Front Mol. Neurosci. 2016, 9, 137. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.F.; Huang, G.B.; Xu, M.D.; Gao, F.; Lin, S.; Huang, J.; Wang, J.; Li, Y.Q.; Wu, C.H.; Yao, S.; et al. Anti-Depression Effects of Ketogenic Diet Are Mediated via the Restoration of Microglial Activation and Neuronal Excitability in the Lateral Habenula. Brain Behav. Immun. 2020, 88, 748–762. [Google Scholar] [CrossRef]
- Piane, M.; Lulli, P.; Farinelli, I.; Simeoni, S.; de Filippis, S.; Patacchioli, F.R.; Martelletti, P. Genetics of Migraine and Pharmacogenomics: Some Considerations. J. Headache. Pain. 2007, 8, 334–339. [Google Scholar] [CrossRef] [PubMed]
- Schürks, M. Genetics of Migraine in the Age of Genome-Wide Association Studies. J. Headache. Pain. 2012, 13, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Headache Classification Committee of the International Headache Society (IHS) The International Classification of Headache Disorders, 3rd Edition. Cephalalgia 2018, 38, 1–211. [CrossRef] [PubMed]
- Albury, C.L.; Stuart, S.; Haupt, L.M.; Griffiths, L.R. Ion Channelopathies and Migraine Pathogenesis. Mol. Genet Genomics 2017, 292, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Al-Karagholi, M.A.-M.; Ghanizada, H.; Nielsen, C.A.W.; Hougaard, A.; Ashina, M. Opening of ATP Sensitive Potassium Channels Causes Migraine Attacks with Aura. Brain 2021, 144, 2322–2332. [Google Scholar] [CrossRef]
- Hsiao, F.J.; Chen, W.T.; Pan, L.L.H.; Liu, H.Y.; Wang, Y.F.; Chen, S.P.; Lai, K.L.; Coppola, G.; Wang, S.J. Dynamic Brainstem and Somatosensory Cortical Excitability during Migraine Cycles. J. Headache. Pain. 2022, 23, 21. [Google Scholar] [CrossRef]
- Glaubic-Łątka, M.; Łatka, D.; Bury, W.; Pierzchała, K. Współczesne Poglądy Na Patofizjologię Migreny. Neurol. Neurochir. Pol. 2004, 38, 307–315. [Google Scholar]
- Hoffmann, U.; Sukhotinsky, I.; Eikermann-Haerter, K.; Ayata, C. Glucose Modulation of Spreading Depression Susceptibility. J. Cereb. Blood Flow. Metab. 2013, 33, 191–195. [Google Scholar] [CrossRef]
- de Almeida Rabello Oliveira, M.; da Rocha Ataíde, T.; de Oliveira, S.L.; de Melo Lucena, A.L.; de Lira, C.E.P.R.; Soares, A.A.; de Almeida, C.B.S.; Ximenes-da-Silva, A. Effects of Short-Term and Long-Term Treatment with Medium- and Long-Chain Triglycerides Ketogenic Diet on Cortical Spreading Depression in Young Rats. Neurosci. Lett. 2008, 434, 66–70. [Google Scholar] [CrossRef]
- Barbanti, P.; Fofi, L.; Aurilia, C.; Egeo, G.; Caprio, M. Ketogenic Diet in Migraine: Rationale, Findings and Perspectives. Neurol. Sci. 2017, 38, 111–115. [Google Scholar] [CrossRef]
- di Lorenzo, C.; Pinto, A.; Ienca, R.; Coppola, G.; Sirianni, G.; di Lorenzo, G.; Parisi, V.; Serrao, M.; Spagnoli, A.; Vestri, A.; et al. A Randomized Double-Blind, Cross-Over Trial of Very Low-Calorie Diet in Overweight Migraine Patients: A Possible Role for Ketones? Nutrients 2019, 11, 1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putananickal, N.; Gross, E.C.; Orsini, A.-L.; Schmidt, S.; Hafner, P.; Gocheva, V.; Nagy, S.; Henzi, B.C.; Rubino, D.; Vogt, D.R.; et al. Efficacy and Safety of Exogenous Beta-Hydroxybutyrate for Preventive Treatment in Episodic Migraine: A Single-Centred, Randomised, Placebo-Controlled, Double-Blind Crossover Trial. Cephalalgia 2022, 42, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sidhu, J.; Goyal, A.; Tsao, J.W. Alzheimer Disease; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK499922/ (accessed on 2 April 2022).
- Forsyth, E.; Ritzline, P.D. An Overview of the Etiology, Diagnosis, and Treatment of Alzheimer Disease. Phys. Ther. 1998, 78, 1325–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006239. [Google Scholar] [CrossRef] [Green Version]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s Disease. Nat. Rev. Dis. Primers. 2015, 1, 15056. [Google Scholar] [CrossRef]
- Apostolova, L.G. Alzheimer Disease. Continuum 2016, 22, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Juszczyk, G.; Mikulska, J.; Kasperek, K.; Pietrzak, D.; Mrozek, W.; Herbet, M. Chronic Stress and Oxidative Stress as Common Factors of the Pathogenesis of Depression and Alzheimer’s Disease; the Role of Antioxidants in Prevention and Treatment. Antioxidants 2021, 10, 1439. [Google Scholar] [CrossRef]
- Caberlotto, L.; Marchetti, L.; Lauria, M.; Scotti, M.; Parolo, S. Integration of Transcriptomic and Genomic Data Suggests Candidate Mechanisms for APOE4-Mediated Pathogenic Action in Alzheimer’s Disease. Sci. Rep. 2016, 6, 32583. [Google Scholar] [CrossRef] [Green Version]
- Klimova, B.; Novotný, M.; Kuca, K.; Valis, M. Effect Of An Extra-Virgin Olive Oil Intake On The Delay Of Cognitive Decline: Role Of Secoiridoid Oleuropein? Neuropsychiatr. Dis. Treat. 2019, 15, 3033–3040. [Google Scholar] [CrossRef] [Green Version]
- Lauretti, E.; Nenov, M.; Dincer, O.; Iuliano, L.; Praticò, D. Extra Virgin Olive Oil Improves Synaptic Activity, Short-term Plasticity, Memory, and Neuropathology in a Tauopathy Model. Aging Cell 2020, 19, e13076. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, A.; Chauhan, V. Beneficial Effects of Walnuts on Cognition and Brain Health. Nutrients 2020, 12, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, G.; Maillard, P.; Himali, J.J.; Beiser, A.S.; Au, R.; Wolf, P.A.; Seshadri, S.; DeCarli, C. Glucose Indices Are Associated with Cognitive and Structural Brain Measures in Young Adults. Neurology 2015, 84, 2329–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerti, L.; Witte, A.V.; Winkler, A.; Grittner, U.; Rujescu, D.; Floel, A. Higher Glucose Levels Associated with Lower Memory and Reduced Hippocampal Microstructure. Neurology 2013, 81, 1746–1752. [Google Scholar] [CrossRef] [PubMed]
- Torosyan, N.; Sethanandha, C.; Grill, J.D.; Dilley, M.L.; Lee, J.; Cummings, J.L.; Ossinalde, C.; Silverman, D.H. Changes in Regional Cerebral Blood Flow Associated with a 45 day Course of the Ketogenic Agent, Caprylidene, in Patients with Mild to Moderate Alzheimer’s Disease: Results of a Randomized, Double-Blinded, Pilot Study. Exp. Gerontol. 2018, 111, 118–121. [Google Scholar] [CrossRef]
- Ota, M.; Matsuo, J.; Ishida, I.; Takano, H.; Yokoi, Y.; Hori, H.; Yoshida, S.; Ashida, K.; Nakamura, K.; Takahashi, T.; et al. Effects of a Medium-Chain Triglyceride-Based Ketogenic Formula on Cognitive Function in Patients with Mild-to-Moderate Alzheimer’s Disease. Neurosci. Lett. 2019, 690, 232–236. [Google Scholar] [CrossRef]
- Myette-Côté, É.; St-Pierre, V.; Beaulieu, S.; Castellano, C.-A.; Fortier, M.; Plourde, M.; Bocti, C.; Fulop, T.; Cunnane, S.C. The Effect of a 6-Month Ketogenic Medium-Chain Triglyceride Supplement on Plasma Cardiometabolic and Inflammatory Markers in Mild Cognitive Impairment. Prostaglandins Leukot Essent Fatty Acids. 2021, 169, 102236. [Google Scholar] [CrossRef]
- Phillips, M.C.L.; Deprez, L.M.; Mortimer, G.M.N.; Murtagh, D.K.J.; McCoy, S.; Mylchreest, R.; Gilbertson, L.J.; Clark, K.M.; Simpson, P.V.; McManus, E.J.; et al. Randomized Crossover Trial of a Modified Ketogenic Diet in Alzheimer’s Disease. Alzheimer’s Res. Ther. 2021, 13, 51. [Google Scholar] [CrossRef]
- Cacabelos, R. Parkinson’s Disease: From Pathogenesis to Pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef]
- Bellou, V.; Belbasis, L.; Tzoulaki, I.; Evangelou, E.; Ioannidis, J.P.A. Environmental Risk Factors and Parkinson’s Disease: An Umbrella Review of Meta-Analyses. Parkinsonism Relat. Disord. 2016, 23, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Aarsland, D.; Kvaløy, J.T.; Andersen, K.; Larsen, J.P.; Tang, M.X.; Lolk, A.; Kragh-Sørensen, P.; Marder, K. The Effect of Age of Onset of PD on Risk of Dementia. J. Neurol. 2007, 254, 38–45. [Google Scholar] [CrossRef]
- Sveinbjornsdottir, S. The Clinical Symptoms of Parkinson’s Disease. J. Neurochem. 2016, 139, 318–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, H.; Massey, L.A.; Lees, A.J.; Brown, P.; Day, B.L. Hypokinesia without Decrement Distinguishes Progressive Supranuclear Palsy from Parkinson’s Disease. Brain 2012, 135, 1141–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankovic, J. Parkinson’s Disease: Clinical Features and Diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; Liu, J.; Tan, Y.; Chen, S. Freezing of Gait in Parkinson’s Disease: Pathophysiology, Risk Factors and Treatments. Transl. Neurodegener. 2020, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giladi, N.; Nieuwboer, A. Understanding and Treating Freezing of Gait in Parkinsonism, Proposed Working Definition, and Setting the Stage. Mov. Disord. 2008, 23, S423–S425. [Google Scholar] [CrossRef] [PubMed]
- Lotankar, S.; Prabhavalkar, K.S.; Bhatt, L.K. Biomarkers for Parkinson’s Disease: Recent Advancement. Neurosci. Bull. 2017, 33, 585–597. [Google Scholar] [CrossRef]
- Mazzoni, P.; Shabbott, B.; Cortes, J.C. Motor Control Abnormalities in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009282. [Google Scholar] [CrossRef] [Green Version]
- Boden, G.; Sargrad, K.; Homko, C.; Mozzoli, M.; Stein, T.P. Effect of a Low-Carbohydrate Diet on Appetite, Blood Glucose Levels, and Insulin Resistance in Obese Patients with Type 2 Diabetes. Ann. Intern. Med. 2005, 142, 403. [Google Scholar] [CrossRef]
- Yasuda, T.; Nakata, Y.; Mochizuki, H. α-Synuclein and Neuronal Cell Death. Mol. Neurobiol. 2013, 47, 466. [Google Scholar] [CrossRef] [Green Version]
- Aureli, C.; Cassano, T.; Masci, A.; Francioso, A.; Martire, S.; Cocciolo, A.; Chichiarelli, S.; Romano, A.; Gaetani, S.; Mancini, P.; et al. 5-S-Cysteinyldopamine Neurotoxicity: Influence on the Expression of α-Synuclein and ERp57 in Cellular and Animal Models of Parkinson’s Disease. J. Neurosci. Res. 2014, 92, 347–358. [Google Scholar] [CrossRef]
- VanItallie, T.B.; Nonas, C.; di Rocco, A.; Boyar, K.; Hyams, K.; Heymsfield, S.B. Treatment of Parkinson Disease with Diet-Induced Hyperketonemia: A Feasibility Study. Neurology 2005, 64, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Kashiwaya, Y.; Takeshima, T.; Mori, N.; Nakashima, K.; Clarke, K.; Veech, R.L. D-Beta -Hydroxybutyrate Protects Neurons in Models of Alzheimer’s and Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2000, 97, 5440–5444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paoli, A.; Bianco, A.; Damiani, E.; Bosco, G. Ketogenic Diet in Neuromuscular and Neurodegenerative Diseases. Biomed. Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chia, S.J.; Tan, E.-K.; Chao, Y.-X. Historical Perspective: Models of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmanouilidou, E.; Minakaki, G.; Keramioti, M.V.; Xylaki, M.; Balafas, E.; Chrysanthou-Piterou, M.; Kloukina, I.; Vekrellis, K. GABA Transmission via ATP-Dependent K+ Channels Regulates α-Synuclein Secretion in Mouse Striatum. Brain 2016, 139, 871–890. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Wang, M.; Ma, Z. Therapeutic Potential of ATP-Sensitive Potassium Channels in Parkinson’s Disease. Brain Res. Bull. 2021, 169, 1–7. [Google Scholar] [CrossRef]
- Lubomski, M.; Tan, A.H.; Lim, S.Y.; Holmes, A.J.; Davis, R.L.; Sue, C.M. Parkinson’s Disease and the Gastrointestinal Microbiome. J. Neurol. 2020, 267, 2507–2523. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef] [Green Version]
- Scheperjans, F.; Derkinderen, P.; Borghammer, P. The Gut and Parkinson’s Disease: Hype or Hope? J. Parkinsons Dis. 2018, 8, S31–S39. [Google Scholar] [CrossRef] [Green Version]
- Forsyth, C.B.; Shannon, K.M.; Kordower, J.H.; Voigt, R.M.; Shaikh, M.; Jaglin, J.A.; Estes, J.D.; Dodiya, H.B.; Keshavarzian, A. Increased Intestinal Permeability Correlates with Sigmoid Mucosa Alpha-Synuclein Staining and Endotoxin Exposure Markers in Early Parkinson’s Disease. PLoS ONE 2011, 6, e28032. [Google Scholar] [CrossRef] [Green Version]
- Phillips, M.C.L.; Murtagh, D.K.J.; Gilbertson, L.J.; Asztely, F.J.S.; Lynch, C.D.P. Low-Fat versus Ketogenic Diet in Parkinson’s Disease: A Pilot Randomized Controlled Trial. Mov. Disord. 2018, 33, 1306–1314. [Google Scholar] [CrossRef]
- Crosby, L.; Davis, B.; Joshi, S.; Jardine, M.; Paul, J.; Neola, M.; Barnard, N.D. Ketogenic Diets and Chronic Disease: Weighing the Benefits Against the Risks. Front Nutr. 2021, 8, 702802. [Google Scholar] [CrossRef] [PubMed]
- Holscher, H.D. Dietary Fiber and Prebiotics and the Gastrointestinal Microbiota. Gut Microbes 2017, 8, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Turner, Z.; Doerrer, S.C.; Stanfield, A.; Kossoff, E.H. Complications During Ketogenic Diet Initiation: Prevalence, Treatment, and Influence on Seizure Outcomes. Pediatr. Neurol. 2017, 68, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.; Swaminathan, A.; Paseka, J.; Hanson, C. Efficacy and Safety of a Ketogenic Diet in Children and Adolescents with Refractory Epilepsy—A Review. Nutrients 2020, 12, 1809. [Google Scholar] [CrossRef]
- Cai, Q.-Y.; Zhou, Z.-J.; Luo, R.; Gan, J.; Li, S.-P.; Mu, D.-Z.; Wan, C.-M. Safety and Tolerability of the Ketogenic Diet Used for the Treatment of Refractory Childhood Epilepsy: A Systematic Review of Published Prospective Studies. World J. Pediatrics 2017, 13, 528–536. [Google Scholar] [CrossRef]
- McNally, M.A.; Pyzik, P.L.; Rubenstein, J.E.; Hamdy, R.F.; Kossoff, E.H. Empiric Use of Potassium Citrate Reduces Kidney-Stone Incidence with the Ketogenic Diet. Pediatrics 2009, 124, e300–e304. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author (Year) | Intervention Period | Diet | Group | Results |
|---|---|---|---|---|
| Kvernelnad et al. [158] (2015) | 12 weeks | MAD | 13 adults | >50% reduction of seizure frequency in 31% (4/13) adults |
| IJff et al. [159] (2016) | 4 months | KD | 28 (20 on MCT); 22 CAU a | Cognitive activation, less anxiety and mood problems, increased productivity were observed in patients treated with the KD |
| Kim et al. [160] (2016) | 6 months | KD | 51 | 39% (20/51) KD patients had >50% seizure reduction, 31% (16/51) of them were seizure-free |
| MAD | 53 | 36% (19/53) had >50% reduction in seizures, 23% (12/53) were seizure free | ||
| Sharma et al. [161] (2016) | 3 months | MAD | 41 on MAD, 40 controls | 56.1% (23/41) of the children on the diet had >50% seizure reduction, 14.6% (6/41) were seizure free compared to 5% (2/40) controls; 19.5% (8/41) had >90% seizure reduction |
| Ashrafi et al. [162] (2017) | 4 months | KD (formula-based powder) | 22 | 27.3% (6/22) had >90% reduction in seizures and 40.9% (9/22) had 50–90% reduction in seizures |
| Lambrechts et al. [163] (2017) | 4 months | KD | 26 KD | >50% reduction in seizure frequency in 50% (13/26) of KD, 11.5% (3/26) had >90% seizure reduction and another 11.5% (3/26) were seizure free |
| 22 CAU a | 18.2% (4/22) were responders; 9.1% (2/22) were seizure free and 4.5% (1/22) had >90% seizure reduction. | |||
| Baby et al. [164] (2018) | 3 months | KD | 54 | 59.4% (44/74) reported >50% seizure reduction. More than 90% reduction was noted in 33.7% children (25/74). 8.1% (6/74) became seizure free |
| 6 months | 45 | |||
| 12 months | 30 | |||
| Kverneland et al. [165] (2018) | 12 weeks | MAD | 24 on diet, 32 control group (habitual diet); adults | >25% seizure reduction among those who completed the intervention |
| Guzel et al. [166] (2019) | 1 month | KD | 369 | 65.8% (243/369) of the patients observed >50% decrease in seizure frequency; 35.5% (131/369) were seizure-free |
| 3 months | 314 | 74.7% (235/314), of the patients observed >50% decrease in seizure frequency; 39.8% (125/314) were seizure-free | ||
| 6 months | 225 | 70.6% (159/225) of the patients observed >50% decrease in seizure frequency; 38.2% (86/225) were seizure-free | ||
| 12 months | 160 | 83.1% (133/160) of the patients observed >50% decrease in seizure frequency; 43.1% (69/160) were seizure-free | ||
| Bjurulf et al. [167] (2020) | 7 months | KD with potassium citrate | 22 | >50% reduction in seizure frequency in 40.9% (9/22) patients supplementing potassium citrate and 27.6% (8/29) participants without potassium citrate |
| KD without potassium citrate | 29 | |||
| Gupta et al. [168] (2021) | 12 weeks | LGIT | 30 | >50% reduction in seizure frequency in 73.3% (22/30) LGIT patients |
| MAD | 30 | >50% reduction in seizure frequency in 43.4% (13/30) MAD patients | ||
| Lakshminarayanan et al. [169] (2021) | 3 months | LGIT | 20 on diet, 20 control group | 30% (6/20) patients observed >50% reduction in seizure frequency |
| Poorshiri et al. [170] (2021) | 6 months | KD | 24 | 45.8% patients from KD group observed >50% decrease in seizure frequency |
| MAD | 11 | 45.5% from MAD group observed >50% decrease in seizure frequency |
| Author (Year) | Duration | Group | Intervention | Control | Results |
|---|---|---|---|---|---|
| Di Lorenzo et al. (2019) [193] | 1 month | 35 episodic migraine patients; 29 completed the study | VLCKD | very low-calorie non-ketogenic diet | reduction in migraine episodes |
| Putananical et al. (2022) [194] | 12 weeks | 41 episodic migraine patients | exogenous administration of β-HB | placebo | no clinically significant amelioration of migraine frequency or intensity |
| Authors | Duration | Group | Diet | Results |
|---|---|---|---|---|
| Torosyan et al. [207] (2018) | 45 days | 16 | Caprylidene (ketogenic agent) administration | Increased blood flow in certain brain regions in patients lacking an APOEɛ4 allele |
| Ota et al. [208] (2019) | 12 weeks | 20 | MCT based ketogenic formula | After 8 weeks, significant improvement in the immediate and delayed logical memory tests compared to their baseline scores were observed; at 12 weeks patients improved in the digit-symbol coding test and immediate logical memory test compared to their baseline scores |
| Fortier et al. [77] (2021) | 6 months | 83 | ketogenic MCT drink | Free and cued recall verbal fluency, Boston Naming Test, and the Trail-Making Test improved significantly in the kMCT group compared to placebo |
| Myette-Côté et al. [209] (2021) | 6 months | 39 | ketogenic MCT drink | No clinically relevant adverse effect on the blood markers. After intervention plasma IL-8 significant increase have been observed |
| Philips et al. [210] (2021) | two 12-week treatment periods | 26 | KD | Improved daily function and quality of life |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietrzak, D.; Kasperek, K.; Rękawek, P.; Piątkowska-Chmiel, I. The Therapeutic Role of Ketogenic Diet in Neurological Disorders. Nutrients 2022, 14, 1952. https://doi.org/10.3390/nu14091952
Pietrzak D, Kasperek K, Rękawek P, Piątkowska-Chmiel I. The Therapeutic Role of Ketogenic Diet in Neurological Disorders. Nutrients. 2022; 14(9):1952. https://doi.org/10.3390/nu14091952
Chicago/Turabian StylePietrzak, Diana, Kamila Kasperek, Paweł Rękawek, and Iwona Piątkowska-Chmiel. 2022. "The Therapeutic Role of Ketogenic Diet in Neurological Disorders" Nutrients 14, no. 9: 1952. https://doi.org/10.3390/nu14091952
APA StylePietrzak, D., Kasperek, K., Rękawek, P., & Piątkowska-Chmiel, I. (2022). The Therapeutic Role of Ketogenic Diet in Neurological Disorders. Nutrients, 14(9), 1952. https://doi.org/10.3390/nu14091952