Impact of Nutrition on Cerebral Circulation and Cognition in the Metabolic Syndrome



Abstract
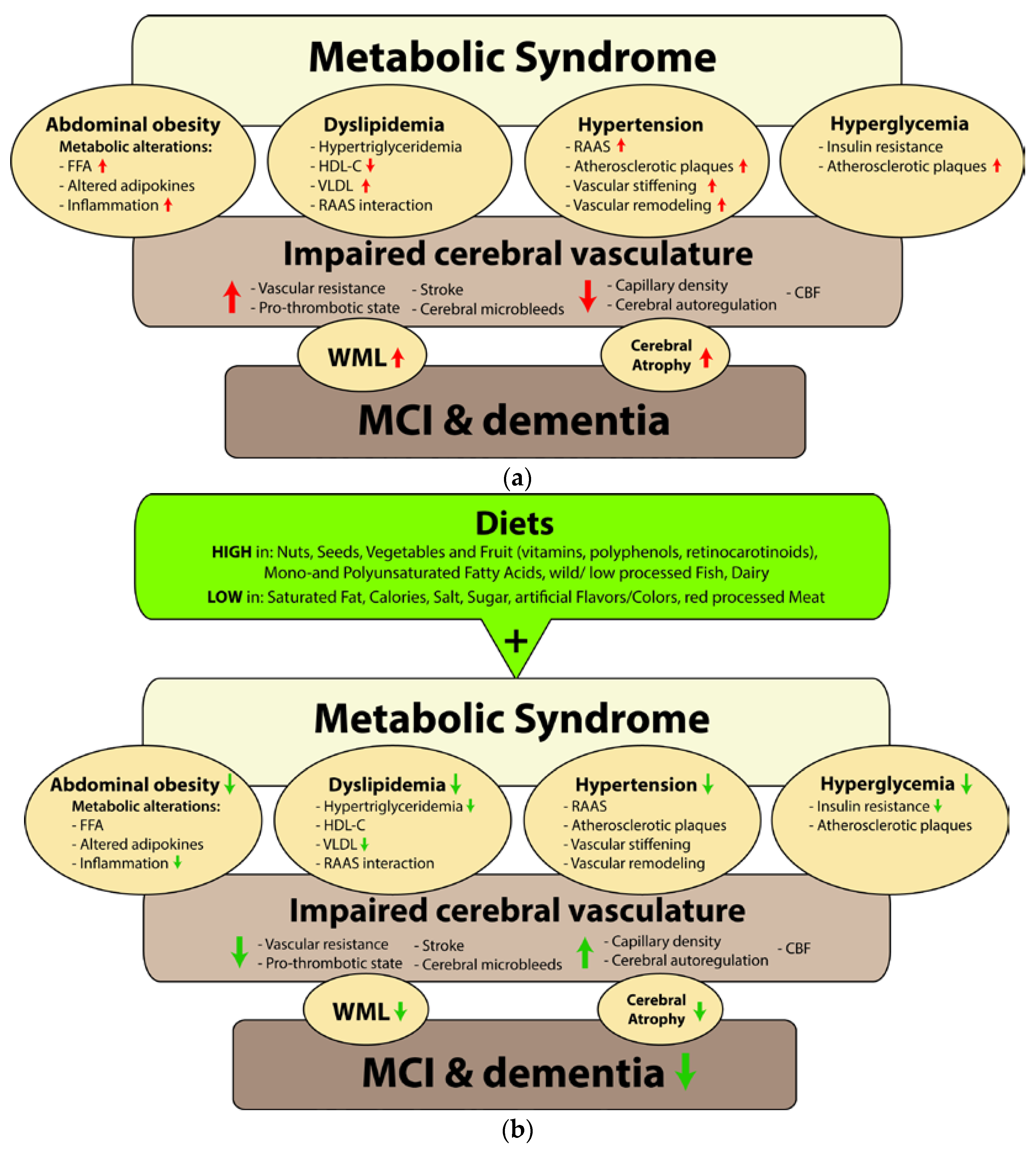
:1. Introduction
2. Search Strategy and Selection of the Papers
3. Individual MetS Components in Relation to Cerebral Circulation
3.1. Abdominal Obesity
3.2. Dyslipidemia
3.3. Hypertension
3.4. Hyperglycemia
3.5. Clustering of MetS Components
4. Relation between MetS, Cerebral Circulation, and Cognition

{kind=link}
| Effects of MetS on Vasculature and Circulation | Effects of MetS on Cognition |
|---|---|
| ↓ Capillary density [86,105] | ↓ Immediate memory function [22] b |
| ↓ Cerebral arterial vasodilation response [86] | ↓ Fluid intelligence [92] e |
| ↓ Capillary recruitment [105] | ↓ Global cognition [92] e, [106] f, [107] and in female [108] a |
| ↑ Intima media thickness [57,85] a | ↓ Information processing speed [93,109,110] |
| ↑ Arterial stiffness [57,85] a | ↓ Executive function [109,110] a, [93] and in male [94] a |
| ↓ Cerebral blood flow (CBF) [22] b and in male [15] | ↓ Attention [109] a and [93] |
| ↑ Amount of atherosclerosis [111] c | ↓ In recall performance [106] f |
| ↓ Vasomotor reactivity (VMR) [112] d | ↓ Visuospatial function [113] a |
| ↓ CBF in medial + lateral aspects of frontal & parietal lobe gray matter (GM) and lateral areas of the temporal & occipital lobe GM [22] b | ↓ Memory function [113] a and in male [94] a |
5. Impact of Nutrition on MetS, Cerebral Circulation, and Cognition
5.1. Abdominal Obesity
5.2. Dyslipidemia
5.3. Hypertension
5.4. Hyperglycemia
6. Conclusions

Acknowledgments
Conflicts of Interest
References
- McNeill, A.M.; Katz, R.; Girman, C.J.; Rosamond, W.D.; Wagenknecht, L.E.; Barzilay, J.I.; Tracy, R.P.; Savage, P.J.; Jackson, S.A. Metabolic syndrome and cardiovascular disease in older people: The cardiovascular health study. J. Am. Geriatr. Soc. 2006, 54, 1317–1324. [Google Scholar] [CrossRef] [PubMed]
- International Diabetes Federation. The IDF Consensus Worldwide Definition of the Metabolic Syndrome. Available online: http://www.idf.org/webdata/docs/IDF_Meta_def_final.pdf (accessed on 14 August 2015).
- Strange, R.C.; Shipman, K.E.; Ramachandran, S. Metabolic syndrome: A review of the role of vitamin D in mediating susceptibility and outcome. World J. Diabetes 2015, 6, 896–911. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M. Role of insulin resistance in human disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M. The metabolic syndrome. In Atlas of Atherosclerosis and Metabolic Syndrome; Springer: New York, NY, USA, 2011; pp. 1–26. [Google Scholar]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Farooqui, T.; Panza, F.; Frisardi, V. Metabolic syndrome as a risk factor for neurological disorders. Cell. Mol. Life Sci. 2012, 69, 741–762. [Google Scholar] [CrossRef] [PubMed]
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on Detection, Evaluation, and Treatment of high blood cholesterol in adults (Adult Treatment Panel III). JAMA 2001, 285, 2486–2497. [CrossRef] [PubMed]
- Grundy, S.M.; Cleeman, J.I.; Daniels, S.R.; Donato, K.A.; Eckel, R.H.; Franklin, B.A.; Gordon, D.J.; Krauss, R.M.; Savage, P.J.; Smith, S.C.; et al. Diagnosis and management of the metabolic syndrome an American Heart Association/National Heart, Lung, and Blood Institute scientific statement. Circulation 2005, 112, 2735–2752. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Hansen, B.; Smith, S.C.; Cleeman, J.I.; Kahn, R.A. Clinical management of metabolic syndrome report of the American Heart Association/National Heart, Lung, and Blood Institute/American Diabetes Association conference on scientific issues related to management. Circulation 2004, 109, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Koren-Morag, N.; Goldbourt, U.; Tanne, D. Relation between the metabolic syndrome and ischemic stroke or transient ischemic attack a prospective cohort study in patients with atherosclerotic cardiovascular disease. Stroke 2005, 36, 1366–1371. [Google Scholar] [CrossRef] [PubMed]
- Hummasti, S.; Hotamisligil, G.S. Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ. Res. 2010, 107, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Safar, M.E.; Thomas, F.; Blacher, J.; Nzietchueng, R.; Bureau, J.-M.; Pannier, B.; Benetos, A.; et al. Metabolic syndrome and age-related progression of aortic stiffness. J. Am. Coll. Cardiol. 2006, 47, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Sipilä, K.; Moilanen, L.; Nieminen, T.; Reunanen, A.; Jula, A.; Salomaa, V.; Kaaja, R.; Kukkonen-Harjula, K.; Lehtimäki, T.; Kesäniemi, Y.A.; et al. Metabolic syndrome and carotid intima media thickness in the Health 2000 Survey. Atherosclerosis 2009, 204, 276–281. [Google Scholar] [CrossRef] [PubMed]
- Jennings, J.R.; Heim, A.F.; Kuan, D.C.-H.; Gianaros, P.J.; Muldoon, M.F.; Manuck, S.B. Use of total cerebral blood flow as an imaging biomarker of known cardiovascular risks. Stroke 2013, 44, 2480–2485. [Google Scholar] [CrossRef] [PubMed]
- Faraci, F.M.; Heistad, D.D. Regulation of large cerebral arteries and cerebral microvascular pressure. Circ. Res. 1990, 66, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, K. Metabolic syndrome and cognitive disorders: Is the sum greater than its parts? Alzheimer Dis. Assoc. Disord. 2007, 21, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Whitmer, R.A.; Gunderson, E.P.; Connor, E.B.; Quesenberry, C.P., Jr.; Yaffe, K. Obesity in middle age and future risk of dementia: A 27 year longitudinal population based study. BMJ 2005, 330, 1360–1362. [Google Scholar] [CrossRef] [PubMed]
- Arnoldussen, I.A.; Kiliaan, A.J.; Gustafson, D.R. Obesity and dementia: Adipokines interact with the brain. Eur. Neuropsychopharmacol. 2014, 24, 1982–1999. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N.; Maestre, G.E.; Arizaga, A.R.; Friedland, R.P.; Galasko, D.; Hall, K.; Luchsinger, J.A.; Ogunniyi, A.; Perry, E.K.; Potocnik, F.; et al. Alzheimer’s disease and vascular dementia in developing countries: Prevalence, management, and risk factors. Lancet Neurol. 2008, 7, 812–826. [Google Scholar] [CrossRef]
- Biessels, G.J.; Staekenborg, S.; Brunner, E.; Brayne, C.; Scheltens, P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol. 2006, 5, 64–74. [Google Scholar] [CrossRef]
- Birdsill, A.C.; Carlsson, C.M.; Willette, A.A.; Okonkwo, O.C.; Johnson, S.C.; Xu, G.; Oh, J.M.; Gallagher, C.L.; Koscik, R.L.; Jonaitis, E.M.; et al. Low cerebral blood flow is associated with lower memory function in metabolic syndrome. Obesity 2013, 21, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Mauro, C.; de Rosa, V.; Marelli-Berg, F.; Solito, E. Metabolic syndrome and the immunological affair with the blood-brain barrier. Front. Immunol. 2014, 5, 677. [Google Scholar] [PubMed]
- Minich, D.M.; Bland, J.S. Dietary management of the metabolic syndrome beyond macronutrients. Nutr. Rev. 2008, 66, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Panagiotakos, D.B.; Pitsavos, C.; Chrysohoou, C.; Skoumas, J.; Tousoulis, D.; Toutouza, M.; Toutouzas, P.; Stefanadis, C. Impact of lifestyle habits on the prevalence of the metabolic syndrome among Greek adults from the ATTICA study. Am. Heart J. 2004, 147, 106–112. [Google Scholar] [CrossRef]
- Esposito, K.; Ciotola, M.; Giugliano, D. Mediterranean diet and the metabolic syndrome. Mol. Nutr. Food Res. 2007, 51, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.E.; Prevost, A.T.; Whichelow, M.J.; Cox, B.D.; Day, N.E.; Wareham, N.J. A cross-sectional study of dietary patterns with glucose intolerance and other features of the metabolic syndrome. Br. J. Nutr. 2000, 83, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Paletas, K.; Athanasiadou, E.; Sarigianni, M.; Paschos, P.; Kalogirou, A.; Hassapidou, M.; Tsapas, A. The protective role of the Mediterranean diet on the prevalence of metabolic syndrome in a population of Greek obese subjects. J. Am. Coll. Nutr. 2010, 29, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Feldeisen, S.E.; Tucker, K.L. Nutritional strategies in the prevention and treatment of metabolic syndrome. Appl. Physiol. Nutr. Metab. 2007, 32, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Organization, W.H. Guideline: Sugars Intake for Adults and Children; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Dildy, T. Evidence for and against dietary recommendations to prevent cardiovascular disease. Tex. Heart Inst. J. 2015, 42, 234–236. [Google Scholar] [CrossRef] [PubMed]
- Muzio, F.; Mondazzi, L.; Harris, W.S.; Sommariva, D.; Branchi, A. Effects of moderate variations in the macronutrient content of the diet on cardiovascular disease risk factors in obese patients with the metabolic syndrome. Am. J. Clin. Nutr. 2007, 86, 946–951. [Google Scholar] [PubMed]
- Doherty, G.H. Obesity and the ageing brain: Could leptin play a role in neurodegeneration? Curr. Gerontol. Geriatr. Res. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Flodmark, C.-E.; Lissau, I.; Moreno, L.A.; Pietrobelli, A.; Widhalm, K. New insights into the field of children and adolescents’ obesity: The European perspective. Int. J. Obes. 2004, 28, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Després, J.-P.; Arsenault, B.J.; Côté, M.; Cartier, A.; Lemieux, I. Abdominal obesity: The cholesterol of the 21st century? Can. J. Cardiol. 2008, 24, 7D–12D. [Google Scholar] [CrossRef]
- Park, Y.-W.; Zhu, S.; Palaniappan, L.; Heshka, S.; Carnethon, M.R.; Heymsfield, S.B. The metabolic syndrome: Prevalence and associated risk factor findings in the US population from the Third National Health and Nutrition Examination Survey, 1988–1994. Arch. Intern. Med. 2003, 163, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M. Metabolic syndrome: A multiplex cardiovascular risk factor. J. Clin. Endocrinol. Metab. 2007, 92, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, Y.; Funahashi, T.; Nakamura, T. The concept of metabolic syndrome: Contribution of visceral fat accumulation and its molecular mechanism. J. Atheroscler. Thromb. 2010, 18, 629–639. [Google Scholar] [CrossRef]
- Trayhurn, P.; Beattie, J.H. Physiological role of adipose tissue: White adipose tissue as an endocrine and secretory organ. Proc. Nutr. Soc. 2001, 60, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Kiliaan, A.J.; Arnoldussen, I.A.; Gustafson, D.R. Adipokines: A link between obesity and dementia? Lancet Neurol. 2014, 13, 913–923. [Google Scholar] [CrossRef]
- Guerre-Millo, M. Adipose tissue and adipokines: For better or worse. Diabetes Metab. 2004, 30, 13–19. [Google Scholar] [CrossRef]
- Pan, W.; Kastin, A.J. Adipokines and the blood-brain barrier. Peptides 2007, 28, 1317–1330. [Google Scholar] [CrossRef] [PubMed]
- Spranger, J.; Verma, S.; Göhring, I.; Bobbert, T.; Seifert, J.; Sindler, A.L.; Pfeiffer, A.; Hileman, S.M.; Tschöp, M.; Banks, W.A. Adiponectin does not cross the blood-brain barrier but modifies cytokine expression of brain endothelial cells. Diabetes 2006, 55, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Lago, F.; Gómez, R.; Gómez-Reino, J.J.; Dieguez, C.; Gualillo, O. Adipokines as novel modulators of lipid metabolism. Trends. Biochem. Sci. 2009, 34, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Esteve-Ràfols, M. Adipose tissue: Cell heterogeneity and functional diversity. Endocrinol. Nutr. 2014, 61, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Shibata, R.; Murohara, T.; Ouchi, N. Role of anti-inflammatory adipokines in obesity-related diseases. Trends Endocrinol. Metab. 2014, 25, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Ryo, M.; Nakamura, T.; Kihara, S.; Kumada, M.; Shibazaki, S.; Takahashi, M.; Nagai, M.; Matsuzawa, Y.; Funahashi, T. Adiponectin as a biomarker of the metabolic syndrome. Circ. J. 2004, 68, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Van Gaal, L.F.; Mertens, I.L.; Christophe, E. Mechanisms linking obesity with cardiovascular disease. Nature 2006, 444, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Neto-Ferreira, R.; Rocha, V.N.; de Carvalho, J.J.; Vilanova, L.C.; Barbosa-da-Silva, S.; Souza-Mello, V. Metabolic syndrome: From human organ disease to fetal programming. J. Metab. Syndr. 2014, 3, 133. [Google Scholar] [CrossRef]
- Dandona, P.; Aljada, A.; Chaudhuri, A.; Mohanty, P.; Garg, R. Metabolic syndrome a comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation 2005, 111, 1448–1454. [Google Scholar] [CrossRef] [PubMed]
- Bastard, J.-P.; Maachi, M.; Lagathu, C.; Kim, M.J.; Caron, M.; Vidal, H.; Capeau, J.; Feve, B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur. Cytokine Netw. 2006, 17, 4–12. [Google Scholar] [PubMed]
- Mathieu, P.; Lemieux, I.; Despres, J. Obesity, inflammation, and cardiovascular risk. Clin. Pharmacol. Ther. 2010, 87, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Tesauro, M.; Cardillo, C. Obesity, blood vessels and metabolic syndrome. Acta Physiol. 2011, 203, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, C.G.; Stehouwer, C.D. Vascular complications in diabetes mellitus: The role of endothelial dysfunction. Clin. Sci. 2005, 109, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M. Obesity, metabolic syndrome, and cardiovascular disease. J. Clin. Endocrinol. Metab. 2004, 89, 2595–2600. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, K.; Tanaka, R.; Tanaka, Y.; Miyamoto, N.; Shimada, Y.; Ueno, Y.; Urabe, T.; Hattori, N. Visceral fat accumulation is associated with cerebral small vessel disease. Eur. J. Neurol. 2014, 21, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Czernichow, S.; Bertrais, S.; Blacher, J.; Oppert, J.-M.; Galan, P.; Ducimetière, P.; Hercberg, S.; Safar, M.; Zureik, M. Metabolic syndrome in relation to structure and function of large arteries: A predominant effect of blood pressure: A report from the SU. VI. MAX. Vascular Study. Am. J. Hypertens. 2005, 18, 1154–1160. [Google Scholar] [CrossRef] [PubMed]
- De Jongh, R.T.; Serné, E.H.; IJzerman, R.G.; de Vries, G.; Stehouwer, C.D.A. Free fatty acid levels modulate microvascular function relevance for obesity-associated insulin resistance, hypertension, and microangiopathy. Diabetes 2004, 53, 2873–2882. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.C.; Brunzell, J.D. Abdominal obesity and dyslipidemia in the metabolic syndrome: Importance of type 2 diabetes and familial combined hyperlipidemia in coronary artery disease risk. J. Clin. Endocrinol. Metab. 2004, 89, 2601–2607. [Google Scholar] [CrossRef] [PubMed]
- Moller, D.E.; Kaufman, K.D. Metabolic syndrome: A clinical and molecular perspective. Annu. Rev. Med. 2005, 56, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Grundy, M.; Scott, M. Hypertriglyceridemia, atherogenic dyslipidemia, and the metabolic syndrome. Am. J. Cardiol. 1998, 81, 18B–25B. [Google Scholar] [CrossRef]
- Lorenzo, C.; Okoloise, M.; Williams, K.; Stern, M.P.; Haffner, S.H. The metabolic syndrome as predictor of type 2 diabetes the San Antonio heart study. Diabetes Care 2003, 26, 3153–3159. [Google Scholar] [CrossRef] [PubMed]
- Hokanson, J.E.; Austin, M.A. Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: A metaanalysis of population-based prospective studies. J. Cardiovasc. Risk 1996, 3, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J.; Mar, R.; Pajukanta, P. Genetics of atherosclerosis. Annu. Rev. Genom. Hum. Genet. 2004, 5, 189–218. [Google Scholar] [CrossRef] [PubMed]
- Von Eckardstein, A.; Hersberger, M.; Rohrer, L. Current understanding of the metabolism and biological actions of HDL. Curr. Opin. Clin. Nutr. Metab. Care 2005, 8, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Girman, C.J.; Rhodes, T.; Mercuri, M.; Pyörälä, K.; Kjekshus, J.; Pedersen, T.R.; Beere, P.A.; Gotto, A.M.; Clearfield, M.; Clearfield, M. The metabolic syndrome and risk of major coronary events in the Scandinavian Simvastatin Survival Study (4S) and the Air Force/Texas coronary atherosclerosis prevention study (AFCAPS/TexCAPS). Am. J. Cardiol. 2004, 93, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Putnam, K.; Shoemaker, R.; Yiannikouris, F.; Cassis, L.A. The renin-angiotensin system: A target of and contributor to dyslipidemias, altered glucose homeostasis, and hypertension of the metabolic syndrome. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1219–H1230. [Google Scholar] [CrossRef] [PubMed]
- Mancia, G.; de Backer, G; Dominiczak, A.; Cifkova, R.; Fagard, R.; Germano, G.; Grassi, G.; Heagerty, A.M.; Kjeldsen, S.E.; Laurent, S.; et al. 2007 Guidelines for the management of arterial hypertension: The task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur. Heart J. 2007, 28, 1462–1536. [Google Scholar] [CrossRef] [PubMed]
- Chobanian, A.V.; Bakris, G.L.; Black, H.R.; Cushman, W.C.; Green, L.A.; Izzo, J.L., Jr.; Jones, D.W.; Materson, B.J.; Oparil, S.; Wright, J.T., Jr.; et al. Seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure. Hypertension 2003, 42, 1206–1252. [Google Scholar] [CrossRef] [PubMed]
- Orellana, C.; Parra, F.; Brito, R. Hypertension in Metabolic Syndrome. In Advances in Hypertension Research; Nova Science Publishers Inc.: New York, NY, USA, 2014; p. 153. [Google Scholar]
- Vaughan, D.E. Angiotensin, fibrinolysis, and vascular homeostasis. Am. J. Cardiol. 2001, 87, 18–24. [Google Scholar] [CrossRef]
- Alessi, M.-C.; Juhan-Vague, I. PAI-1 and the metabolic syndrome links, causes, and consequences. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2200–2207. [Google Scholar] [CrossRef] [PubMed]
- Eringa, E.C.; Bakker, W.; Smulders, Y.M.; Serné, E.H.; Yudkin, J.S.; Stehouwer, C.D.A. Regulation of vascular function and insulin sensitivity by adipose tissue: Focus on perivascular adipose tissue. Microcirculation 2007, 14, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, G.E.; Beske, S.D.; Ballard, T.P.; Davy, K.P. Sympathetic neural activation in visceral obesity. Circulation 2002, 106, 2533–2536. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Davisson, R.L. Hypertension and cerebrovascular dysfunction. Cell Metab. 2008, 7, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Alistair, D.G. Hypertensive cerebral small vessel disease and stroke. Brain Pathol. 2002, 12, 358–370. [Google Scholar] [CrossRef]
- FitzGerald, M.J.T.; Gruener, G.; Mtui, E. Clinical Neuroanatomy and Neuroscience; Elsevier Health Sciences: London, UK, 2011. [Google Scholar]
- Baumbach, G.L.; Heistad, D.D. Cerebral circulation in chronic arterial hypertension. Hypertension 1988, 12, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Intengan, H.D.; Schiffrin, E.L. Vascular remodeling in hypertension roles of apoptosis, inflammation, and fibrosis. Hypertension 2001, 38, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Tessier, D.; Carpentier, A. The metabolic syndrome. Pathol. Biol. 2006, 54, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Rusinek, H.; Convit, A. Obesity: Cerebral damage in obesity-associated metabolic syndrome. Nat. Rev. Endocrinol. 2014, 10, 642–644. [Google Scholar] [CrossRef] [PubMed]
- Aronson, D.; Rayfield, E.J. How hyperglycemia promotes atherosclerosis: Molecular mechanisms. Cardiovasc. Diabetol. 2002, 1. [Google Scholar] [CrossRef] [Green Version]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Martini, S.R.; Kent, T.A. Hyperglycemia in acute ischemic stroke: A vascular perspective. J. Cereb. Blood Flow Metab. 2006, 27, 435–451. [Google Scholar] [CrossRef] [PubMed]
- Scuteri, A.; Najjar, S.S.; Muller, D.C.; Andres, R.; Hougaku, H.; Metter, J.; Lakatta, E.G. Metabolic syndrome amplifies the age-associated increases in vascular thickness and stiffness. J. Am. Coll. Cardiol. 2004, 43, 1388–1395. [Google Scholar] [CrossRef] [PubMed]
- Nazzaro, P.; Schirosi, G.; Mezzapesa, D.; Petruzzellis, M.; Pascazio, L.; Serio, G.; de Benedittis, L.; Federico, F. Effect of clustering of metabolic syndrome factors on capillary and cerebrovascular impairment. Eur. J. Intern. Med. 2013, 24, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef] [PubMed]
- Sala, M.; de Roos, A.; van den Berg, A.; Altmann-Schneider, I.; Slagboom, P.E.; Westendorp, R.G.; van Buchem, M.A.; de Craen, A.J.M.; van der Grond, J. Microstructural brain tissue damage in metabolic syndrome. Diabetes Care 2014, 37, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Tiehuis, A.M.; van der Graaf, Y.; Mali, W.P.T.M.; Vincken, K.; Muller, M.; Geerlings, I. Metabolic syndrome, prediabetes, and brain abnormalities on MRI in patients with manifest arterial disease: The SMART-MR study. Diabetes Care 2014, 37, 2515–2521. [Google Scholar] [CrossRef] [PubMed]
- Frisardi, V.; Solfrizzi, V.; Seripa, D.; Capurso, C.; Santamato, A.; Sancarlo, D.; Vendemiale, G.; Pilotto, A.; Panza, F. Metabolic-cognitive syndrome: A cross-talk between metabolic syndrome and Alzheimer’s disease. Ageing Res. Rev. 2010, 9, 399–417. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Gustafson, D.R. Adiposity, type 2 diabetes, and Alzheimer’s disease. J. Alzheimer’s Dis. 2009, 16, 693–704. [Google Scholar]
- Dik, M.G.; Jonker, C.; Comijs, H.C.; Deeg, D.J.H.; Kok, A.; Yaffe, K.; Penninx, B.W. Contribution of metabolic syndrome components to cognition in older individuals. Diabetes Care 2007, 30, 2655–2660. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, E.; Dekker, J.M.; Nijpels, G.; Kessels, R.P.C.; Kappelle, L.J.; de Haan, E.H.F.; Heine, R.J.; Stehouwer, C.D.A.; Biessels, G.J. Cognitive functioning in elderly persons with type 2 diabetes and metabolic syndrome: The Hoorn study. Dement. Geriatr. Cogn. Disord. 2008, 26, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Cavalieri, M.; Ropele, S.; Petrovic, K.; Pluta-Fuerst, A.; Homayoon, N.; Enzinger, C.; Grazer, A.; Katschnig, P.; Schwingenschuh, P.; Berghold, A. Metabolic syndrome, brain magnetic resonance imaging, and cognition. Diabetes Care 2010, 33, 2489–2495. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S.; Mosley, P.H.; Catellier, D.J.; Coker, L.H. Fourteen-year longitudinal study of vascular risk factors, APOE genotype, and cognition: The ARIC MRI Study. Alzheimer’s Dement. 2009, 5, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, K.; Haan, M.; Blackwell, T.; Cherkasova, E.; Whitmer, R.A.; West, N. Metabolic syndrome and cognitive decline in elderly Latinos: Findings from the Sacramento Area Latino Study of Aging study. J. Am. Geriatr. Soc. 2007, 55, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Seiler, S.; Cavalieri, M.; Schmidt, R. Vascular cognitive impairment—An ill-defined concept with the need to define its vascular component. J. Neurol. Sci. 2012, 322, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.; Caracciolo, B.; Brayne, C.; Gauthier, S.; Jelic, V.; Fratiglioni, L.F. Mild cognitive impairment: A concept in evolution. J. Intern. Med. 2014, 275, 214–228. [Google Scholar] [CrossRef] [PubMed]
- Solfrizzi, V.; Scafato, E.; Capurso, C.; D’Introno, A.; Colacicco, A.M.; Frisardi, V.; Vendemiale, G.; Baldereschi, M.; Crepaldi, G.; di Carlo, A.; et al. Metabolic syndrome, mild cognitive impairment, and progression to dementia. The Italian Longitudinal Study on Aging. Neurobiol. Aging 2011, 32, 1932–1941. [Google Scholar] [CrossRef] [PubMed]
- Razay, G.; Vreugdenhil, A.; Wilcock, G. The metabolic syndrome and Alzheimer disease. Arch. Neurol. 2007, 64, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Vanhanen, M.; Koivisto, K.; Moilanen, L.; Helkala, E.L.; Hänninen, T.; Soininen, H.; Kervinen, K.; Kesäniemi, Y.A.; Laakso, M.; Kuusisto, J. Association of metabolic syndrome with Alzheimer disease: A population-based study. Neurology 2006, 67, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Ferris, A.H.; Cai, W.; Kahn, C.R. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef] [PubMed]
- Solfrizzi, V.; Scafato, E.; Capurso, C.; D’Introno, A.; Colacicco, A.M.; Frisardi, V.; Vendemiale, G.; Baldereschi, M.; Crepaldi, G.; di Carlo, A.; et al. Metabolic syndrome and the risk of vascular dementia: The Italian Longitudinal Study on Ageing. J. Neurol. Neurosurg. Psychiatry 2010, 81, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Raffaitin, C.; Gin, H.; Empana, J.-P.; Helmer, C.; Berr, C.; Tzourio, C.; Portet, F.; Dartigues, J.F.; Alpérovitch, A.; Barberger-Gateau, P. Metabolic syndrome and risk for incident Alzheimer’s disease or vascular dementia: The three-city study. Diabetes Care 2009, 32, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Francischetti, E.A.; Tibirica, E.; da Silva, E.G.; Rodrigues, E.; Celoria, B.M.; de Abreu, V.G. Skin capillary density and microvascular reactivity in obese subjects with and without metabolic syndrome. Microvasc. Res. 2011, 81, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Hassenstab, J.J.; Sweat, V.; Bruehl, H.; Convit, A. Metabolic syndrome is associated with learning and recall impairment in middle age. Dement. Geriatr. Cogn. Disord. 2010, 29, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Vieira, J.R.; Elkind, M.S.V.; Moon, Y.P.; Rundek, T.; Boden-Albala, B.; Paik, M.C.; Sacco, R.L.; Wright, C.B. The metabolic syndrome and cognitive performance: The Northern Manhattan Study. Neuroepidemiology 2011, 37, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Komulainen, P.; Lakka, T.A.; Kivipelto, M.; Hassinen, M.; Helkala, E.-L.; Haapala, I.; Nissinen, A.; Rauramaa, R. Metabolic syndrome and cognitive function: A population-based follow-up study in elderly women. Dement. Geriatr. Cogn. Disord. 2006, 23, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Reijmer, Y.D.; van den Berg, E.; Dekker, J.M.; Nijpels, G.; Stehouwer, C.D.A.; Kappelle, L.J.; Biessels, G.J. The metabolic syndrome, atherosclerosis and cognitive functioning in a non-demented population: The Hoorn Study. Atherosclerosis 2011, 219, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Segura, B.; Jurado, M.A.; Freixenet, N.; Albuin, C.; Muniesa, J.; Junqué, C. Mental slowness and executive dysfunctions in patients with metabolic syndrome. Neurosci. Lett. 2009, 462, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.S.; Yi, Q.; Gerstein, H.; Lonn, E.; Jacobs, R.; Vuksan, V.; Teo, K.; Davis, B.; Montague, P.; Yusuf, S.; et al. Relationship of metabolic syndrome and fibrinolytic dysfunction to cardiovascular disease. Circulation 2003, 108, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulos, S.; Boden-Albala, B.; Choi, J.H.; Carrera, E.; Doyle, M.; Perez, T.; Marshall, R.S. Metabolic syndrome and cerebral vasomotor reactivity. Eur. J. Neurol. 2010, 17, 1457–1462. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; van Raamt, F.; Visseren, F.L.J.; Kalmijn, S.; Geerlings, M.I.; Mali, W.P.T.M.; van der Graaf, Y. Metabolic syndrome and cognition in patients with manifest atherosclerotic disease: The SMART study. Neuroepidemiology 2009, 34, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Crichton, G.E.; Elias, M.F.; Buckley, J.D.; Murphy, K.J.; Bryan, J.; Frisardi, V. Metabolic syndrome, cognitive performance, and dementia. J. Alzheimer’s Dis. 2012, 30, S77–S87. [Google Scholar]
- Van den Berg, E.; Biessels, G.J.; de Craen, J.M.; Gussekloo, J.; Westendorp, R.G.J. The metabolic syndrome is associated with decelerated cognitive decline in the oldest old. Neurology 2007, 69, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Katsumata, Y.; Todoriki, H.; Higashiuesato, Y.; Yasura, S.; Willcox, D.C.; Ohya, Y.; Willcox, B.J.; Dodge, H.H. Metabolic syndrome and cognitive decline among the oldest old in Okinawa: In search of a mechanism. The KOCOA Project. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2011, 67, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Siervo, M.; Harrison, S.L.; Jagger, C.; Robinson, L.; Stephan, B.C.M. Metabolic syndrome and longitudinal changes in cognitive function: A systematic review and meta-analysis. J. Alzheimer’s Dis. 2014, 41, 151–161. [Google Scholar]
- Fitzpatrick, A.L.; Kuller, L.H.; Lopez, O.L.; Diehr, P.; O’Meara, E.S.; Longstreth, W.T., Jr.; Luchsinger, J.A. Midlife and late-life obesity and the risk of dementia: Cardiovascular health study. Arch. Neurol. 2009, 66, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Qiu, C.; Gatz, M.; Pedersen, N.L.; Johansson, B.; Fratiglioni, L. Mid- and late-life diabetes in relation to the risk of dementia: A population-based twin study. Diabetes 2009, 58, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol. 2010, 120, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Convit, A. Links between cognitive impairment in insulin resistance: An explanatory model. Neurobiol. Aging 2005, 26, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Drake, C.T.; Iadecola, C. The role of neuronal signaling in controlling cerebral blood flow. Brain Lang. 2007, 102, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Peiser, C.; McGregor, G.P.; Lang, R.E. Binding and internalization of leptin by porcine choroid plexus cells in culture. Neurosci. Lett. 2000, 283, 209–212. [Google Scholar] [CrossRef]
- Zlokovic, B.V.; Jovanovic, S.; Miao, W.; Samara, S.; Verma, S.; Farrell, C.L. Differential regulation of leptin transport by the choroid plexus and blood-brain barrier and high affinity transport systems for entry into hypothalamus and across the blood-cerebrospinal fluid barrier 1. Endocrinology 2000, 141, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; O’connor, J.; Moynagh, P. Dissection of tumor-necrosis factor-α inhibition of long-term potentiation (LTP) reveals a p38 mitogen-activated protein kinase-dependent mechanism which maps to early—But not late—Phase LTP. Neuroscience 2004, 124, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Marsland, A.L.; Gianaros, P.J.; Abramowitch, S.M.; Manuck, S.B.; Hariri, A.R. Interleukin-6 covaries inversely with hippocampal grey matter volume in middle-aged adults. Biol. Psychiatry 2008, 64, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, K.; Kanaya, A.; Lindquist, K.; Simonsick, E.M.; Harris, T.; Shorr, R.I.; Tylavsky, F.A.; Newman, A.B. The metabolic syndrome, inflammation, and risk of cognitive decline. JAMA 2004, 292, 2237–2242. [Google Scholar] [CrossRef] [PubMed]
- Volek, J.S.; Phinney, S.D.; Forsythe, C.E.; Quann, E.E.; Wood, R.J.; Puglisi, M.J.; Kraemer, W.J.; Bibus, D.M.; Fernandez, M.L.; Feinman, R.D. Carbohydrate restriction has a more favorable impact on the metabolic syndrome than a low fat diet. Lipids 2009, 44, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Monzillo, L.U.; Hamdy, O.; Horton, E.S.; Ledbury, S.; Mullooly, C.; Jarema, C.; Porter, S.; Ovalle, K.; Moussa, A.; Mantzoros, C.S. Effect of lifestyle modification on adipokine levels in obese subjects with insulin resistance. Obes. Res. 2003, 11, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-S.; Lee, W.-J.; Funahashi, T.; Tanaka, S.; Matsuzawa, Y.; Chao, C.-L.; Chen, C.-L.; Tai, T.Y; Chuang, L.-M. Weight reduction increases plasma levels of an adipose-derived anti-inflammatory protein, adiponectin. J. Clin. Endocrinol. Metab. 2001, 86, 3815–3819. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, I.S.; Choue, R. Obesity, inflammation and diet. Pediatr. Gastroenterol. Hepatol. Nutr. 2013, 16, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Marfella, R.; Ciotola, M.; di Palo, C.; Giugliano, F.; Giugliano, G.; D’Armiento, M.; D’Andrea, F.; Giugliano, D. Effect of a Mediterranean-style diet on endothelial dysfunction and markers of vascular inflammation in the metabolic syndrome: A randomized trial. JAMA 2004, 292, 1440–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojeda, P.; Bobe, A.; Dolan, K.; Leone, V.; Martinez, K. Nutritional modulation of gut microbiota—The impact on metabolic disease pathophysiology. J. Nutri. Biochem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Gaskins, H.R.; McIntosh, M.K. Influence of dietary fat on intestinal microbes, inflammation, barrier function and metabolic outcomes. J. Nutr. Biochem. 2014, 25, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Volek, J.S.; Fernandez, M.L.; Feinman, R.D.; Phinney, S.D. Dietary carbohydrate restriction induces a unique metabolic state positively affecting atherogenic dyslipidemia, fatty acid partitioning, and metabolic syndrome. Prog. Lipid Res. 2008, 47, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Abate, N.; Chandalia, M. Diet composition and the metabolic syndrome: What is the optimal fat intake? Am. J. Med. 2002, 113, 25–29. [Google Scholar] [CrossRef]
- Eskelinen, M.H.; Ngandu, T.; Helkala, E.-L.; Helkala, J.; Nissinen, A.; Soininen, H.; Kivipelto, M. Fat intake at midlife and cognitive impairment later in life: A population-based CAIDE study. Int. J. Geriatr. Psychiatry 2008, 23, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Solfrizzi, V.; Colacicco, A.M.; D’Introno, A.; Capurso, C.; Torres, F.; Rizzo, C.; Capurso, A.; Panza, F. Dietary intake of unsaturated fatty acids and age-related cognitive decline: A 8.5-year follow-up of the Italian longitudinal study on aging. Neurobiol. Aging 2006, 27, 1694–1704. [Google Scholar] [CrossRef] [PubMed]
- McKeown, N.M.; Meigs, J.B.; Liu, S.; Saltzman, E.; Wilson, P.W.F.; Jacques, P.F. Carbohydrate nutrition, insulin resistance, and the prevalence of the metabolic syndrome in the Framingham Offspring Cohort. Diabetes Care 2004, 27, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Manson, J.E.; Stampfer, M.J.; Holmes, M.D.; Hu, F.B.; Hankinson, S.E.; Willett, W.C. Dietary glycemic load assessed by food-frequency questionnaire in relation to plasma high-density-lipoprotein cholesterol and fasting plasma triacylglycerols in postmenopausal women. Am. J. Clin. Nutr. 2001, 73, 560–566. [Google Scholar] [PubMed]
- Ford, E.S.; Liu, S. Glycemic index and serum high-density lipoprotein cholesterol concentration among US adults. Arch. Intern. Med. 2001, 161, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Galisteo, M.; Duarte, J.; Zarzuelo, A. Effects of dietary fibers on disturbances clustered in the metabolic syndrome. J. Nutr. Biochem. 2008, 19, 71–84. [Google Scholar] [CrossRef] [PubMed]
- James, S.L.; Muir, J.G.; Curtis, S.L.; Gibson, P.R. Dietary fibre: A roughage guide. Intern. Med. J. 2003, 33, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Padayachee, A.; Day, L.; Howell, K.; Gidley, M.J. Complexity and health functionality of plant cell wall fibres from fruits and vegetables. Crit. Rev. Food Sci. Nutr. 2015. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, E.; Houben, T.; Fu, J.; Shiri-Sverdlov, R.; Hofker, M.H. The immunity-diet-microbiota axis in the development of metabolic syndrome. Curr. Opin. Lipidol. 2015, 26, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Canfora, E.E.; Jocken, J.W.; Blaak, E.E. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 2015, 11, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, A.; van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.W.M.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Psaltopoulou, T.; Naska, A.; Orfanos, P.; Trichopoulos, D.; Mountokalakis, T.; Trichopoulou, A. Olive oil, the Mediterranean diet, and arterial blood pressure: The Greek European prospective investigation into cancer and nutrition (EPIC) study. Am. J. Clin. Nutr. 2004, 80, 1012–1018. [Google Scholar] [PubMed]
- Ard, J.D.; Coffman, C.J.; Lin, P.-H.; Svetkey, L.P. One-year follow-up study of blood pressure and dietary patterns in dietary approaches to stop hypertension (DASH)-sodium participants. Am. J. Hypertens. 2004, 17, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Azadbakht, L.; Mirmiran, P.; Esmaillzadeh, A.; Azizi, T.; Azizi, F. Beneficial effects of a Dietary Approaches to Stop Hypertension eating plan on features of the metabolic syndrome. Diabetes Care 2005, 28, 2823–2831. [Google Scholar] [CrossRef] [PubMed]
- Brader, L.; Uusitupa, M.; Dragsted, L.O.; Hermansen, K. Effects of an isocaloric healthy Nordic diet on ambulatory blood pressure in metabolic syndrome: A randomized SYSDIET sub-study. Eur. J. Clin. Nutr. 2014, 68, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, I.M.; Grim, C.E.; Kotchen, T.A. Dietary calcium lowers the age-related rise in blood pressure in the United States: The NHANES III survey. J. Clin. Hypertens. 2003, 5, 122–126. [Google Scholar] [CrossRef]
- Ballard, K.D.; Mah, E.; Guo, Y.; Pei, R.; Volek, J.S.; Bruno, R.S. Low-fat milk ingestion prevents postprandial hyperglycemia-mediated impairments in vascular endothelial function in obese individuals with metabolic syndrome. J. Nutr. 2013, 143, 1602–1610. [Google Scholar] [CrossRef] [PubMed]
- Calton, E.K.; James, A.P.; Pannu, P.K.; Soares, M.J. Certain dietary patterns are beneficial for the metabolic syndrome: Reviewing the evidence. Nutr. Res. 2014, 34, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Pribis, P.; Shukitt-Hale, B. Cognition: The new frontier for nuts and berries. Am. J. Clin. Nutr. 2014, 100 (Suppl. S1), 347S–352S. [Google Scholar] [CrossRef] [PubMed]
- Bajerska, J.; Wozniewicz, M.; Suwalska, A.; Jeszka, J. Eating patterns are associated with cognitive function in the elderly at risk of metabolic syndrome from rural areas. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 3234–3245. [Google Scholar] [PubMed]
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.-I.; Corella, D.; Arós, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J.; et al. Primary prevention of cardiovascular disease with a Mediterranean diet. N. Engl. J. Med. 2013, 368, 1279–1290. [Google Scholar] [CrossRef] [PubMed]
- Damasceno, N.R.T.; Sala-Vila, A.; Cofán, M.; Pérez-Heras, A.M.; Fitó, M.; Ruiz-Gutiérrez, V.; Martínez-González, M.-A.; Corella, D.; Arós, F.; Estruch, R.; et al. Mediterranean diet supplemented with nuts reduces waist circumference and shifts lipoprotein subfractions to a less atherogenic pattern in subjects at high cardiovascular risk. Atherosclerosis 2013, 230, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Appel, L.J.; Sacks, F.M.; Carey, V.J.; Obarzanek, E.; Swain, J.F.; Miller, E.R.; Conlin, P.R.; Erlinger, T.P.; Rosner, B.A.; Laranjo, N.M.; et al. Effects of protein, monounsaturated fat, and carbohydrate intake on blood pressure and serum lipids: Results of the OmniHeart randomized trial. JAMA 2005, 294, 2455–2464. [Google Scholar] [CrossRef] [PubMed]
- Ueshima, H.; Stamler, J.; Elliott, P.; Chan, Q.; Brown, I.J.; Carnethon, M.R.; Daviglus, M.L.; He, K.; Moag-Stahlberg, A.; Rodriguez, B.L.; et al. Food omega-3 fatty acid intake of individuals (total, linolenic acid, long-chain) and their blood pressure INTERMAP study. Hypertension 2007, 50, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, M.; Ros, E.; Salas-Salvadó, J.; Covas, M.-I.; Corella, C.; Arós, M.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J.; et al. Omega-3 fatty acid supplements improve the cardiovascular risk profile of subjects with metabolic syndrome, including markers of inflammation and auto-immunity. Acta Cardiol. 2009, 64, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R.; Arnoldi, A.; Cicero, A.F.G. Nutraceuticals for blood pressure control. Ann. Med. 2015, 47, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Luft, F.C.; Miller, J.Z.; Grim, C.E.; Fineberg, N.S.; Christian, J.C.; Daugherty, S.A.; Weinberger, M.H. Salt sensitivity and resistance of blood pressure. Age and race as factors in physiological responses. Hypertension 1991, 17 (Suppl. S1), I102–I108. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, I.; Cubeddu, L. Increased blood pressure reactivity to dietary salt in patients with the metabolic syndrome. J. Hum. Hypertens. 2007, 21, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gu, D.; Huang, J.; Rao, D.C.; Jaquish, C.E.; Hixson, J.E.; Chen, C.-S.; Chen, J.; Lu, F.; Hu, D.; et al. Metabolic syndrome and salt sensitivity of blood pressure in non-diabetic people in China: A dietary intervention study. Lancet 2009, 373, 829–835. [Google Scholar] [CrossRef]
- Adrogué, H.J.; Madias, N.E. Sodium surfeit and potassium deficit: Keys to the pathogenesis of hypertension. J. Am. Soc. Hypertens. 2014, 8, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Whelton, P.K.; He, J. Potassium in preventing and treating high blood pressure. Semin. Nephrol. 1999, 19, 494–499. [Google Scholar] [PubMed]
- Gu, D.; He, J.; Wu, X.; Duan, X.; Whelton, P.K. Effect of potassium supplementation on blood pressure in Chinese: A randomized, placebo-controlled trial. J. Hypertens. 2001, 19, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Liu, T.; Kuklina, E.V.; Flanders, W.D.; Hong, Y.; Gillespie, C.; Chang, M.-H.; Gwinn, M.; Dowling, N.; Khoury, M.J.; et al. Sodium and potassium intake and mortality among US adults: Prospective data from the third national health and nutrition examination survey. Arch. Intern. Med. 2011, 171, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, G.; Rivellese, A. Dietary treatment of the metabolic syndrome—The optimal diet. Br. J. Nutr. 2000, 83 (Suppl. S1), S143–S148. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.; Wolever, T.M.; Taylor, R.H.; Barker, H.; Fielden, H.; Baldwin, J.M.; Bowling, A.C.; Newman, H.C.; Jenkins, A.L.; Goff, D.V. Glycemic index of foods: A physiological basis for carbohydrate exchange. Am. J. Clin. Nutr. 1981, 34, 362–366. [Google Scholar] [PubMed]
- Holt, S.; Miller, J.; Petocz, P. An insulin index of foods: The insulin demand generated by 1000-kJ portions of common foods. Am. J. Clin. Nutr. 1997, 66, 1264–1276. [Google Scholar] [PubMed]
- Nimptsch, K.; Brand-Miller, J.C.; Franz, M.; Sampson, L.; Willett, W.C.; Giovannucci, E. Dietary insulin index and insulin load in relation to biomarkers of glycemic control, plasma lipids, and inflammation markers. Am. J. Clin. Nutr. 2011, 94, 182–190. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Liu, K.; Daviglus, M.L.; Morris, S.J.; Loria, C.M.; van Horn, L.; Jacobs, D.R., Jr.; Savage, P.J. Magnesium intake and incidence of metabolic syndrome among young adults. Circulation 2006, 113, 1675–1682. [Google Scholar] [CrossRef] [PubMed]
- Rumawas, M.E.; McKeown, N.M.; Rogers, G.; Meigs, J.B.; Wilson, P.W.F.; Jacques, P.F. Magnesium intake is related to improved insulin homeostasis in the Framingham offspring cohort. J. Am. Coll. Nutr. 2006, 25, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Xun, P.; Liu, K.; Loria, C.; Yokota, K.; Jacobs, D.R., Jr.; He, K. Magnesium intake in relation to systemic inflammation, insulin resistance, and the incidence of diabetes. Diabetes Care 2010, 33, 2604–2610. [Google Scholar] [CrossRef] [PubMed]
- Mooren, F.; Krüger, K.; Völker, K.; Golf, S.W.; Wadepuhl, M.; Kraus, A. Oral magnesium supplementation reduces insulin resistance in non-diabetic subjects—A double-blind, placebo-controlled, randomized trial. Diabetes Obes. Metab. 2011, 13, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; He, K.; Levitan, E.B.; Manson, J.E.; Liu, S. Effects of oral magnesium supplementation on glycaemic control in type 2 diabetes: A meta-analysis of randomized double-blind controlled trials. Diabet. Med. 2006, 23, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Hadjistavri, L.S.; Sarafidis, P.A.; Georgianos, P.I.; Tziolas, I.M.; Aroditis, C.P.; Hitoglou-Makedou, A.; Zebekakis, P.E.; Pikilidou, M.I.; Lasaridis, A.N. Beneficial effects of oral magnesium supplementation on insulin sensitivity and serum lipid profile. Med. Sci. Rev. 2010, 16, CR307–CR312. [Google Scholar]
- Deer, J.; Koska, J.; Ozias, M.; Reaven, P. Dietary models of insulin resistance. Metabolism 2015, 64, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Cherniack, E.P. Polyphenols: Planting the seeds of treatment for the metabolic syndrome. Nutrition 2011, 27, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jin, Y.; Choi, Y.; Park, T. Resveratrol exerts anti-obesity effects via mechanisms involving down-regulation of adipogenic and inflammatory processes in mice. Biochem. Pharmacol. 2011, 81, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Rossi, L.; Mazzitelli, S.; Arciello, M.; Capo, C.R.; Rotilio, G. Benefits from dietary polyphenols for brain aging and Alzheimer’s disease. Neurochem. Res. 2008, 33, 2390–2400. [Google Scholar] [CrossRef] [PubMed]
- Cherniack, E.P. A berry thought-provoking idea: The potential role of plant polyphenols in the treatment of age-related cognitive disorders. Br. J. Nutr. 2012, 108, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Sohrab, G.; Hosseinpour-Niazi, S.; Hejazi, J.; Yuzbashian, E.; Mirmiran, P.; Azizi, F. Dietary polyphenols and metabolic syndrome among Iranian adults. Int. J. Food Sci. Nutr. 2013, 64, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Karatas, S.; Hekimsoy, K.; Dinc, G.; Onur, E.; Ozmen, B. Vitamin D levels in overweight/obese adults with and without metabolic syndrome. J. Endocrinol. Metab. 2013, 3, 47–56. [Google Scholar] [CrossRef]
- Vitezova, A.; Zillikens, M.C.; van Herpt, T.T.W.; Sijbrands, E.J.G.; Hofman, A.; Uitterlinden, A.G.; Franco, O.H.; Kiefte-de Jong, J.C. Vitamin D status and metabolic syndrome in the elderly: The Rotterdam Study. Eur. J. Endocrinol. 2015, 172, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Salonen, M.K.; Wasenius, N.; Kajantie, E.; Lano, A.; Lahti, J.; Heinonen, K.; Räikkönen, K.; Eriksson, J.G. Physical activity, body composition and metabolic syndrome in young adults. PLoS ONE 2015, 10, e0126737. [Google Scholar] [CrossRef] [PubMed]
- Babio, N.; Bulló, M.; Salas-Salvadó, J. Mediterranean diet and metabolic syndrome: The evidence. Public Health Nutr. 2009, 12, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mellendijk, L.; Wiesmann, M.; Kiliaan, A.J. Impact of Nutrition on Cerebral Circulation and Cognition in the Metabolic Syndrome. Nutrients 2015, 7, 9416-9439. https://doi.org/10.3390/nu7115477
Mellendijk L, Wiesmann M, Kiliaan AJ. Impact of Nutrition on Cerebral Circulation and Cognition in the Metabolic Syndrome. Nutrients. 2015; 7(11):9416-9439. https://doi.org/10.3390/nu7115477
Chicago/Turabian StyleMellendijk, Laura, Maximilian Wiesmann, and Amanda J. Kiliaan. 2015. "Impact of Nutrition on Cerebral Circulation and Cognition in the Metabolic Syndrome" Nutrients 7, no. 11: 9416-9439. https://doi.org/10.3390/nu7115477
APA StyleMellendijk, L., Wiesmann, M., & Kiliaan, A. J. (2015). Impact of Nutrition on Cerebral Circulation and Cognition in the Metabolic Syndrome. Nutrients, 7(11), 9416-9439. https://doi.org/10.3390/nu7115477