Paternal High Fat Diet in Rats Leads to Renal Accumulation of Lipid and Tubular Changes in Adult Offspring

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Kidney Triglyceride Measurement
2.3. Plasma Lipid, Electrolytes, Creatinine, Albumin and Cystatin C Measurements
2.4. Quantitative Polymerase Chain Reaction (q-PCR)
2.5. Histology
2.6. Statistical Analysis
3. Results
3.1. Effect of HFD on Fathers
3.2. Offspring Body Weight, Tissue Mass and Kidney Triglyceride at Week 27
3.3. Plasma Creatinine, Albumin, Electrolyte and Cystatin C Concentrations
3.4. Renal Gene Expression
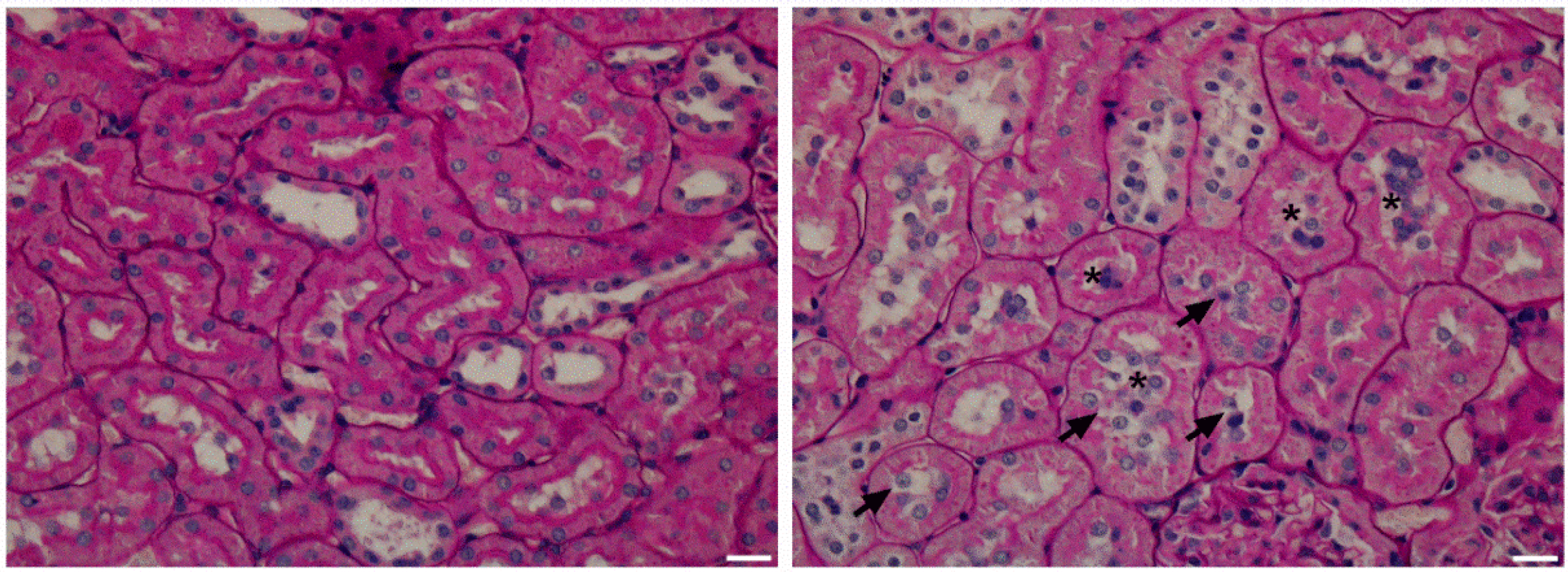
3.5. Kidney histology
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Acat 1 | Acetyl-CoA acetyltransferase 1 |
| Acaca | Acetyl-CoA carboxylase alpha |
| Cd36 | Cd36 molecule |
| CKD | Chronic kidney disease |
| Cpt1a | Carnitine palmitoyltransferase 1A |
| Ep | Epididymal |
| Fasn | Fatty acid synthase |
| GFR | Glomerular filtration rate |
| Hadh | Hydroxyacyl-CoA dehydrogenase |
| HFD | High fat diet |
| Hprt 1 | Hypoxanthine phosphoribosyltransferase 1 |
| IL-6 | Interleukin 6 |
| Kim1 | Kidney injury molecule 1 |
| Ldlr | Low density lipoprotein receptor |
| Lpl | Lipoprotein lipase |
| Mcp1 | Monocyte chemoattractant protein-1 |
| NEFA | Non-esterified fatty acid |
| Ngal | Neutrophil gelatinase-associated lipocalin |
| PAS | Periodic acid-Schiff |
| Pgc1α | Peroxisome proliferator-activated receptor gamma, coactivator 1 alpha |
| Pparg | Peroxisome proliferator-activated receptor gamma |
| q-PCR | Quantitative polymerase chain reaction |
| Rp | Retroperitoneal |
| SD | Sprague Dawley |
| Srebp | Sterol regulatory element binding protein |
| Tgf β1 | Transforming growth factor beta 1 |
| Tnf-α | Tumor necrosis factor alpha |
| WAT | White adipose tissue |
References
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the global burden of disease study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef]
- Levey, A.S.; Coresh, J. Chronic kidney disease. Lancet 2012, 379, 165–180. [Google Scholar] [CrossRef]
- Savino, A.; Pelliccia, P.; Chiarelli, F.; Mohn, A. Obesity-related renal injury in childhood. Horm. Res. Paediatr. 2010, 73, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J. Early life influences on obesity risk: Maternal overnutrition and programming of obesity. Expert Rev. Endocrinol. Metab. 2009, 4, 625–637. [Google Scholar] [CrossRef]
- Samuelsson, A.-M.; Morris, A.; Igosheva, N.; Kirk, S.L.; Pombo, J.M.; Coen, C.W.; Poston, L.; Taylor, P.D. Evidence for sympathetic origins of hypertension in juvenile offspring of obese rats. Hypertension 2010, 55, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Fullston, T.; Ohlsson Teague, E.M.; Palmer, N.O.; DeBlasio, M.J.; Mitchell, M.; Corbett, M.; Print, C.G.; Owens, J.A.; Lane, M. Paternal obesity initiates metabolic disturbances in two generations of mice with incomplete penetrance to the f2 generation and alters the transcriptional profile of testis and sperm microrna content. FASEB 2013, 27, 4226–4243. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.-F.; Lin, R.C.; Laybutt, D.R.; Barres, R.; Owens, J.A.; Morris, M.J. Chronic high-fat diet in fathers programs [bgr]-cell dysfunction in female rat offspring. Nature 2010, 467, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.-F.; Lin, R.C.; Maloney, C.A.; Youngson, N.A.; Owens, J.A.; Morris, M.J. Paternal high-fat diet consumption induces common changes in the transcriptomes of retroperitoneal adipose and pancreatic islet tissues in female rat offspring. FASEB 2014, 28, 1830–1841. [Google Scholar] [CrossRef] [PubMed]
- Moritz, K.M.; Dodic, M.; Wintour, E.M. Kidney development and the fetal programming of adult disease. BioEssays 2003, 25, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Dorey, E.S.; Pantaleon, M.; Weir, K.A.; Moritz, K.M. Adverse prenatal environment and kidney development: Implications for programing of adult disease. Reproduction 2014, 147, R189–R198. [Google Scholar] [CrossRef] [PubMed]
- Vik, K.L.; Romundstad, P.; Carslake, D.; Smith, G.D.; Nilsen, T.I. Comparison of father-offspring and mother-offspring associations of cardiovascular risk factors: Family linkage within the population-based hunt study, norway. Int. J. Epidemiol. 2014, 43, 760–771. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, K.; Ye, Y.L.; Yuan, S.; He, Q.Q. Parental weight status and offspring cardiovascular disease risks: A cross-sectional study of chinese children. Prev. Chronic Dis. 2015, 12, 140384. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tsuprykov, O.; Yang, X.; Hocher, B. Paternal programming of offspring cardiometabolic diseases in later life. J. Hyperten. 2016, 34, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Xiao, X.M.; Li, J.; Reichetzeder, C.; Wang, Z.N.; Hocher, B. Paternal body mass index (bmi) is associated with offspring intrauterine growth in a gender dependent manner. PLoS ONE 2012, 7, e36329. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, V.; Youngson, N.A.; Maloney, C.A.; Morris, M.J. Parental programming: How can we improve study design to discern the molecular mechanisms? BioEssays 2013, 35, 787–793. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Yang, L. Kidney injury molecule-1. Curr. Opin. Crit. Care 2010, 16, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Cruz, D.N.; de Cal, M.; Garzotto, F.; Perazella, M.A.; Lentini, P.; Corradi, V.; Piccinni, P.; Ronco, C. Plasma neutrophil gelatinase-associated lipocalin is an early biomarker for acute kidney injury in an adult icu population. Intensive Care Med. 2010, 36, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.; Ørntoft, T. Normalization of real-time quantitative rt-pcr data: A model based variance estimation approach to identify genes suited for normalization-applied to bladder-and colon-cancer data-sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, V.Y.; Faubel, S.; Siegmund, B.; Lucia, M.S.; Ljubanovic, D.; Edelstein, C.L. Neutrophil-independent mechanisms of caspase-1- and il-18-mediated ischemic acute tubular necrosis in mice. J. Clin. Invest. 2002, 110, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, X.; Song, Y.; Caballero, B.; Cheskin, L. Association between obesity and kidney disease: A systematic review and meta-analysis. Kidney Int. 2008, 73, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Nitsch, D.; Sandling, J.K.; Byberg, L.; Larsson, A.; Tuvemo, T.; Syvanen, A.C.; Koupil, I.; Leon, D.A. Fetal, developmental, and parental influences on cystatin c in childhood: The uppsala family study. Am. J. Kidney Dis. 2011, 57, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Blydt-Hansen, T.D.; Katori, M.; Lassman, C.; Ke, B.; Coito, A.J.; Iyer, S.; Buelow, R.; Ettenger, R.; Busuttil, R.W.; Kupiec-Weglinski, J.W. Gene transfer-induced local heme oxygenase-1 overexpression protects rat kidney transplants from ischemia/reperfusion injury. J. Am. Soc. Nephrol. 2003, 14, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Solez, K.; Morel-Maroger, L.; Sraer, J.D. The morphology of “acute tubular necrosis” in man: Analysis of 57 renal biopsies and a comparison with the glycerol model. Medicine 1979, 58, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S. Mechanisms and consequences of hypertriglyceridemia and cellular lipid accumulation in chronic kidney disease and metabolic syndrome. Histol. Histopathol. 2011, 26, 1599–1610. [Google Scholar] [PubMed]
- Zager, R.A.; Burkhart, K.M.; Johnson, A.C.; Sacks, B.M. Increased proximal tubular cholesterol content: Implications for cell injury and “acquired cytoresistance”. Kidney Int. 1999, 56, 1788–1797. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.C.M.; Yabu, J.M.; Hanson, S.; Shah, V.O.; Zager, R.A. Experimental glomerulopathy alters renal cortical cholesterol, sr-b1, abca1, and hmg coa reductase expression. Am. J. Pathol. 2003, 162, 283–291. [Google Scholar] [CrossRef]
- Mount, P.; Davies, M.; Choy, S.W.; Cook, N.; Power, D. Obesity-related chronic kidney disease-the role of lipid metabolism. Metabolites 2015, 5, 720–732. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Hao, L.; Fu, X.; Huang, M.; Li, R. Severe hypertriglyceridemia and hypercholesterolemia accelerating renal injury: A novel model of type 1 diabetic hamsters induced by short-term high-fat/high-cholesterol diet and low-dose streptozotocin. BMC Nephrol. 2015, 16, 51. [Google Scholar] [CrossRef]
- Wahl, P.; Ducasa, G.M.; Fornoni, A. Systemic and renal lipids in kidney disease development and progression. Am. J. Physiol. Ren. Physiol. 2016, 310, F433–F445. [Google Scholar] [CrossRef] [PubMed]
- Grove, K.J.; Voziyan, P.A.; Spraggins, J.M.; Wang, S.; Paueksakon, P.; Harris, R.C.; Hudson, B.G.; Caprioli, R.M. Diabetic nephropathy induces alterations in the glomerular and tubule lipid profiles. J. Lipid Res. 2014, 55, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.M.; Alexander, B.T.; Roach, L.; Haggerty, D.; Marbury, D.C.; Hutchens, Z.M.; Flynn, E.R.; Maric-Bilkan, C. Exposure to maternal overnutrition and a high-fat diet during early postnatal development increases susceptibility to renal and metabolic injury later in life. Am. J. Physiol. Ren. Physiol. 2012, 302, F774–F783. [Google Scholar] [CrossRef] [PubMed]
- Bobulescu, I.A. Renal lipid metabolism and lipotoxicity. Curr. Opin. Nephrol. Hypertens. 2010, 19, 393. [Google Scholar] [CrossRef] [PubMed]
- Wu, I.; Parikh, C.R. Screening for kidney diseases: Older measures versus novel biomarkers. Clin. J. Am. Soc. Nephrol. 2008, 3, 1895–1901. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, S.; Sleight, P.; Pogue, J.; Bosch, J.; Davies, R.; Dagenais, G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The heart outcomes prevention evaluation study investigators. N. Eng. J. Med. 2000, 342, 145–153. [Google Scholar]
- Luyckx, V.A.; Bertram, J.F.; Brenner, B.M.; Fall, C.; Hoy, W.E.; Ozanne, S.E.; Vikse, B.E. Effect of fetal and child health on kidney development and long-term risk of hypertension and kidney disease. Lancet 2013, 382, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Painter, R.C.; Roseboom, T.J.; Van Montfrans, G.A.; Bossuyt, P.M.; Krediet, R.T.; Osmond, C.; Barker, D.J.; Bleker, O.P. Microalbuminuria in adults after prenatal exposure to the dutch famine. J. Am. Soc. Nephrol. 2005, 16, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Ritz, E.; Amann, K.; Koleganova, N.; Benz, K. Prenatal programming—Effects on blood pressure and renal function. Nat. Rev. Nephrol. 2011, 7, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Hoy, W.E.; Rees, M.; Kile, E.; Mathews, J.D.; Wang, Z. A new dimension to the barker hypothesis: Low birthweight and susceptibility to renal disease. Kidney Int. 1999, 56, 1072–1077. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.W.; Yamamoto, K.T.; Henry, R.K.; De Roos, A.J.; Flynn, J.T. Prenatal risk factors for childhood ckd. J. Am. Soc. Nephrol. 2014, 25, 2105–2111. [Google Scholar] [CrossRef] [PubMed]
- Flynn, E.R.; Alexander, B.T.; Lee, J.; Hutchens, Z.M., Jr.; Maric-Bilkan, C. High-fat/fructose feeding during prenatal and postnatal development in female rats increases susceptibility to renal and metabolic injury later in life. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R278–R285. [Google Scholar] [CrossRef] [PubMed]
- Youngson, N.A.; Lecomte, V.; Maloney, C.A.; Leung, P.; Liu, J.; Hesson, L.B.; Luciani, F.; Krause, L.; Morris, M.J. Obesity-induced sperm DNA methylation changes at satellite repeats are reprogrammed in rat offspring. Asian J. Androl. 2016, 18, 1–7. [Google Scholar]


{kind=link}
| Paternal Diet | Control (n = 8) | HFD (n = 8) |
|---|---|---|
| Body weight (g) | 567.7 ± 12.1 | 504.1 ± 11.3 ** |
| Fat mass (g) | 14.56 ± 1.08 | 10.94 ± 1.14 * |
| Liver mass (g) | 16.23 ± 0.75 | 13.71 ± 0.65 * |
| Kidney mass (g) | 1.55 ± 0.09 | 1.55 ± 0.05 |
| Kidney triglyceride (μg/mg) | 2.99 ± 0.47 | 5.35 ± 0.84 * |
| Plasma triglyceride (mmol/L) | 0.94 ± 0.13 | 0.90 ± 0.07 |
| Plasma NEFA (mmol/L) | 1.28 ± 0.22 | 1.18 ± 0.10 |
| Paternal Diet | Control (n = 8) | HFD (n = 8) |
|---|---|---|
| Creatinine (μmol/L) | 27.78 ± 1.60 | 25.50 ± 1.35 |
| Albumin (g/L) | 28.69 ± 0.37 | 29.07 ± 0.73 |
| Cystatin C (pg/ml) | 295.6 ± 28.0 | 278.4 ± 15.0 |
| Sodium (mmol/L) | 141.8 ± 0.83 | 142.5 ± 0.78 |
| Potassium (mmol/L) | 3.33 ± 0.14 | 3.46 ± 0.15 |
| Calcium (mmol/L) | 0.34 ± 0.03 | 0.31 ± 0.01 |
| Lactate (mmol/L) | 1.72 ± 0.25 | 1.23 ± 0.14 |
| Paternal Diet | Control (n = 8) | HFD (n = 8) |
|---|---|---|
| Cd36 | 0.65 ± 0.15 | 0.57 ± 0.11 |
| Ldlr | 0.87 ± 0.06 | 0.88 ± 0.09 |
| Srebp1 | 0.94 ± 0.05 | 0.90 ± 0.04 |
| Srebp2 | 0.90 ± 0.08 | 0.85 ± 0.06 |
| Acaca | 1.05 ± 0.21 | 0.86 ± 0.12 |
| Fasn | 1.24 ± 0.14 | 1.10 ± 0.15 |
| Acat 1 | 0.81 ± 0.02 | 0.95 ± 0.06 * |
| Lpl | 1.01 ± 0.17 | 0.87 ± 0.07 |
| Hadh | 0.95 ± 0.13 | 0.82 ± 0.03 |
| Pgc1α | 0.99 ± 0.08 | 0.91 ± 0.03 |
| Pparg | 1.10 ± 0.15 | 1.16 ± 0.05 |
| Cpt1a | 1.03 ± 0.12 | 0.83 ± 0.03 |
| IL-6 | 1.52 ± 0.21 | 1.88 ± 0.49 |
| Tnf-α | 0.82 ± 0.13 | 0.90 ± 0.13 |
| Mcp1a | 1.18 ± 0.09 | 0.92 ± 0.11 |
| Tgf β1 | 1.08 ± 0.06 | 1.01 ± 0.09 |
| Kim 1 | 1.32 ± 0.45 | 0.92 ± 0.15 |
| Ngal | 0.98 ± 0.11 | 0.96 ± 0.09 |
| (%) | Cells Sloughing in Lumen | Absence/Rupture Brush Border | Apoptotic Cells | Debris |
|---|---|---|---|---|
| Control Father | 38.72 ± 2.46 | 22.77 ± 2.16 | 0.85 ± 0.40 | 12.02 ± 0.61 |
| HFD Father | 46.86 ± 2.30 * | 27.03 ± 1.72 * | 1.71 ± 0.74 | 13.81 ± 1.15 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chowdhury, S.S.; Lecomte, V.; Erlich, J.H.; Maloney, C.A.; Morris, M.J. Paternal High Fat Diet in Rats Leads to Renal Accumulation of Lipid and Tubular Changes in Adult Offspring. Nutrients 2016, 8, 521. https://doi.org/10.3390/nu8090521
Chowdhury SS, Lecomte V, Erlich JH, Maloney CA, Morris MJ. Paternal High Fat Diet in Rats Leads to Renal Accumulation of Lipid and Tubular Changes in Adult Offspring. Nutrients. 2016; 8(9):521. https://doi.org/10.3390/nu8090521
Chicago/Turabian StyleChowdhury, Sabiha S., Virginie Lecomte, Jonathan H. Erlich, Christopher A. Maloney, and Margaret J. Morris. 2016. "Paternal High Fat Diet in Rats Leads to Renal Accumulation of Lipid and Tubular Changes in Adult Offspring" Nutrients 8, no. 9: 521. https://doi.org/10.3390/nu8090521
APA StyleChowdhury, S. S., Lecomte, V., Erlich, J. H., Maloney, C. A., & Morris, M. J. (2016). Paternal High Fat Diet in Rats Leads to Renal Accumulation of Lipid and Tubular Changes in Adult Offspring. Nutrients, 8(9), 521. https://doi.org/10.3390/nu8090521