Butyrate Inhibits Cancerous HCT116 Colon Cell Proliferation but to a Lesser Extent in Noncancerous NCM460 Colon Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Cell Count/Growth Assay
2.3. Apoptosis Analysis
2.4. DNA Fragmentation Assay
2.5. Western Blotting Analysis
2.6. Immunfluorescent Staining
2.7. Statistical Analysis
3. Results
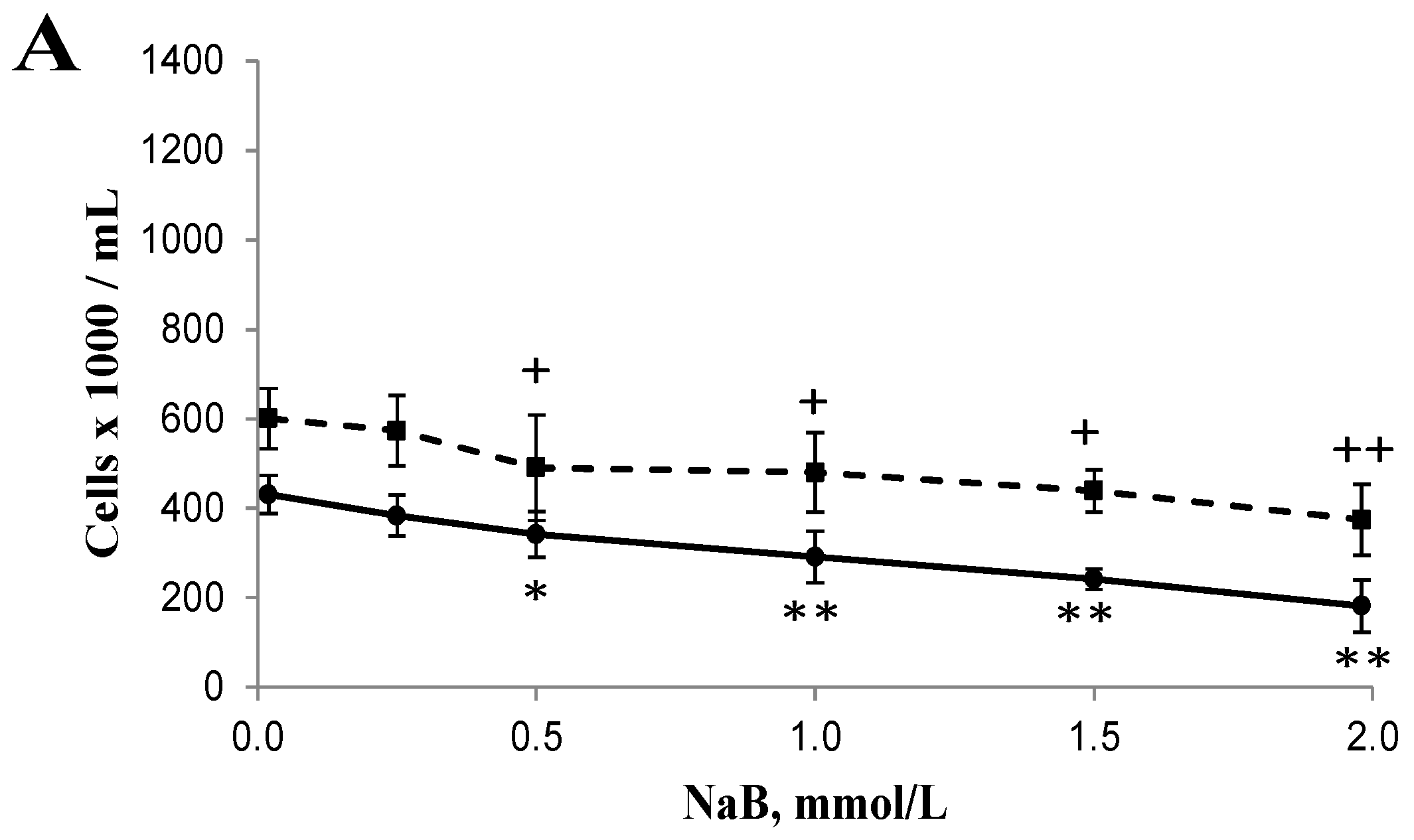
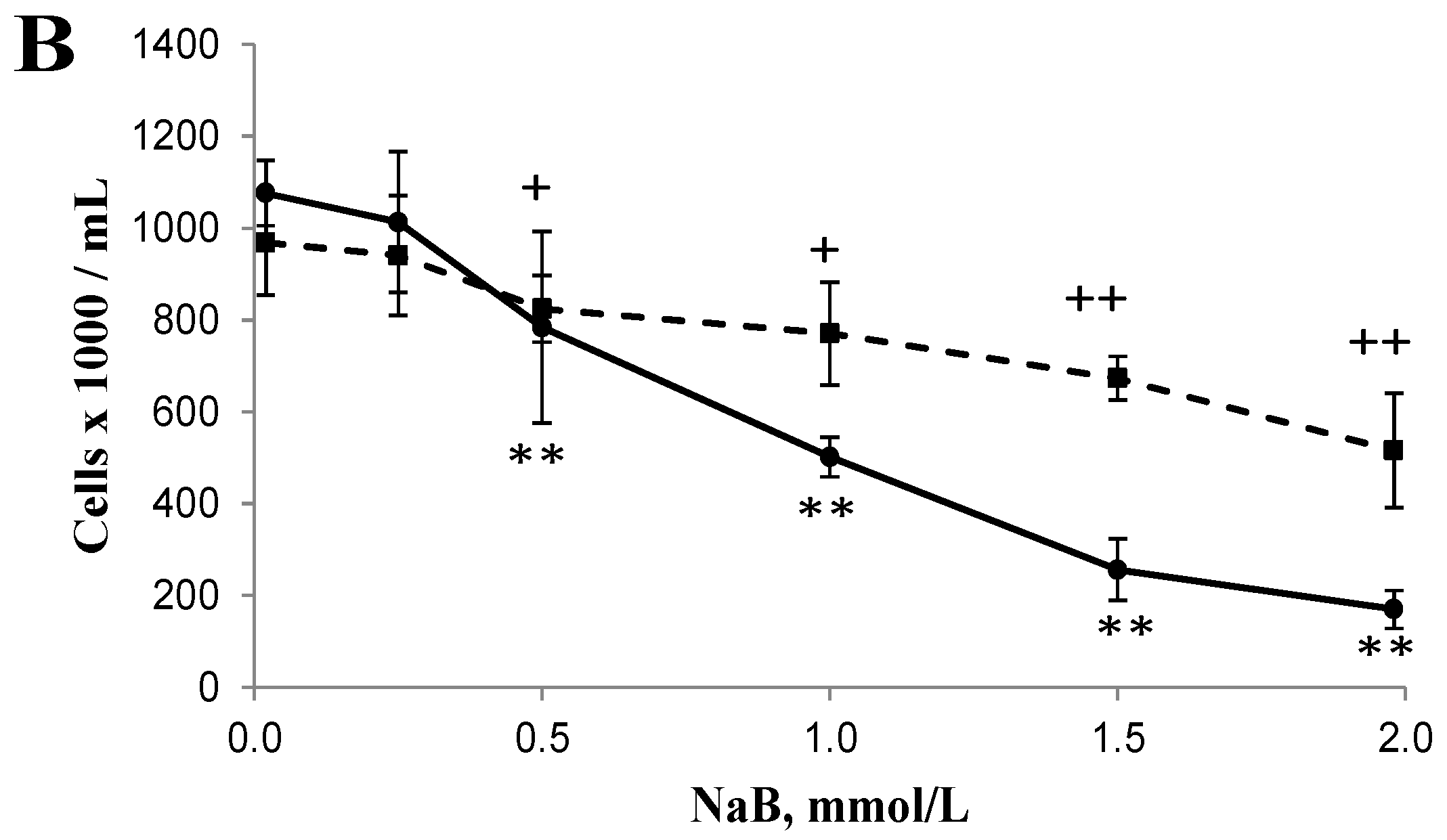
3.1. Differential Effects of Butyrate (NaB) on Cell Growth
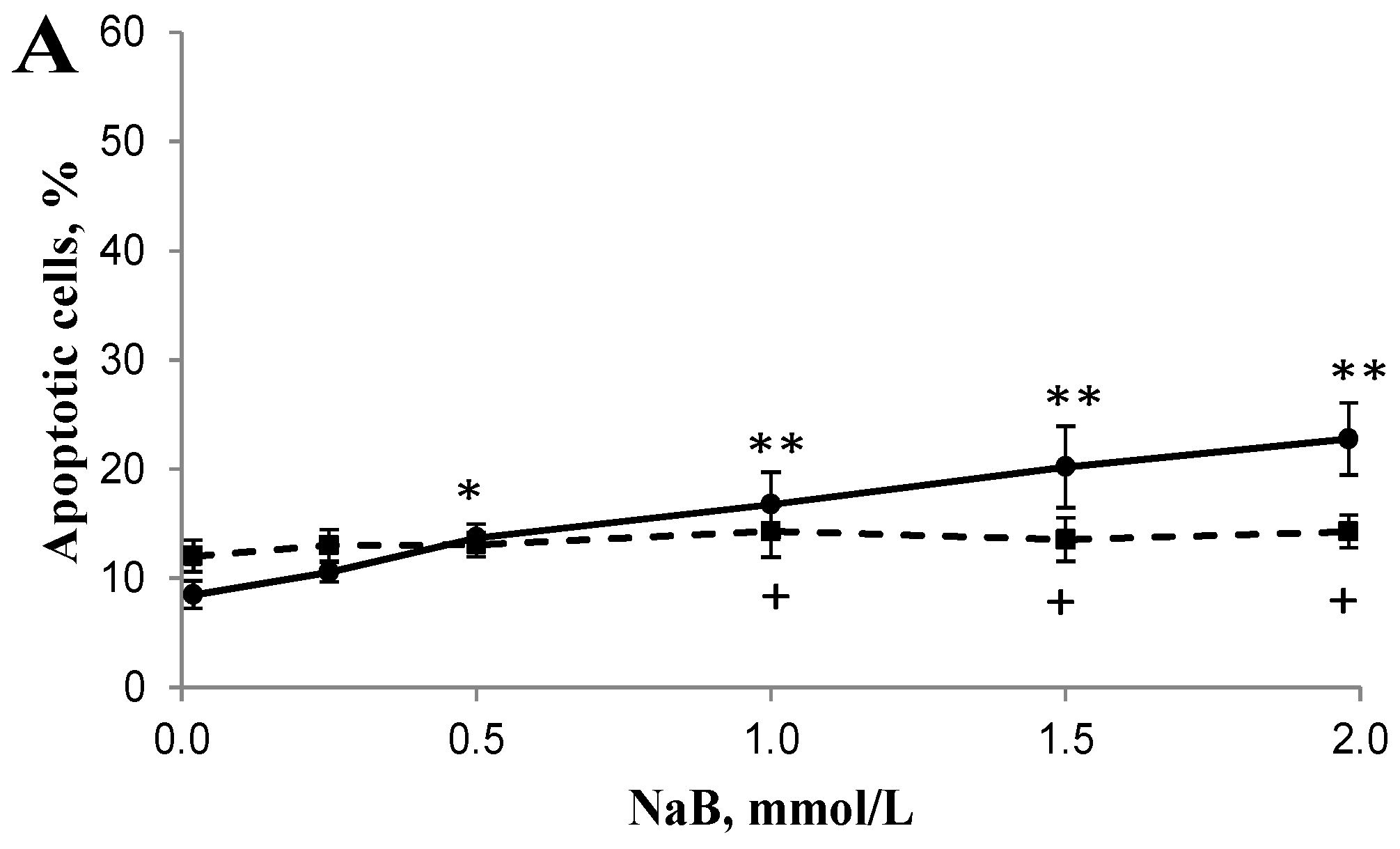
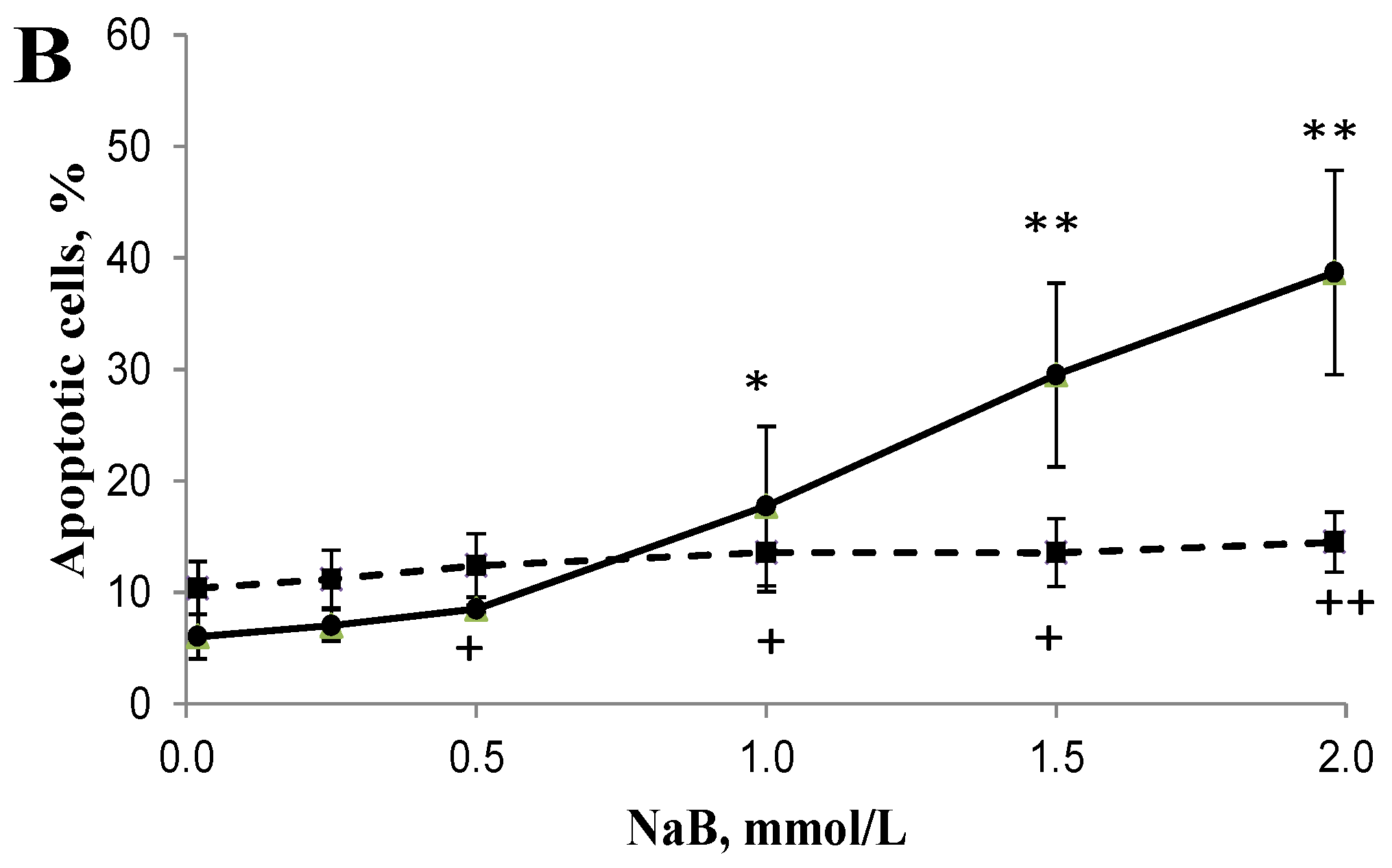
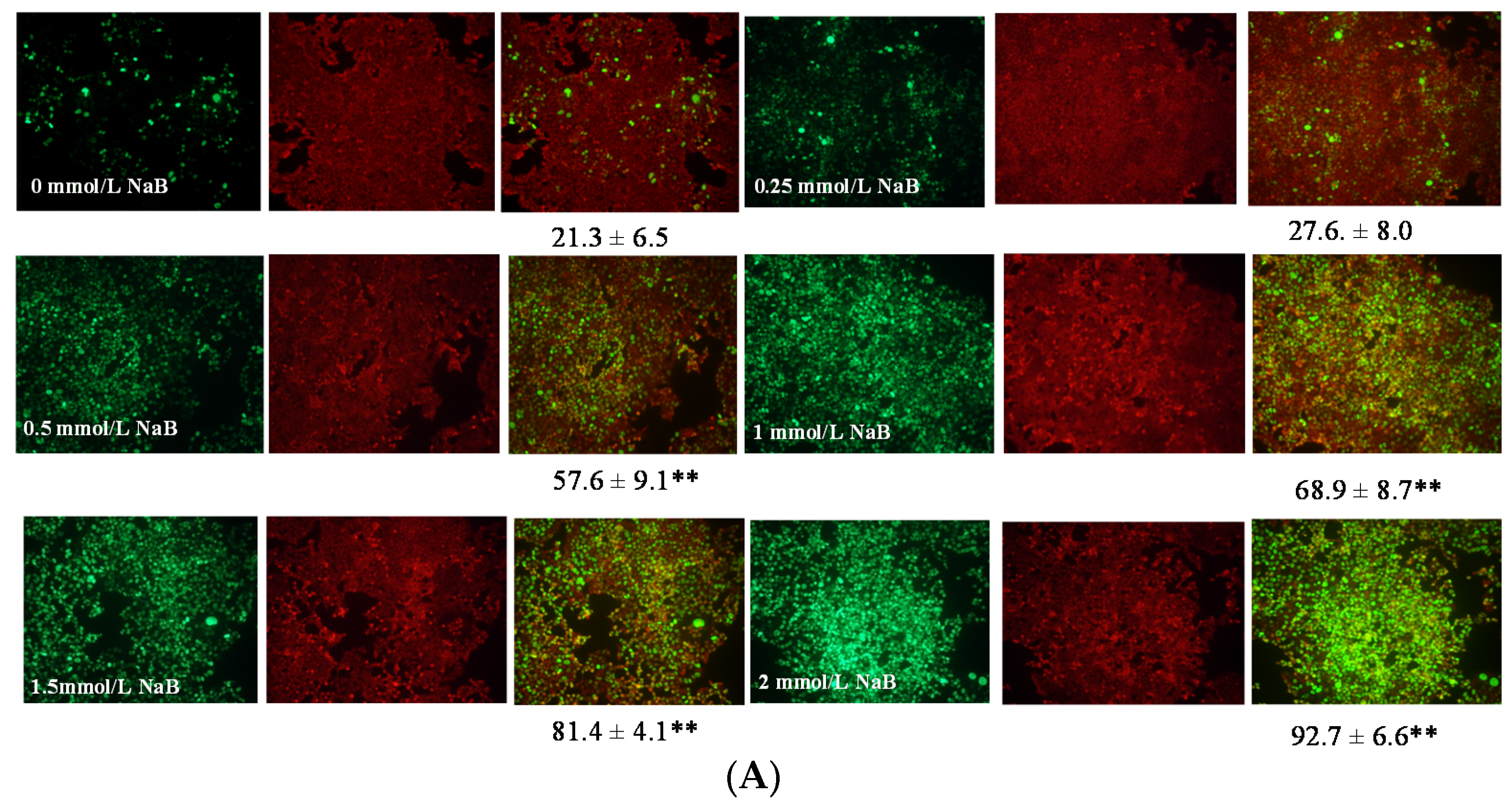
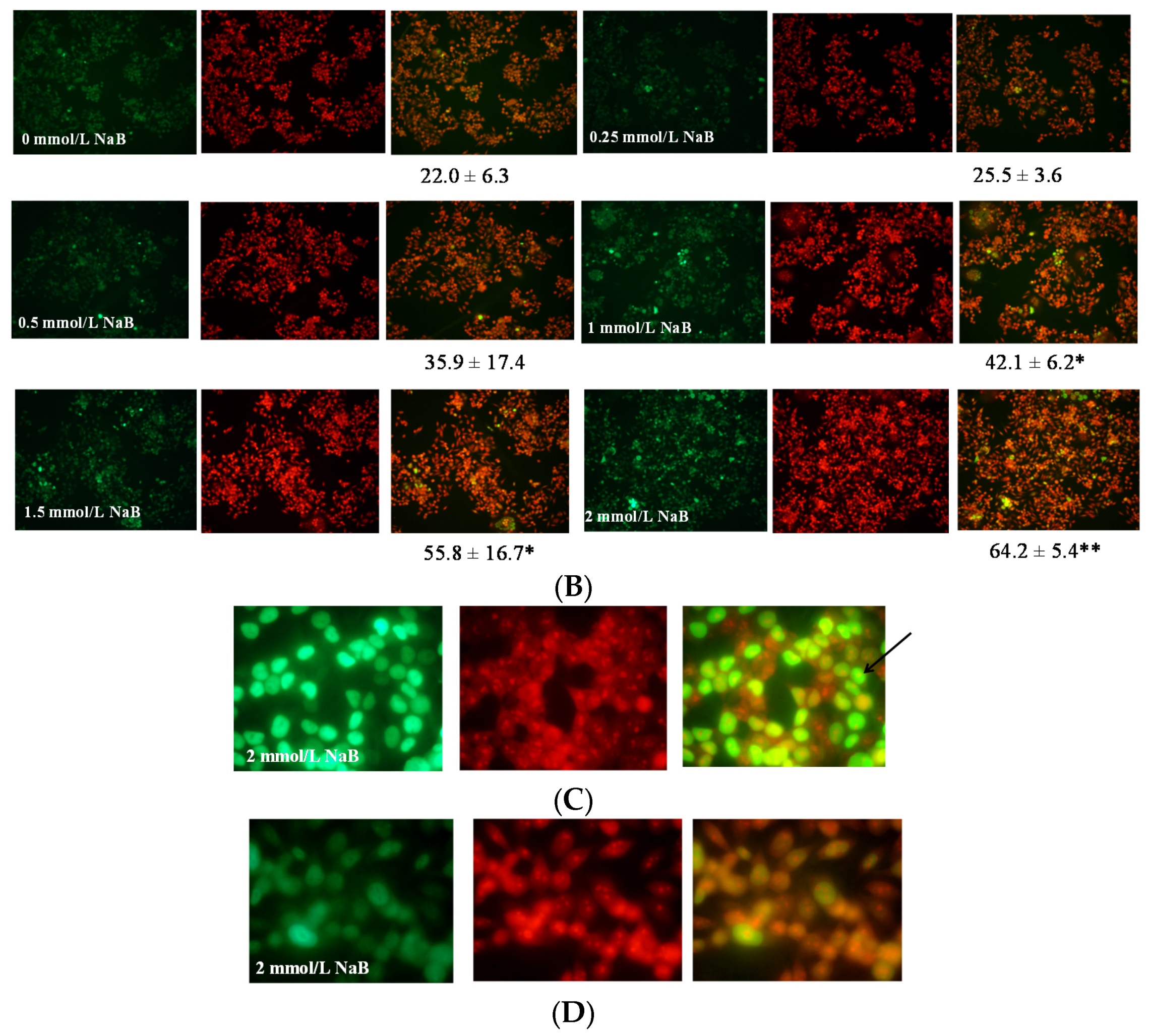
3.2. Differential Effects of Butyrate (NaB) on Apoptosis
3.3. Differential Effects of Butyrate (NaB) on DNA Fragmentation
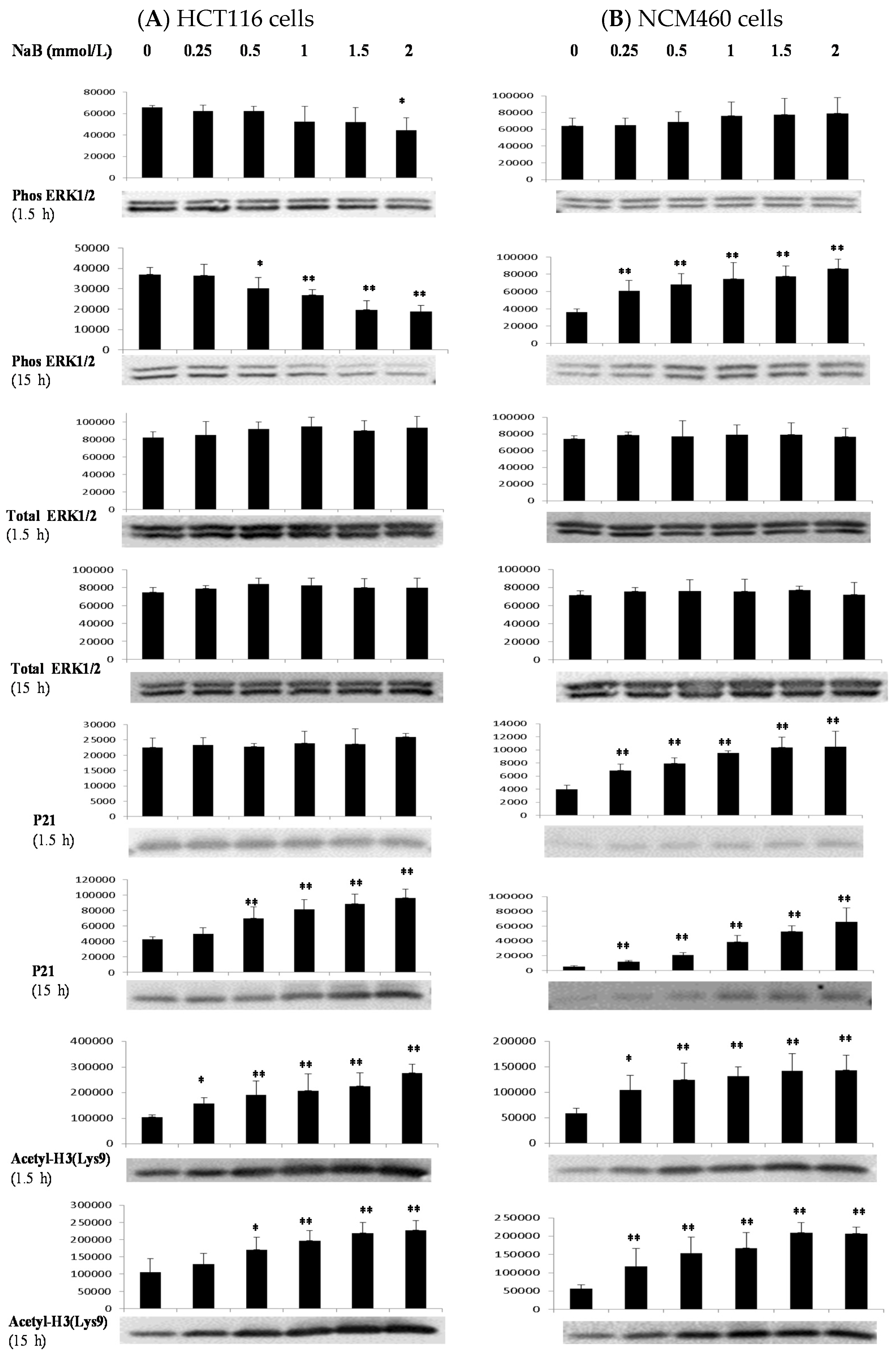
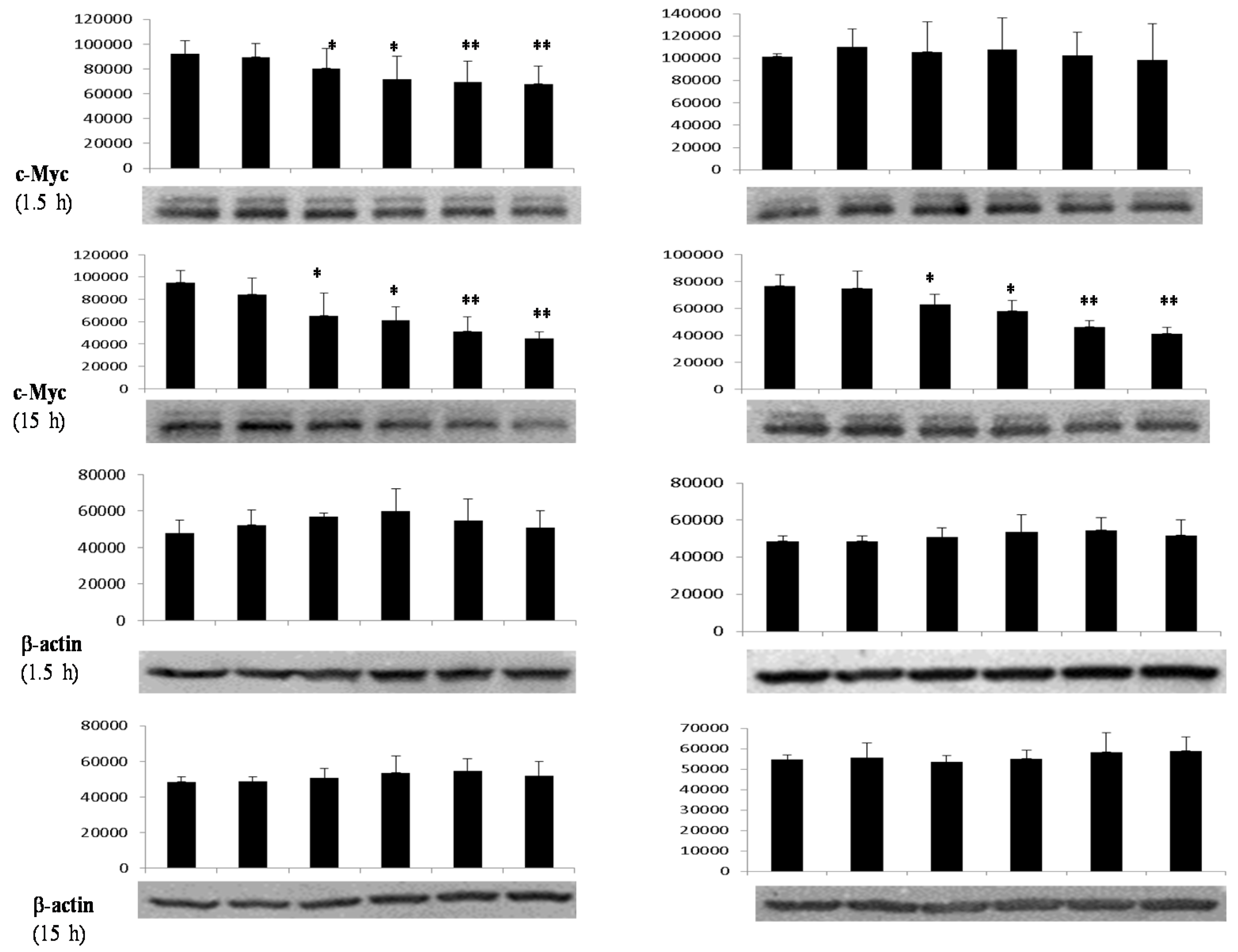
3.4. Differential Effects of Butyrate (NaB) on Signaling Molecules
3.5. Differential Effects of Butyrate (NaB) on p21 Protein Level and Cellular Localization
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- American Cancer Society. Cancer Facts & Figures; American Cancer Society: Atlanta, GA, USA, 2016. [Google Scholar]
- Simon, K. Colorectal cancer development and advances in screening. J. Clin. Interv. Aging 2016, 11, 967–976. [Google Scholar]
- Perrin, P.; Pierre, F.; Patry, Y.; Champ, M.; Berreur, M.; Pradal, G.; Bornet, F.; Meflah, K.; Menanteau, J. Only fibres promoting a stable butyrate producing colonic ecosystem decrease the rate of aberrant crypt foci in rats. Gut 2001, 48, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Lazarova, D.L.; Bordonaro, M. Mechanisms linking dietary fiber, gut microbiota and colon cancer prevention. World J. Gastrointest. Oncol. 2014, 6, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Lazarova, D.L.; Bordonaro, M. Vimentin, colon cancer progression and resistance to butyrate and other HDACis. J. Cell. Mol. Med. 2016, 20, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Bultman, S.J. The microbiome and its potential as a cancer preventive intervention. Semin. Oncol. 2016, 43, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S.E.; O’Donnell, A.U.; Perman, J.A. Influence of frequent and long-term bean consumption on colonic function and fermentation. Am. J. Clin. Nutr. 1985, 41, 909–918. [Google Scholar] [PubMed]
- Fleming, S.E.; Marthinsen, D.; Kuhnlein, H. Colonic function and fermentation in men consuming high fiber diets. J. Nutr. 1983, 113, 2535–2544. [Google Scholar] [PubMed]
- Leonel, A.J.; Alvarez-Leite, J.I. Butyrate: Implications for intestinal function. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.G.; Rao, S.; Weir, T.L.; O’Malia, J.; Bazan, M.; Brown, R.J.; Ryan, E.P. Metabolomics and metabolic pathway networks from human colorectal cancers, adjacent mucosa, and stool. Cancer Metab. 2016, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Davis, C.D. Down-regulation of proliferating cell nuclear antigen gene expression occurs during cell cycle arrest induced by human fecal water in colonic HT-29 cells. J. Nutr. 2003, 133, 2682–2687. [Google Scholar] [PubMed]
- Hu, G.X.; Chen, G.R.; Xu, H.; Ge, R.S.; Lin, J. Activation of the AMP activated protein kinase by short-chain fatty acids is the main mechanism underlying the beneficial effect of a high fiber diet on the metabolic syndrome. Med. Hypotheses 2010, 74, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Ocker, M.; Schneider-Stock, R. Histone deacetylase inhibitors: Signalling towards p21cip1/waf1. Int. J. Biochem. Cell Biol. 2007, 39, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Bi, G.; Jiang, G. The molecular mechanism of HDAC inhibitors in anticancer effects. Cell. Mol. Immunol. 2006, 3, 285–290. [Google Scholar] [PubMed]
- Chen, H.P.; Zhao, Y.T.; Zhao, T.C. Histone deacetylases and mechanisms of regulation of gene expression. Crit. Rev. Oncog. 2015, 20, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.M.; Kazlauskas, A. Growth-factor-dependent mitogenesis requires two distinct phases of signalling. Nat. Cell Biol. 2001, 3, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Karslioglu, E.; Kleinberger, J.W.; Salim, F.G.; Cox, A.E.; Takane, K.K.; Scott, D.K.; Stewart, A.F. cMyc is a principal upstream driver of beta-cell proliferation in rat insulinoma cell lines and is an effective mediator of human beta-cell replication. Mol. Endocrinol. 2011, 25, 1760–1772. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. Res. 2015, 35, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L.; Ye, X.; Goufman, E.; Shianov, P.; Hay, N.; Najmabadi, F.; Tyner, A.L. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc. Natl. Acad. Sci. USA 2001, 98, 4510–4515. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, M.; Zhu, H.; Zhang, W.; He, S.; Hu, C.; Quan, L.; Bai, J.; Xu, N. Suppression of p21 by c-Myc through members of miR-17 family at the post-transcriptional level. Int. J. Oncol. 2010, 37, 1315–1321. [Google Scholar] [PubMed]
- Moyer, M.P.; Manzano, L.A.; Merriman, R.L.; Stauffer, J.S.; Tanzer, L.R. NCM460, a normal human colon mucosal epithelial cell line. In Vitro Cell. Dev. Biol. Anim. 1996, 32, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Brattain, M.G.; Fine, W.D.; Khaled, F.M.; Thompson, J.; Brattain, D.E. Heterogeneity of malignant cells from a human colonic carcinoma. Cancer Res. 1981, 41, 1751–1756. [Google Scholar] [PubMed]
- Megna, B.W.; Carney, P.R.; Nukaya, M.; Geiger, P.; Kennedy, G.D. Indole-3-carbinol induces tumor cell death: Function follows form. J. Surg. Res. 2016, 204, 47–54. [Google Scholar] [CrossRef] [PubMed]
- De Meij, T.G.; Larbi, I.B.; van der Schee, M.P.; Lentferink, Y.E.; Paff, T.; Terhaar Sive Droste, J.S.; Mulder, C.J.; van Bodegraven, A.A.; de Boer, N.K. Electronic nose can discriminate colorectal carcinoma and advanced adenomas by fecal volatile biomarker analysis: Proof of principle study. Int. J. Cancer 2014, 134, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Keates, A.C.; Kuhnt-Moore, S.; Moyer, M.P.; Kelly, C.P.; Pothoulakis, C. Signal transduction pathways mediating neurotensin-stimulated interleukin-8 expression in human colonocytes. J. Biol. Chem. 2001, 276, 44464–44471. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.R. In situ detection of mycoplasma contamination in cell cultures by fluorescent Hoechst 33258 stain. Exp. Cell Res. 1977, 104, 255–262. [Google Scholar] [CrossRef]
- Schmid, I.; Krall, W.J.; Uittenbogaart, C.H.; Braun, J.; Giorgi, J.V. Dead cell discrimination with 7-amino-actinomycin D in combination with dual color immunofluorescence in single laser flow cytometry. Cytometry 1992, 13, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Briske-Anderson, M. Prolonged butyrate treatment inhibits the migration and invasion potential of HT1080 tumor cells. J. Nutr. 2005, 135, 291–295. [Google Scholar] [PubMed]
- Saldanha, S.N.; Kala, R.; Tollefsbol, T.O. Molecular mechanisms for inhibition of colon cancer cells by combined epigenetic-modulating epigallocatechin gallate and sodium butyrate. Exp. Cell Res. 2014, 324, 40–53. [Google Scholar] [CrossRef] [PubMed]
- Gospodinov, A.; Popova, S.; Vassileva, I.; Anachkova, B. The inhibitor of histone deacetylases sodium butyrate enhances the cytotoxicity of mitomycin C. Mol. Cancer Ther. 2012, 11, 2116–2126. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.T. Secondary necrosis: The natural outcome of the complete apoptotic program. FEBS Lett. 2010, 584, 4491–4499. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.K.; Moriwaki, K.; De Rosa, M.J. Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol. Biol. 2013, 979, 65–70. [Google Scholar] [PubMed]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Meloche, S.; Pouysségur, J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 2007, 26, 3227–3239. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Park, J.I. Growth arrest signaling of the Raf/MEK/ERK pathway in cancer. Front. Biol. (Beijing) 2014, 9, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Squires, M.S.; Nixon, P.M.; Cook, S.J. Cell-cycle arrest by PD184352 requires inhibition of extracellular signal-regulated kinases (ERK) 1/2 but not ERK5/BMK1. Biochem. J. 2002, 366, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Davido, D.J.; Richter, F.; Boxberger, F.; Stahl, A.; Menzel, T.; Lührs, H.; Löffler, S.; Dusel, G.; Rapp, U.R.; Scheppach, W. Butyrate and propionate downregulate ERK phosphorylation in HT-29 colon carcinoma cells prior to differentiation. Eur. J. Cancer Prev. 2001, 10, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Romanov, V.S.; Pospelov, V.A.; Pospelova, T.V. Cyclin-dependent kinase inhibitor p21(Waf1): Contemporary view on its role in senescence and oncogenesis. Biochemistry (Mosc.) 2012, 77, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Parveen, A.; Akash, M.S.; Rehman, K.; Kyunn, W.W. Dual Role of p21 in the Progression of Cancer and Its Treatment. Crit. Rev. Eukaryot. Gene Expr. 2016, 26, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.Y.; Meng, S.; Shei, A.; Hodin, R.A. p21(WAF1) is required for butyrate-mediated growth inhibition of human colon cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 6791–6796. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.X.; Lee, J. Dietary regulation of histone acetylases and deacetylases for the prevention of metabolic diseases. Nutrients 2012, 4, 1868–1886. [Google Scholar] [CrossRef] [PubMed]
- Nian, H.; Delage, B.; Pinto, J.T.; Dashwood, R.H. Allyl mercaptan, a garlic-derived organosulfur compound, inhibits histone deacetylase and enhances Sp3 binding on the P21WAF1 promoter. Carcinogenesis 2008, 29, 1816–1824. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Vincent, T.L. An evolutionary model of carcinogenesis. Cancer Res. 2003, 63, 6212–6220. [Google Scholar] [PubMed]
- Rajamanickam, S.; Agarwal, R. Natural products and colon cancer: Current status and future prospects. Drug Dev. Res. 2008, 69, 460–471. [Google Scholar] [CrossRef] [PubMed]









© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, H.; Taussig, D.P.; Cheng, W.-H.; Johnson, L.K.; Hakkak, R. Butyrate Inhibits Cancerous HCT116 Colon Cell Proliferation but to a Lesser Extent in Noncancerous NCM460 Colon Cells. Nutrients 2017, 9, 25. https://doi.org/10.3390/nu9010025
Zeng H, Taussig DP, Cheng W-H, Johnson LK, Hakkak R. Butyrate Inhibits Cancerous HCT116 Colon Cell Proliferation but to a Lesser Extent in Noncancerous NCM460 Colon Cells. Nutrients. 2017; 9(1):25. https://doi.org/10.3390/nu9010025
Chicago/Turabian StyleZeng, Huawei, David P. Taussig, Wen-Hsing Cheng, LuAnn K. Johnson, and Reza Hakkak. 2017. "Butyrate Inhibits Cancerous HCT116 Colon Cell Proliferation but to a Lesser Extent in Noncancerous NCM460 Colon Cells" Nutrients 9, no. 1: 25. https://doi.org/10.3390/nu9010025
APA StyleZeng, H., Taussig, D. P., Cheng, W. -H., Johnson, L. K., & Hakkak, R. (2017). Butyrate Inhibits Cancerous HCT116 Colon Cell Proliferation but to a Lesser Extent in Noncancerous NCM460 Colon Cells. Nutrients, 9(1), 25. https://doi.org/10.3390/nu9010025