Vitamin E Modifies High-Fat Diet-Induced Increase of DNA Strand Breaks, and Changes in Expression and DNA Methylation of Dnmt1 and MLH1 in C57BL/6J Male Mice



Abstract
:1. Introduction
2. Materials and Methods
2.1. SCGE (Single Cell Gel Electrophoresis) Assay
2.2. Gene Expression Analysis
2.3. Methylation Analysis
2.4. Statistical Analyses
3. Results
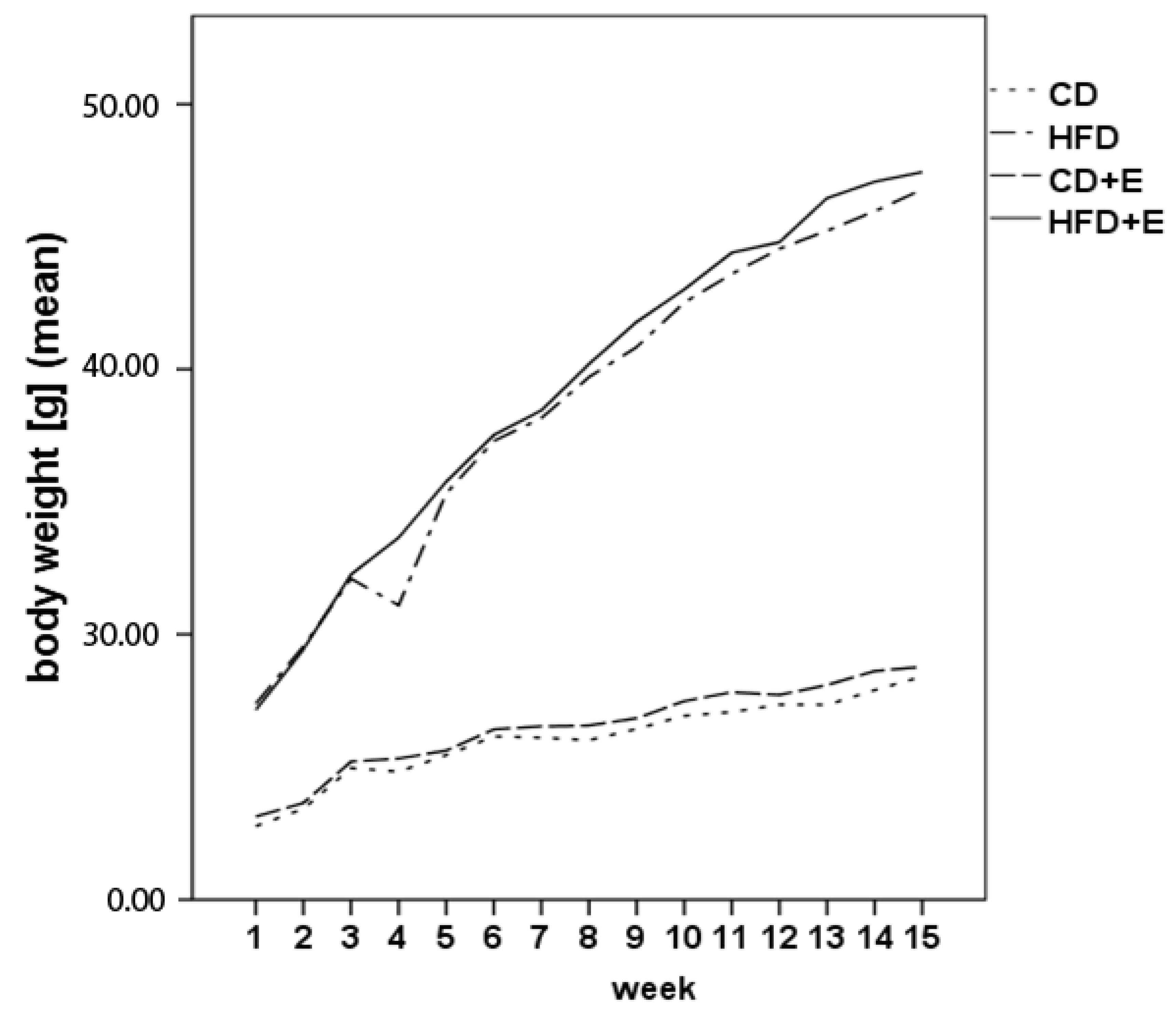
3.1. Body Weight, Food Intake and Vitamin E Uptake
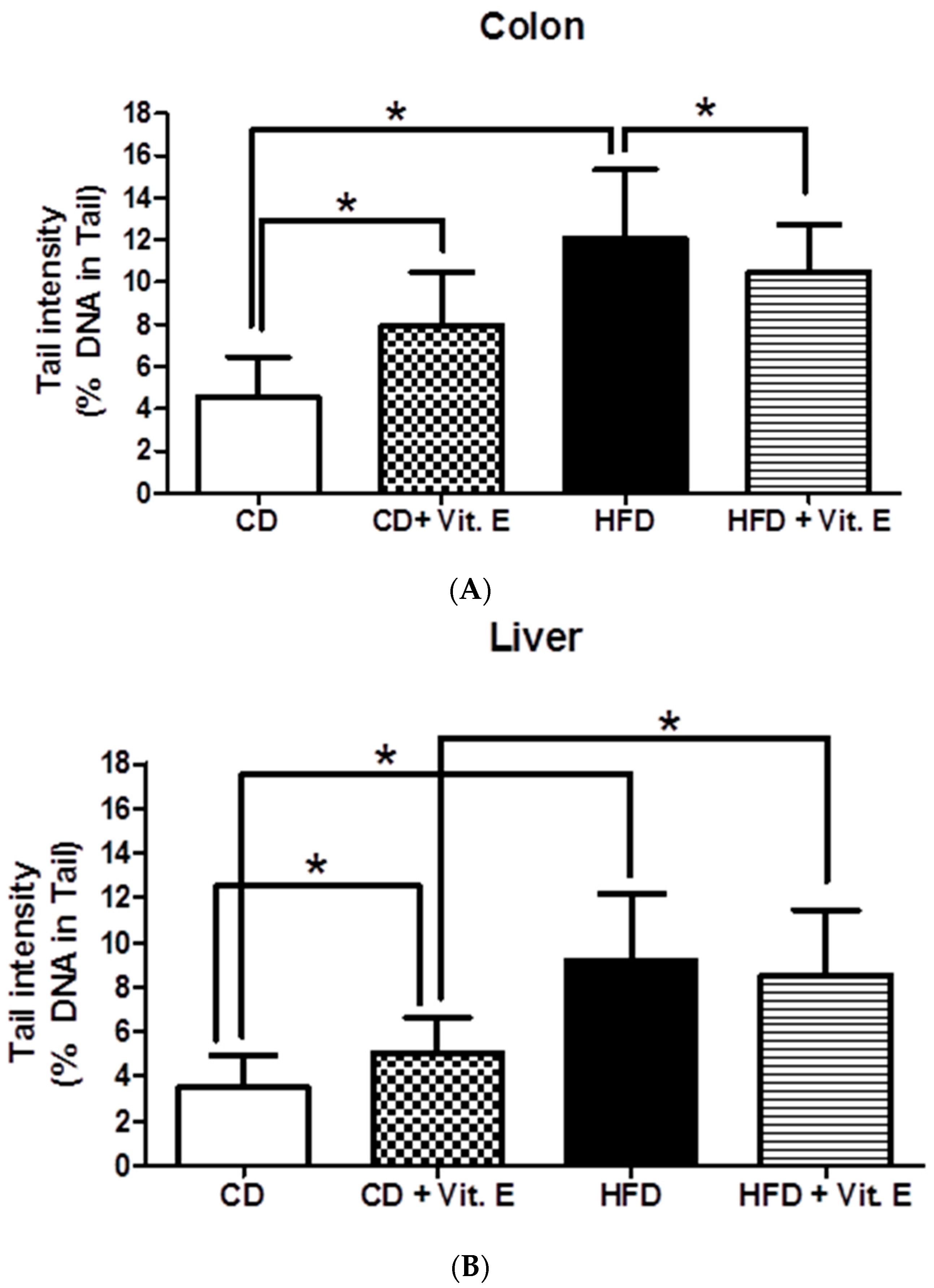
3.2. SCGE Experiments from Colon and Liver Cells
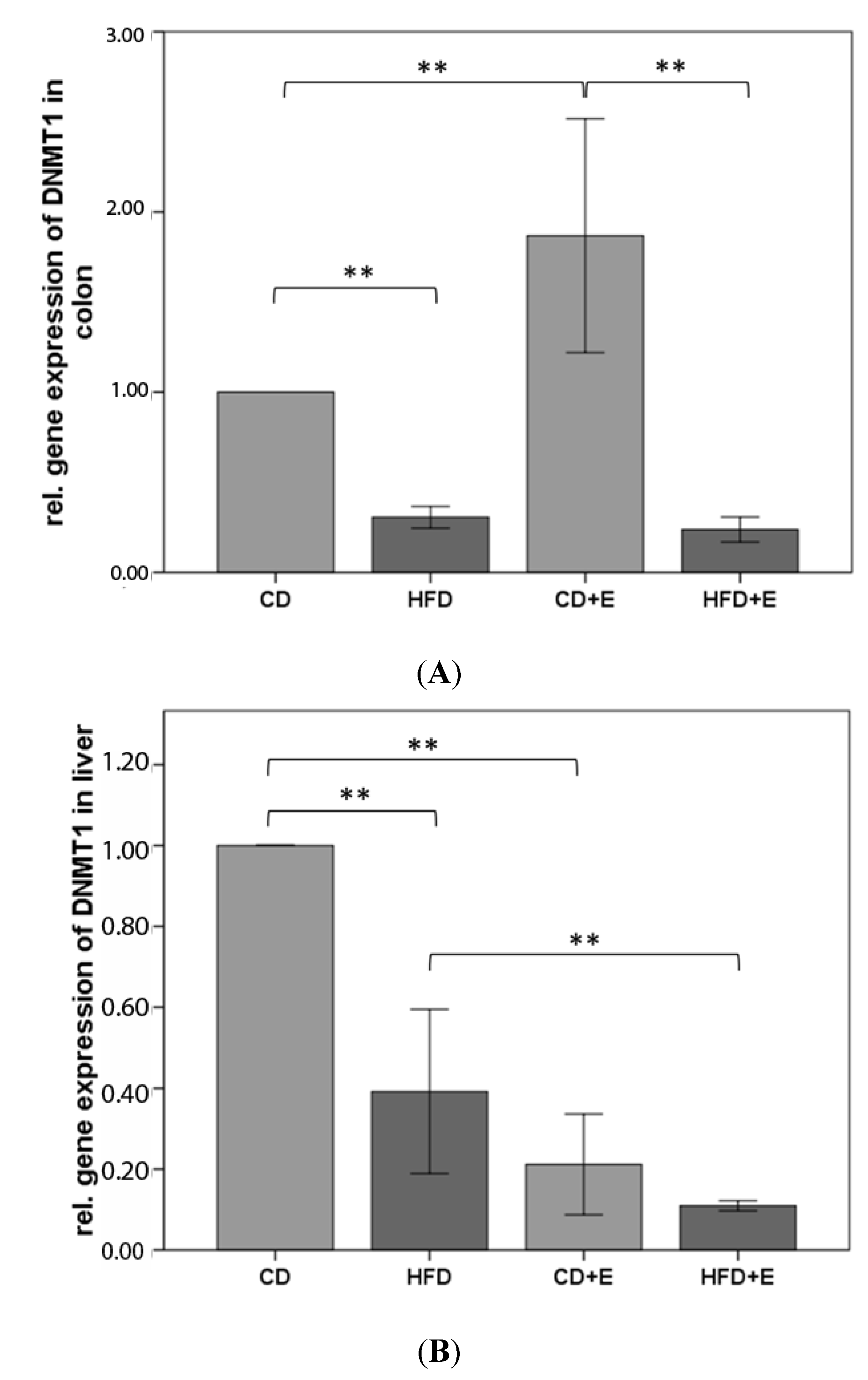
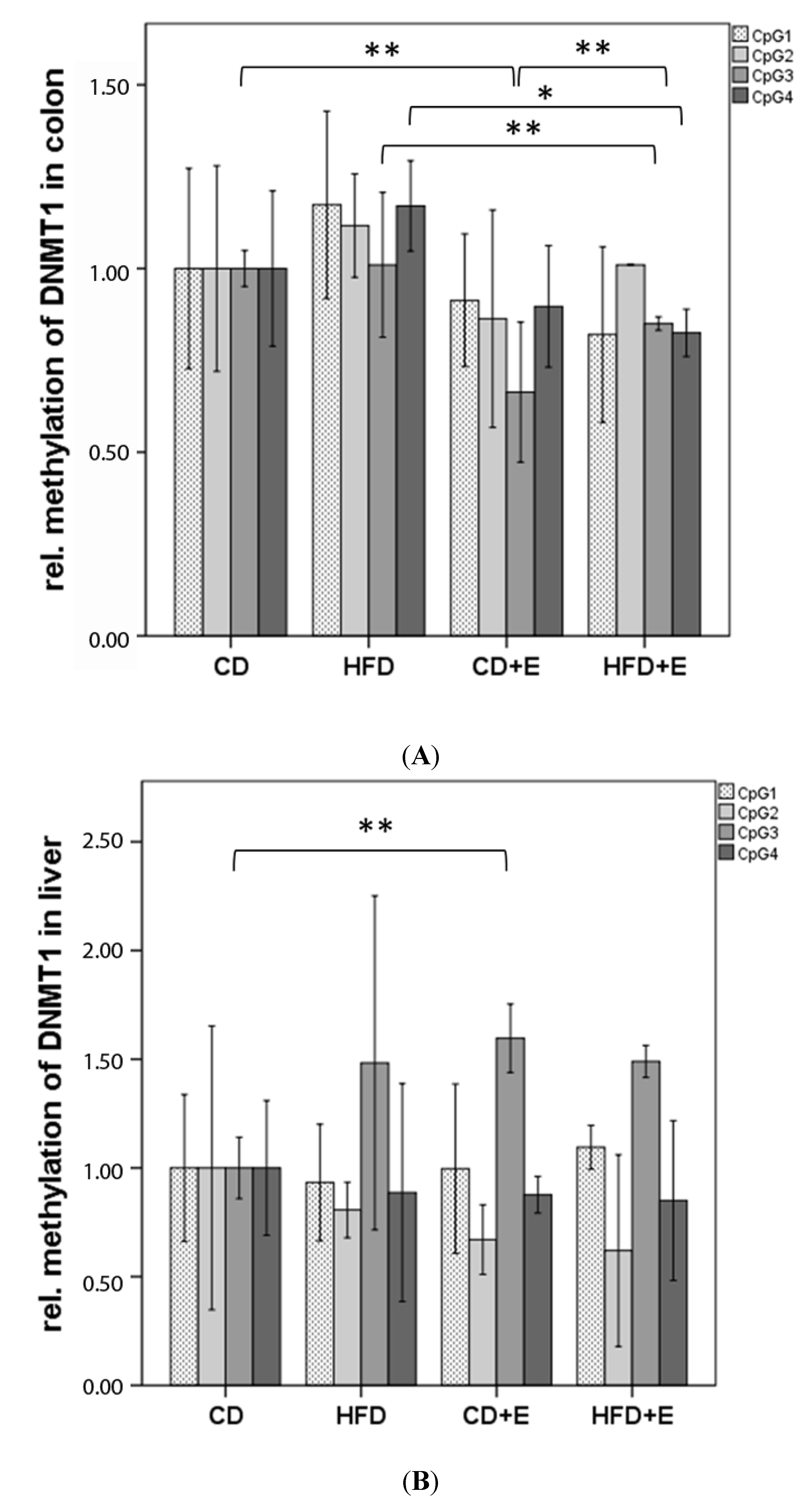
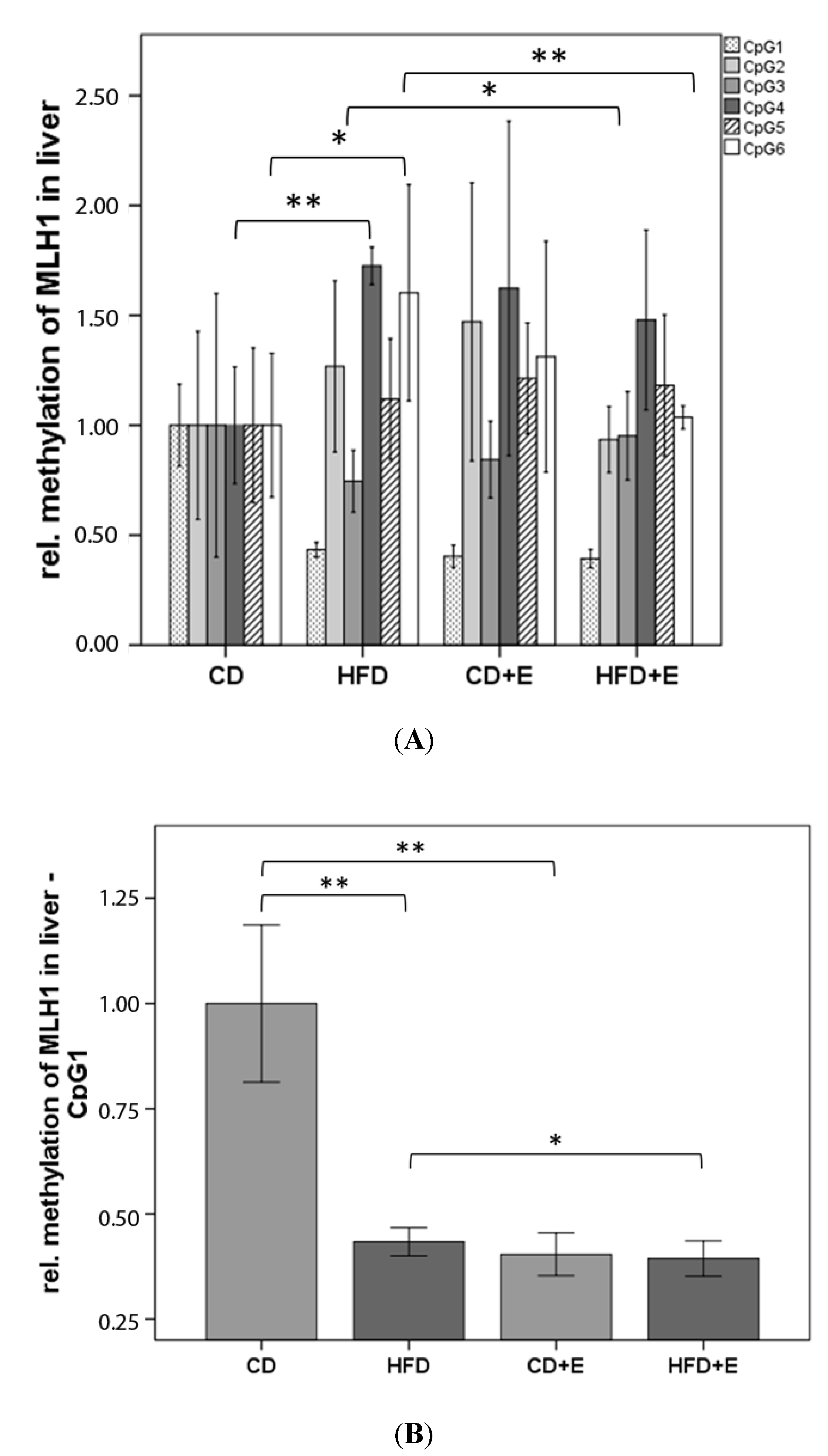
3.3. Relative Gene Expression (Figure 3) and CpG Methylation (Figure 4) of Dnmt1 in Colon and Liver Cells
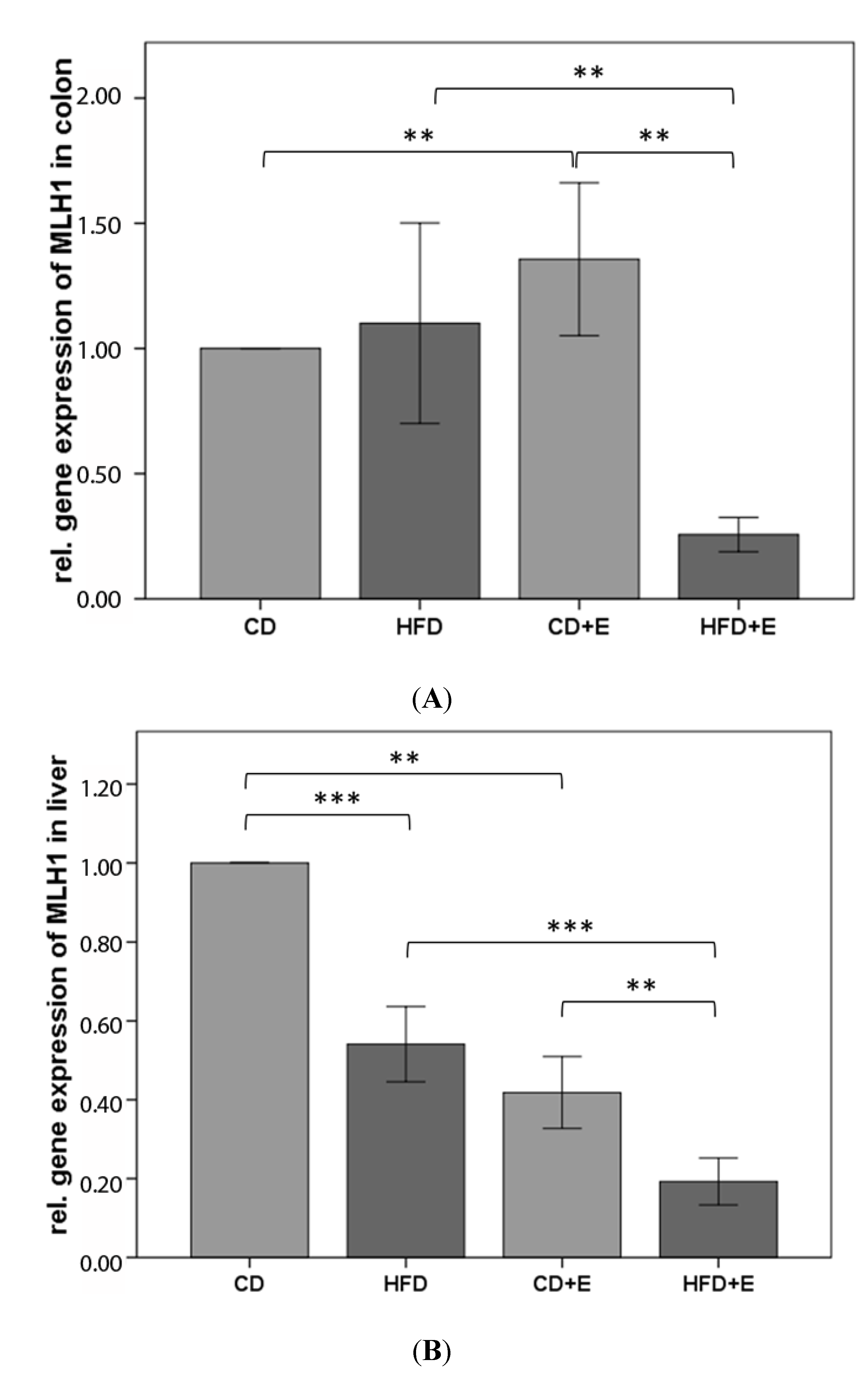
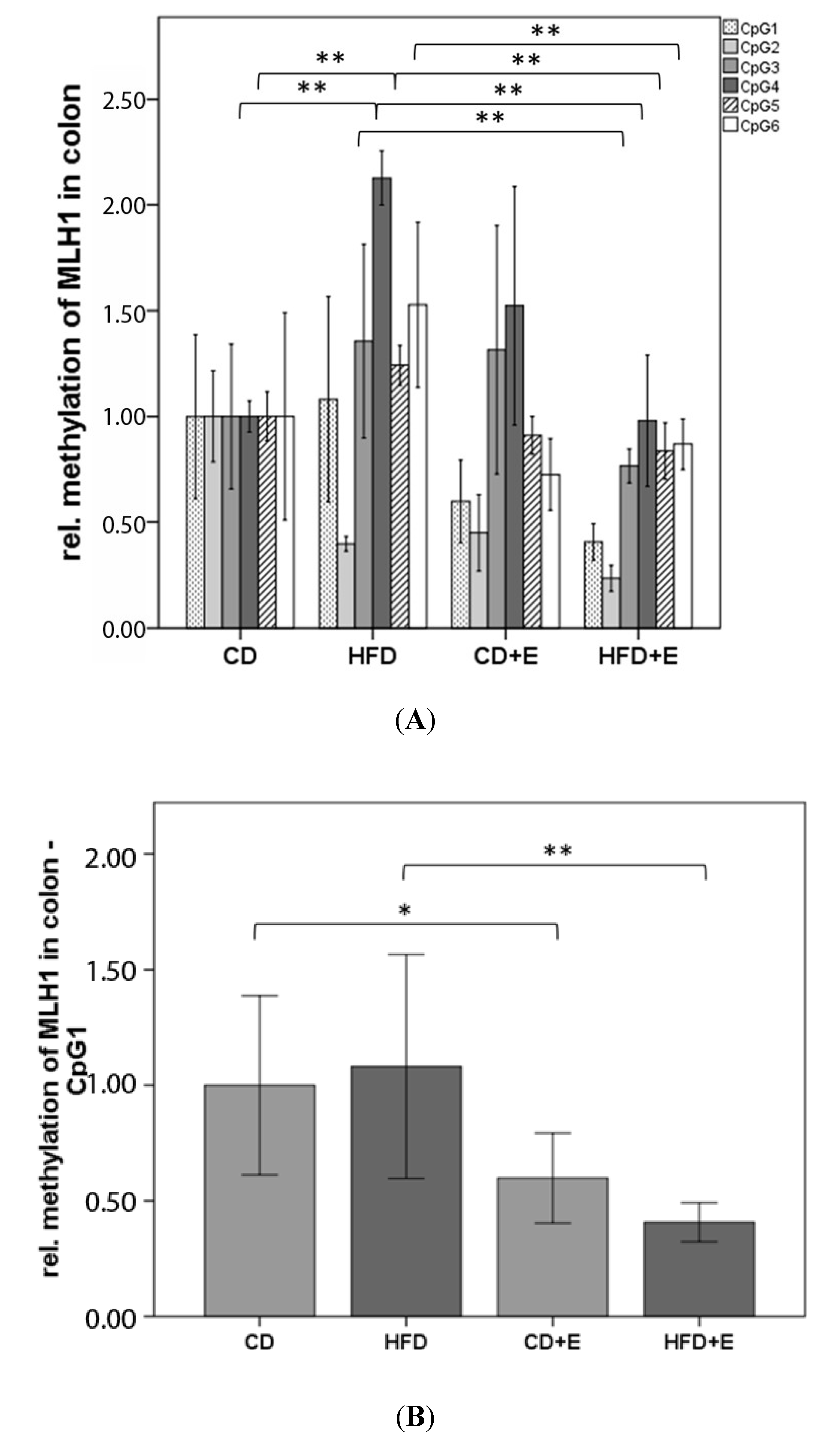
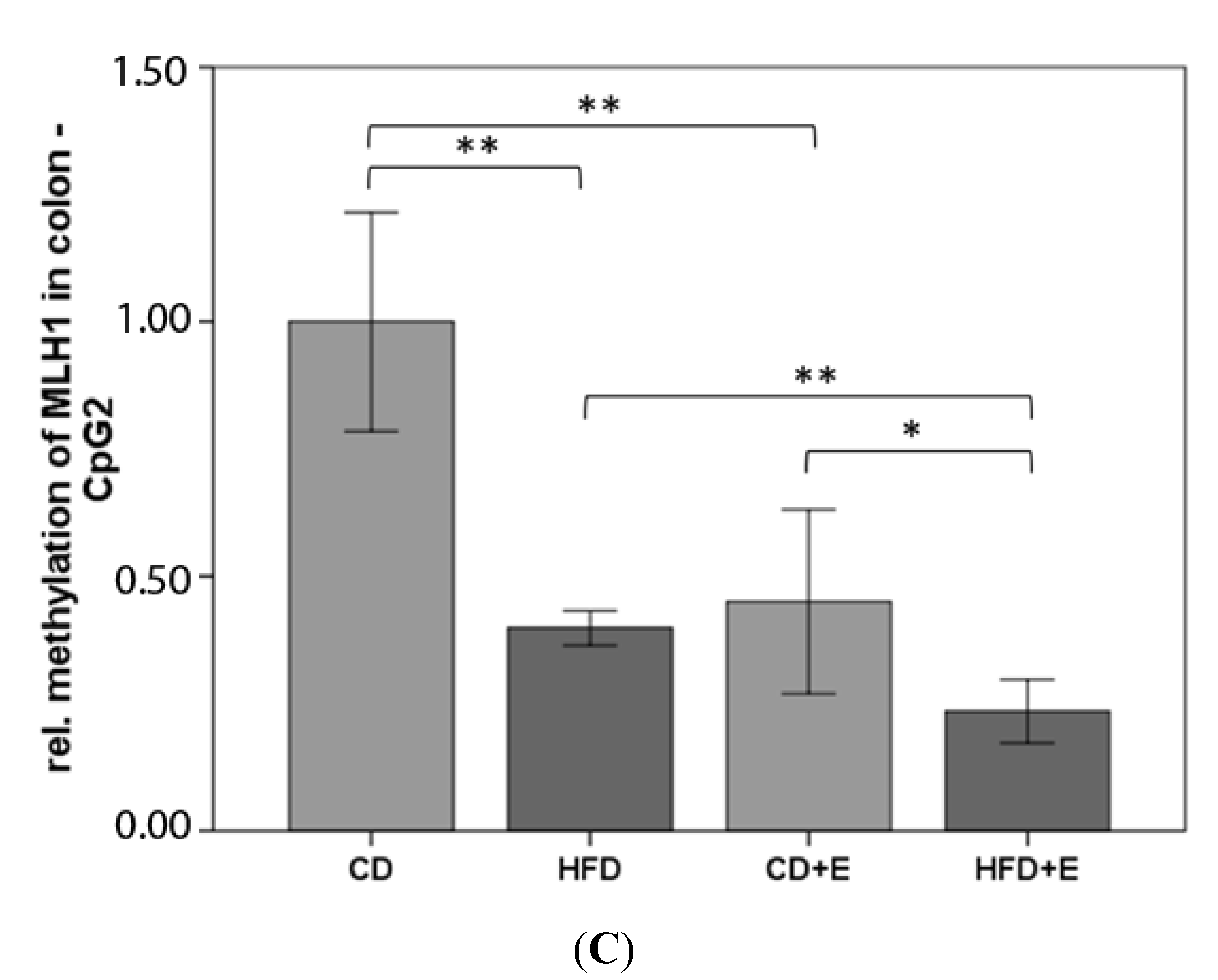
3.4. Relative Gene Expression (Figure 5) and CpG Methylation (Figure 6 and Figure 7) of MLH1 in Colon and Liver Cells
4. Discussion
4.1. Vitamin E Protects DNA Damage Caused by HFD
4.2. Vitamin E Supplementation Affects Specific CpG Sites of Dnmt1, Resulting in Altered Relative Gene Expression of Dnmt1
4.3. Vitamin E Supplementation Affects Specific CpG Sites of MLH1, Inducing a Lower Gene Expression of MLH1 with High-Fat Diet
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| α-TTP | α-tocopherol transfer protein |
| CAT | catalase |
| CD | control diet |
| CD + E | control diet plus vitamin E |
| cDNA | complementary DNA |
| CRP | C reactive protein |
| CYPs | cytochrome P450s |
| DIO | diet induced obesity |
| Dnmt1 | DNA methyltransferase 1 |
| FFAs | free fatty acids |
| GAPDH | glycerinaldehyd-3-phosphat-Dehydrogenase |
| GPX | glutathione peroxidase |
| H | histone |
| HFD | high fat diet |
| HFD + E | high fat diet plus vitamin E |
| IL-6 | interleukin-6 |
| LDL | low-density lipoprotein |
| LMPA | low melting point agarose |
| MBD2 | methyl-CpG-binding domain protein 2 |
| MCP-1 | monocyte chemoattractant protein-1 |
| MGMT | O6-methylguanine-deoxyribonucleic acidmethyltransferase |
| MLH1 | MutL homolog 1 |
| MMR | DNA mismatch repair |
| MSI | microsatellite instability |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NAFLD | non-alcoholic fatty liver diseases |
| NMPA | normal melting point agarose |
| Nrf2 | Nuclear factor-erythroid 2-related factor 2 |
| PAI-1 | plasminogen activator inhibitor-1 |
| PHD | plant homeodomain |
| ROS | reactive oxygen species |
| SAM | S-Adenosylmethionin |
| SCGE | single cell gel electrophoresis |
| SOD | superoxide dismutase |
| TNFα | tumor necrosis factor α |
| T | time point |
| UHRF1 | Ubiquitin-like, Containing PHD and RING Finger Domains, 1 |
| UIM | ubiquitin interacting motif |
References
- WHO Consultation on Obesity. Obesity: Preventing and managing the global epidemic. Report of a WHO consultation. World Health Organ. Tech. Rep. Ser. 2000, 894, 1–253. [Google Scholar]
- Fazel, S.; Luigi, G.; Daglia, M.; Mohammad, S. Role of quercetin as an alternative for obesity treatment: You are what you eat! Food Chem. 2015, 179, 305–310. [Google Scholar]
- Fernández-Sánchez, A.; Madrigal-Santillán, E.; Bautista, M.; Esquivel-Soto, J.; Morales-González, Á.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sánchez-Rivera, G.; Valadez-Vega, C.; Morales-González, J.A. Inflammation, oxidative stress, and obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, E.E.; Flier, J.S. Adipose Tissue as an Endocrine Organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Savini, I.; Catani, M.V.; Evangelista, D.; Gasperi, V.; Avigliano, L. Obesity-associated oxidative stress: Strategies finalized to improve redox state. Int. J. Mol. Sci. 2013, 14, 10497–10538. [Google Scholar] [CrossRef] [PubMed]
- Le Lay, S.; Simard, G.; Martinez, M.C.; Andriantsitohaina, R. Oxidative Stress and Metabolic Pathologies: From an Adipocentric Point of View. Oxid. Med. Cell. Longev. 2014, 2014, 908539. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Thanan, R.; Oikawa, S.; Hiraku, Y.; Ohnishi, S.; Ma, N.; Pinlaor, S.; Yongvanit, P.; Kawanishi, S.; Murata, M. Oxidative Stress and Its Significant Roles in Neurodegenerative Diseases and Cancer. Int. J. Mol. Sci. 2014, 16, 193–217. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Schoneveld, O.; Georgakilas, A.G.; Panayiotidis, M.I. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008, 266, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Donkena, K.V.; Young, C.Y.F.; Tindall, D.J. Oxidative stress and DNA methylation in prostate cancer. Obstet. Gynecol. Int. 2010, 2010, 302051. [Google Scholar] [CrossRef] [PubMed]
- Ziech, D.; Franco, R.; Pappa, A.; Panayiotidis, M.I. Reactive oxygen species (ROS)--Induced genetic and epigenetic alterations in human carcinogenesis. Mutat. Res. 2011, 711, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Chahal, M.; Xu, Y.; Lesniak, D.; Graham, K.; Famulski, K.; Christensen, J.G.; Aghi, M.; Jacques, A.; Murray, D.; Sabri, S.; et al. MGMT modulates glioblastoma angiogenesis and response to the tyrosine kinase inhibitor sunitinib. Neuro Oncol. 2010, 12, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Kovalchuk, O.; Burke, P.; Besplug, J.; Slovack, M.; Filkowski, J.; Pogribny, I. Methylation changes in muscle and liver tissues of male and female mice exposed to acute and chronic low-dose X-ray-irradiation. Mutat. Res. 2004, 548, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Harfe, B.D.; Jinks-Robertson, S. DNA mismatch repair and genetic instability. Annu. Rev. Genet. 2000, 34, 359–399. [Google Scholar] [CrossRef] [PubMed]
- Peltomäki, P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J. Clin. Oncol. 2003, 21, 1174–1179. [Google Scholar] [CrossRef] [PubMed]
- Li, G.-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Hitchler, M.J.; Domann, F.E. An epigenetic perspective on the free radical theory of development. Free Radic. Biol. Med. 2007, 43, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, D.; Thombre, R.; Dhar, A.; Anant, S. DNA methyltransferases: A novel target for prevention and therapy. Front. Oncol. 2014, 4, 80. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Chattopadhyay, B.D.; Paul, B.N. Epigenetic regulation of DNMT1 gene in mouse model of asthma disease. Mol. Biol. Rep. 2013, 40, 2357–2368. [Google Scholar] [CrossRef] [PubMed]
- Hodge, D.R.; Xiao, W.; Clausen, P.A.; Heidecker, G.; Szyf, M.; Farrar, W.L. Interleukin-6 regulation of the human DNA methyltransferase (HDNMT) gene in human erythroleukemia cells. J. Biol. Chem. 2001, 276, 39508–39511. [Google Scholar] [CrossRef] [PubMed]
- Hodge, D.R. Interleukin 6 Supports the Maintenance of p53 Tumor Suppressor Gene Promoter Methylation. Cancer Res. 2005, 65, 4673–4682. [Google Scholar] [CrossRef] [PubMed]
- Hodge, D.R.; Cho, E.; Copeland, T.D.; Guszczynski, T.; Yang, E.; Seth, A.K.; Farrar, W.L. IL-6 enhances the nuclear translocation of DNA cytosine-5-methyltransferase 1 (DNMT1) via phosphorylation of the nuclear localization sequence by the AKT kinase. Cancer Genom. Proteom. 2007, 4, 387–398. [Google Scholar]
- Remely, M.; Ferk, F.; Sterneder, S.; Setavesh, T.; Roth, S.; Kepcija, T.; Noorizadeh, R.; Rebhan, I.; Greunz, M.; Beckmann, J.; et al. EGCG prevents high-fat diet-induced changes in gut microbiota, decreases of DNA strand breaks, and changes in expression and DNA methylation of Dnmt1 and MLH1 in C57BL/6J male mice. Oxid. Med. Cell. Longev. 2017, 2017, 3079148. [Google Scholar] [CrossRef] [PubMed]
- Switzeny, O.J.; Mullner, E.; Wagner, K.H.; Brath, H.; Aumuller, E.; Haslberger, A.G. Vitamin and antioxidant rich diet increases MLH1 promoter DNA methylation in DMT2 subjects. Clin. Epigenetics 2012, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.L. An update on vitamin E, tocopherol and tocotrienol-perspectives. Molecules 2010, 15, 2103–2113. [Google Scholar] [CrossRef] [PubMed]
- Mocchegiani, E.; Costarelli, L.; Giacconi, R.; Malavolta, M.; Basso, A.; Piacenza, F.; Ostan, R.; Cevenini, E.; Gonos, E.S.; Franceschi, C.; et al. Vitamin E-gene interactions in aging and inflammatory age-related diseases: Implications for treatment. A systematic review. Ageing Res. Rev. 2014, 14, 81–101. [Google Scholar] [CrossRef] [PubMed]
- Stone, W.L.; Papas, A.M. Tocopherols and the etiology of colon cancer. J. Natl. Cancer Inst. 1997, 89, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Traussnigg, S.; Kienbacher, C.; Halilbasic, E.; Rechling, C.; Kazemi-Shirazi, L.; Hofer, H.; Munda, P.; Trauner, M. Challenges and Management of Liver Cirrhosis: Practical Issues in the Therapy of Patients with Cirrhosis due to NAFLD and NASH. Dig. Dis. 2015, 33, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Dias, F.M.; Leffa, D.D.; Daumann, F.; de Marques, S.O.; Luciano, T.F.; Possato, J.C.; de Santana, A.A.; Neves, R.X.; Rosa, J.C.; Oyama, L.M.; et al. Acerola (Malpighia emarginata DC.) juice intake protects against alterations to proteins involved in inflammatory and lipolysis pathways in the adipose tissue of obese mice fed a cafeteria diet. Lipids Health Dis. 2014, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Hakkak, R.; Korourian, S.; Pavliv, O.; Evans, T.; Melnyk, S. Effects of obesity on development of oxidative stress and DNA damages in liver of the obese Zucker rat (643.1). FASEB J. 2014, 28, 643.1. [Google Scholar]
- Al-Aubaidy, H.A.; Jelinek, H.F. Oxidative DNA damage and obesity in type 2 diabetes mellitus. Eur. J. Endocrinol. 2011, 164, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R.; Oscoz, A.A.; Brunborg, G.; Gaivão, I.; Giovannelli, L.; Kruszewski, M.; Smith, C.C.; Stetina, R. The comet assay: Topical issues. Mutagenesis 2008, 23, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Papas, K.; Kalbfleisch, J.; Mohon, R. Bioavailability of a novel, water-soluble vitamin E formulation in malabsorbing patients. Dig. Dis. Sci. 2007, 52, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Tice, R.R.; Agurell, E.; Anderson, D.; Burlinson, B.; Hartmann, A.; Kobayashi, H.; Miyamae, Y.; Rojas, E.; Ryu, J.C.; Sasaki, Y.F. Single cell gel/comet assay: Guidelines for in vitro and in vivo genetic toxicology testing. Environ. Mol. Mutagen. 2000, 35, 206–221. [Google Scholar] [CrossRef]
- Sasaki, Y.F.; Kawaguchi, S.; Kamaya, A.; Ohshita, M.; Kabasawa, K.; Iwama, K.; Taniguchi, K.; Tsuda, S. The comet assay with 8 mouse organs: Results with 39 currently used food additives. Mutat. Res. 2002, 519, 103–119. [Google Scholar] [CrossRef]
- Burlinson, B.; Tice, R.R.; Speit, G.; Agurell, E.; Brendler-Schwaab, S.Y.; Collins, A.R.; Escobar, P.; Honma, M.; Kumaravel, T.S.; Nakajima, M.; et al. Fourth International Workgroup on Genotoxicity testing: Results of the in vivo Comet assay workgroup. Mutat. Res. 2007, 627, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Jialal, I. The effects of alpha-tocopherol on critical cells in atherogenesis. Curr. Opin. Lipidol. 1998, 9, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Maddux, B.A.; See, W.; Lawrence, J.C.; Goldfine, A.L.; Goldfine, I.D.; Evans, J.L. Protection Against Oxidative Stress—Induced Insulin Resistance in Rat L6 Muscle Cells by Micromolar Concentrations of -Lipoic Acid. Diabetes 2001, 50, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Paolisso, G.; D’Amore, A.; Galzerano, D.; Balbi, V.; Giugliano, D.; Varricchio, M.; D’Onofrio, F. Daily vitamin E supplements improve metabolic control but not insulin secretion in elderly type II diabetic patients. Diabetes Care 1993, 16, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Jacob, S.; Ruus, P.; Hermann, R.; Tritschler, H.; Maerker, E.; Renn, W.; Augustin, H.; Dietze, G.; Rett, K. Oral administration of rac-α-lipoic acid modulates insulin sensitivity in patients with type-2 diabetes mellitus: A placebo-controlled pilot trial. Free Radic. Biol. Med. 1999, 27, 309–314. [Google Scholar] [CrossRef]
- Manning, P.J.; Sutherland, W.H.F.; Walker, R.J.; Williams, S.M.; De Jong, S.A.; Ryalls, A.R.; Berry, E.A. Effect of high-dose vitamin E on insulin resistance and associated parameters in overweight subjects. Diabetes Care 2004, 27, 2166–2171. [Google Scholar] [CrossRef] [PubMed]
- Picklo, M.J.; Thyfault, J.P. Vitamin E and vitamin C do not reduce insulin sensitivity but inhibit mitochondrial protein expression in exercising obese rats. Appl. Physiol. Nutr. Metab. 2015, 40, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Barbagallo, M.; Dominguez, L.J.; Tagliamonte, M.R.; Resnick, L.M.; Paolisso, G. Effects of Vitamin E and Glutathione on Glucose Metabolism. Hypertension 1999, 34, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Hagiwara, K.; Arai, H.; Inoue, K. Purification and characterization of the alpha-tocopherol transfer protein from rat liver. FEBS Lett. 1991, 288, 41–45. [Google Scholar] [CrossRef]
- Raederstorff, D.; Wyss, A.; Calder, P.C.; Weber, P.; Eggersdorfer, M. Vitamin E function and requirements in relation to PUFA. Br. J. Nutr. 2015, 114, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Krebs und Ernährung - Thieme.de - Thieme Webshop - Siegfried Knasmüller. Available online: https://www.thieme.de/shop/Endokrinologie--Diabetologie/Knasmueller-Krebs-und-Ernaehrung-9783131542113/p/000000000269440101 (accessed on 18 April 2016).
- Aggarwal, B.B.; Sundaram, C.; Prasad, S.; Kannappan, R. Tocotrienols, the vitamin E of the 21st century: Its potential against cancer and other chronic diseases. Biochem. Pharmacol. 2010, 80, 1613–1631. [Google Scholar] [CrossRef] [PubMed]
- Bardowell, S.A.; Duan, F.; Manor, D.; Swanson, J.E.; Parker, R.S. Disruption of mouse cytochrome p450 4f14 (Cyp4f14 gene) causes severe perturbations in vitamin E metabolism. J. Biol. Chem. 2012, 287, 26077–26086. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.; Picinich, S.C.; Yang, Z.; Zhao, Y.; Suh, N.; Kong, A.-N.; Yang, C.S. Cancer-preventive activities of tocopherols and tocotrienols. Carcinogenesis 2010, 31, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Factor, V.M.; Laskowska, D.; Jensen, M.R.; Woitach, J.T.; Popescu, N.C.; Thorgeirsson, S.S. Vitamin E reduces chromosomal damage and inhibits hepatic tumor formation in a transgenic mouse model. Proc. Natl. Acad. Sci. USA 2000, 97, 2196–2201. [Google Scholar] [CrossRef] [PubMed]
- Kakizaki, S.; Takagi, H.; Fukusato, T.; Toyoda, M.; Horiguchi, N.; Sato, K.; Takayama, H.; Nagamine, T.; Mori, M. Effect of alpha-tocopherol on hepatocarcinogenesis in transforming growth factor-alpha (TGF-alpha) transgenic mice treated with diethylnitrosamine. Int. J. Vitam. Nutr. Res. 2001, 71, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q. Natural forms of vitamin E: Metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic. Biol. Med. 2014, 72, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Ames, B.N. Gamma-Tocopherol, but not alpha-tocopherol, decreases proinflammatory eicosanoids and inflammation damage in rats. FASEB J. 2003, 17, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Glauert, H.P. Vitamin E and NF-κB Activation: A Review. Vitam. Horm. 2007, 76, 135–153. [Google Scholar] [PubMed]
- Kim, A.Y.; Park, Y.J.; Pan, X.; Shin, K.C.; Kwak, S.-H.; Bassas, A.F.; Sallam, R.M.; Park, K.S.; Alfadda, A.A.; Xu, A.; et al. Obesity-induced DNA hypermethylation of the adiponectin gene mediates insulin resistance. Nat. Commun. 2015, 6, 7585. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Goto, M.; Matsumoto, A.; Tanaka, I. Inhibition of NF-kappa B transcriptional activity by alpha-tocopheryl succinate. Biofactors 1998, 7, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Upritchard, J.E.; Sutherland, W.H.; Mann, J.I. Effect of supplementation with tomato juice, vitamin E, and vitamin C on LDL oxidation and products of inflammatory activity in type 2 diabetes. Diabetes Care 2000, 23, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Jialal, I. Alpha-tocopherol decreases interleukin-1 beta release from activated human monocytes by inhibition of 5-lipoxygenase. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Lonn, E.; Bosch, J.; Yusuf, S.; Sheridan, P.; Pogue, J.; Arnold, J.M.O.; Ross, C.; Arnold, A.; Sleight, P.; Probstfield, J.; et al. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: A randomized controlled trial. JAMA 2005, 293, 1338–1347. [Google Scholar] [PubMed]
- Sesso, H.D.; Buring, J.E.; Christen, W.G.; Kurth, T.; Belanger, C.; MacFadyen, J.; Bubes, V.; Manson, J.E.; Glynn, R.J.; Gaziano, J.M. Vitamins E and C in the prevention of cardiovascular disease in men: The Physicians’ Health Study II randomized controlled trial. JAMA 2008, 300, 2123–2133. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Wolf, P.; Liu, N.; Link, S.; Smets, M.; La Mastra, F.; Forné, I.; Pichler, G.; Hörl, D.; Fellinger, K.; et al. DNA methylation requires a DNMT1 ubiquitin interacting motif (UIM) and histone ubiquitination. Cell Res. 2015, 25, 911–929. [Google Scholar] [CrossRef] [PubMed]
- Fuks, F.; Burgers, W.A.; Brehm, A.; Hughes-Davies, L.; Kouzarides, T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet. 2000, 24, 88–91. [Google Scholar] [PubMed]
- Shen, W.; Wang, C.; Xia, L.; Fan, C.; Dong, H.; Deckelbaum, R.J.; Qi, K. Epigenetic modification of the leptin promoter in diet-induced obese mice and the effects of N-3 polyunsaturated fatty acids. Sci. Rep. 2014, 4, 5282. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.A.; Milagro, F.I.; Claycombe, K.J.; Schalinske, K.L. Epigenetics in adipose tissue, obesity, weight loss, and diabetes. Adv. Nutr. 2014, 5, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Nakamura, T.; Ogawa, T.; Tanaka, S.; Shiota, K. Transcription of mouse DNA methyltransferase 1 (Dnmt1) is regulated by both E2F-Rb-HDAC-dependent and -independent pathways. Nucleic Acids Res. 2003, 31, 3101–3113. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.T.; Davis, J.N.; Day, M.L. Regulation of DNA Methyltransferase 1 by the pRb/E2F1 Pathway. Cancer Res. 2005, 65, 3624–3632. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.K.; Sindhu, K.K. Oxidative stress and metabolic syndrome. Life Sci. 2009, 84, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Foster, N.R.; Yoon, H.H.; Smyrk, T.C.; Kim, G.P.; Allegra, C.J.; Yothers, G.; Nikcevich, D.A.; Sargent, D.J. Association of obesity with DNA mismatch repair status and clinical outcome in patients with stage II or III colon carcinoma participating in NCCTG and NSABP adjuvant chemotherapy trials. J. Clin. Oncol. 2012, 30, 406–412. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Sequence (5′→3′) | Size (bp) | GC% |
|---|---|---|---|---|
| DNMT1 | FW | Biotin - GTA GGT TGT AGA AGA TAG AAT AGT TTT GA | 29 | 31 |
| RW | CCC ACT CTC TTA CCC TAT ATA ATA CAT | 27 | 37 | |
| Seq | CCC CTC CCA ATT AAT TTC | 18 | 44.4 | |
| Sequence ID: gb|AH009208.2| DNMT1: at reverse strand of chromosome 9: 20907205–20959888 (52684 bp). | ||||
| Sequence to analyze | 7104-CGCGCGCGCGAAAAAGCCGGGGTCTCGT-7131 | 27 | 7 CpGs | |
| MLH1 | FW | AGG GTA TTT TAG TTT TTA TTG GTT GGA GA | 29 | 31 |
| RW | TTA CAC CTC AAT TCC TAA AAT CTC TAT CCC – Biotin | 30 | 37 | |
| Seq | TTT AGT TTT TAG AAA TGA GTT AAT A | 25 | 16 | |
| Sequence ID: ref|XR_379849.3 MLH1: at reverse strand of Chromosome 9: 111228228–111271786 (43559 bp) | ||||
| Sequence to analyze | 19-GAAGAGCGGACCGTGAACTTTGACGCGCAAGCGCG TTGCCTTCTA-GCCTGGTGTCGGGCCGCTG-82 | 64 | 8 CpGs | |
| Chow Intake (g) | Water Intake (mL) | Weight (g) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Month | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | 1 | 2 | 3 | 4 | |
| Intervention | CD | 2.64 ± 0.07 | 2.11 ± 0.01 | 2.08 ± 0.04 | 2.06 ± 0.03 | 5.58 ± 0.21 | 5.29 ± 0.22 | 5.39 ± 0.28 | 4.95 ± 0.56 | 24.66 ± 0.75 | 26.17 ± 0.16 | 27.17 ± 0.18 | 28.31 ± 0.24 |
| CD + E | 2.70 ± 0.08 | 2.70 ± 0.05 | 2.76 ± 0.06 | 2.76 ± 0.51 | 5.80 ± 0.21 | 5.76 ± 0.15 | 5.97 ± 0.17 | 5.55 ± 0.27 | 24.94 ± 0.77 | 26.58 ± 0.15 | 27.75 ± 0.19 | 28.63 ± 0.14 | |
| HFD | 2.56 ± 0.04 | 2.59 ± 0.02 | 2.60 ± 0.02 | 2.56 ± 0.06 | 5.34 ± 0.20 | 4.93 ± 0.24 | 5.10 ± 0.14 | 5.01 ± 0.18 | 32.57 ± 2.09 | 39.00 ± 1.37 | 43.97 ± 1.02 | 47.09 ± 0.83 | |
| HFD + E | 2.51 ± 0.02 | 2.45 ± 0.06 | 2.50 ± 0.05 | 2.54 ± 0.01 | 5.21 ± 0.20 | 4.69 ± 0.06 | 5.05 ± 0.04 | 5.09 ± 0.11 | 32.77 ± 2.3 | 39.49 ± 1.64 | 44.67 ± 1.23 | 47.67 ± 0.49 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Remely, M.; Ferk, F.; Sterneder, S.; Setayesh, T.; Kepcija, T.; Roth, S.; Noorizadeh, R.; Greunz, M.; Rebhan, I.; Wagner, K.-H.; et al. Vitamin E Modifies High-Fat Diet-Induced Increase of DNA Strand Breaks, and Changes in Expression and DNA Methylation of Dnmt1 and MLH1 in C57BL/6J Male Mice. Nutrients 2017, 9, 607. https://doi.org/10.3390/nu9060607
Remely M, Ferk F, Sterneder S, Setayesh T, Kepcija T, Roth S, Noorizadeh R, Greunz M, Rebhan I, Wagner K-H, et al. Vitamin E Modifies High-Fat Diet-Induced Increase of DNA Strand Breaks, and Changes in Expression and DNA Methylation of Dnmt1 and MLH1 in C57BL/6J Male Mice. Nutrients. 2017; 9(6):607. https://doi.org/10.3390/nu9060607
Chicago/Turabian StyleRemely, Marlene, Franziska Ferk, Sonja Sterneder, Tahereh Setayesh, Tatjana Kepcija, Sylvia Roth, Rahil Noorizadeh, Martina Greunz, Irene Rebhan, Karl-Heinz Wagner, and et al. 2017. "Vitamin E Modifies High-Fat Diet-Induced Increase of DNA Strand Breaks, and Changes in Expression and DNA Methylation of Dnmt1 and MLH1 in C57BL/6J Male Mice" Nutrients 9, no. 6: 607. https://doi.org/10.3390/nu9060607
APA StyleRemely, M., Ferk, F., Sterneder, S., Setayesh, T., Kepcija, T., Roth, S., Noorizadeh, R., Greunz, M., Rebhan, I., Wagner, K. -H., Knasmüller, S., & Haslberger, A. (2017). Vitamin E Modifies High-Fat Diet-Induced Increase of DNA Strand Breaks, and Changes in Expression and DNA Methylation of Dnmt1 and MLH1 in C57BL/6J Male Mice. Nutrients, 9(6), 607. https://doi.org/10.3390/nu9060607