A Systematic Overview of Type II and III Toxin-Antitoxin Systems with a Focus on Druggability


Abstract
:1. Introduction
2. Type II TA Systems in Pathogenic Bacteria
3. Application of Antimicrobial Peptides Based on the Type II TA Interface
4. Type III TA Systems in the Human Intestinal Microbiota
5. Closing Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Page, R.; Peti, W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 2016, 12, 208–214. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Park, J.H.; Inouye, M. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 2011, 45, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Lee, B.J. Structure, biology, and therapeutic application of toxin-antitoxin systems in pathogenic bacteria. Toxins 2016, 8, 305. [Google Scholar] [CrossRef]
- Ogura, T.; Hiraga, S. Mini-f plasmid genes that couple host cell division to plasmid proliferation. Proc. Natl. Acad. Sci. USA 1983, 80, 4784–4788. [Google Scholar] [CrossRef] [PubMed]
- Jurenaite, M.; Markuckas, A.; Suziedeliene, E. Identification and characterization of type II toxin-antitoxin systems in the opportunistic pathogen acinetobacter baumannii. J. Bacteriol. 2013, 195, 3165–3172. [Google Scholar] [CrossRef] [PubMed]
- Pandey, D.P.; Gerdes, K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005, 33, 966–976. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, M.; Austin, S. Prevalence and significance of plasmid maintenance functions in the virulence plasmids of pathogenic bacteria. Infect. Immun. 2011, 79, 2502–2509. [Google Scholar] [CrossRef]
- Kwan, B.W.; Valenta, J.A.; Benedik, M.J.; Wood, T.K. Arrested protein synthesis increases persister-like cell formation. Antimicrob. Agents Chemother. 2013, 57, 1468–1473. [Google Scholar] [CrossRef]
- Goeders, N.; Van Melderen, L. Toxin-antitoxin systems as multilevel interaction systems. Toxins 2014, 6, 304–324. [Google Scholar] [CrossRef]
- Mutschler, H.; Gebhardt, M.; Shoeman, R.L.; Meinhart, A. A novel mechanism of programmed cell death in bacteria by toxin-antitoxin systems corrupts peptidoglycan synthesis. PLoS Biol. 2011, 9, e1001033. [Google Scholar] [CrossRef]
- Unoson, C.; Wagner, E.G.H. A small sos-induced toxin is targeted against the inner membrane in escherichia coli. Mol. Microbiol. 2008, 70, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Marra, A. Can virulence factors be viable antibacterial targets? Expert Rev. Anti-Infect. Ther. 2004, 2, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Mann, J.; Li, N. Targeted inhibitor design: Lessons from small molecule drug design, directed evolution, and vaccine research. Chem. Eng. Process. Technol. 2013, 1, 1004. [Google Scholar]
- Goeders, N.; Chai, R.; Chen, B.H.; Day, A.; Salmond, G.P.C. Structure, evolution, and functions of bacterial type III toxin-antitoxin systems. Toxins 2016, 8, 282. [Google Scholar] [CrossRef] [PubMed]
- Lobato-Marquez, D.; Diaz-Orejas, R.; Garcia-del Portillo, F. Toxin-antitoxins and bacterial virulence. FEMS Microbiol. Rev. 2016, 40, 592–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, A.; Brodersen, D.E.; Mitarai, N.; Gerdes, K. Toxins, targets, and triggers: An overview of toxin-antitoxin biology. Mol. Cell 2018, 70, 768–784. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Lord, D.M.; Cheng, H.Y.; Osbourne, D.O.; Hong, S.H.; Sanchez-Torres, V.; Quiroga, C.; Zheng, K.; Herrmann, T.; Peti, W.; et al. A new type v toxin-antitoxin system where mRNA for toxin ghot is cleaved by antitoxin ghos. Nat. Chem. Biol. 2012, 8, 855–861. [Google Scholar] [CrossRef]
- Aakre, C.D.; Phung, T.N.; Huang, D.; Laub, M.T. A bacterial toxin inhibits DNA replication elongation through a direct interaction with the beta sliding clamp. Mol. Cell 2013, 52, 617–628. [Google Scholar] [CrossRef]
- Sala, A.; Bordes, P.; Genevaux, P. Multiple toxin-antitoxin systems in mycobacterium tuberculosis. Toxins 2014, 6, 1002–1020. [Google Scholar] [CrossRef]
- Van Melderen, L.; De Bast, M.S. Bacterial toxin-antitoxin systems: More than selfish entities? PLoS Genet. 2009, 5, e1000437. [Google Scholar] [CrossRef]
- Coussens, N.P.; Daines, D.A. Wake me when it’s over—Bacterial toxin-antitoxin proteins and induced dormancy. Exp. Biol. Med. 2016, 241, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Chukwudi, C.U.; Good, L. The role of the hok/sok locus in bacterial response to stressful growth conditions. Microb. Pathog. 2015, 79, 70–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.X.; Kim, Y.; Hong, S.H.; Ma, Q.; Brown, B.L.; Pu, M.M.; Tarone, A.M.; Benedik, M.J.; Peti, W.; Page, R.; et al. Antitoxin mqsa helps mediate the bacterial general stress response. Nat. Chem. Biol. 2011, 7, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Wood, T.K. Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 2011, 77, 5577–5583. [Google Scholar] [CrossRef] [PubMed]
- Ramage, H.R.; Connolly, L.E.; Cox, J.S. Comprehensive functional analysis of mycobacterium tuberculosis toxin-antitoxin systems: Implications for pathogenesis, stress responses, and evolution. PLoS Genet. 2009, 5, e1000767. [Google Scholar] [CrossRef] [PubMed]
- Fasani, R.A.; Savageau, M.A. Molecular mechanisms of multiple toxin-antitoxin systems are coordinated to govern the persister phenotype. Proc. Natl. Acad. Sci. USA 2013, 110, E2528–E2537. [Google Scholar] [CrossRef]
- Chan, W.T.; Espinosa, M.; Yeo, C.C. Keeping the wolves at bay: Antitoxins of prokaryotic type II toxin-antitoxin systems. Front. Mol. Biosci. 2016, 3, 9. [Google Scholar] [CrossRef]
- Gupta, K.; Tripathi, A.; Sahu, A.; Varadarajan, R. Contribution of the chromosomal ccdab operon to bacterial drug tolerance. J. Bacteriol. 2017, 199, JB.00397-17. [Google Scholar] [CrossRef]
- Song, S.; Wood, T.K. Post-segregational killing and phage inhibition are not mediated by cell death through toxin/antitoxin systems. Front. Microbiol. 2018, 9, 814. [Google Scholar] [CrossRef]
- Goormaghtigh, F.; Fraikin, N.; Putrins, M.; Hallaert, T.; Hauryliuk, V.; Garcia-Pino, A.; Sjodin, A.; Kasvandik, S.; Udekwu, K.; Tenson, T.; et al. Reassessing the role of type II toxin-antitoxin systems in formation of escherichia coli type II persister cells. mBio 2018, 9, e00640-18. [Google Scholar] [CrossRef]
- Kang, S.M.; Kim, D.H.; Lee, K.Y.; Park, S.J.; Yoon, H.J.; Lee, S.J.; Im, H.; Lee, B.J. Functional details of the mycobacterium tuberculosis vapbc26 toxin-antitoxin system based on a structural study: Insights into unique binding and antibiotic peptides. Nucleic Acids Res. 2017, 45, 8564–8580. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.H.; Lee, K.Y.; Lee, S.J.; Park, S.J.; Yoon, H.J.; Kim, S.J.; Lee, B.J. Structural analyses of the mazef4 toxin-antitoxin pair in mycobacterium tuberculosis provide evidence for a unique extracellular death factor. J. Biol. Chem. 2017, 292, 18832–18847. [Google Scholar] [CrossRef] [PubMed]
- Blower, T.R.; Pei, X.Y.; Short, F.L.; Fineran, P.C.; Humphreys, D.P.; Luisi, B.F.; Salmond, G.P.C. A processed noncoding RNA regulates an altruistic bacterial antiviral system. Nat. Struct. Mol. Biol. 2011, 18, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, F.; Short, F.L.; Voss, J.E.; Blower, T.R.; Orme, A.L.; Whittaker, T.E.; Luisi, B.F.; Salmond, G.P.C. Co-evolution of quaternary organization and novel RNA tertiary interactions revealed in the crystal structure of a bacterial protein-RNA toxin-antitoxin system. Nucleic Acids Res. 2015, 43, 9529–9540. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Kumar, S.; Gupta, V.P.; Gourinath, S.; Bhatnagar, S.; Bhatnagar, R. Structural basis of bacillus anthracis moxxt disruption and the modulation of moxt ribonuclease activity by rationally designed peptides. J. Biomol. Struct. Dyn. 2015, 33, 606–624. [Google Scholar] [CrossRef]
- Lee, I.G.; Lee, S.J.; Chae, S.; Lee, K.Y.; Kim, J.H.; Lee, B.J. Structural and functional studies of the mycobacterium tuberculosis vapbc30 toxin-antitoxin system. Protein Sci. 2016, 25, 156–157. [Google Scholar] [CrossRef]
- Kim, D.H.; Kang, S.M.; Park, S.J.; Jin, C.; Yoon, H.J.; Lee, B.J. Functional insights into the streptococcus pneumoniae hicba toxin-antitoxin system based on a structural study. Nucleic Acids Res. 2018, 46, 6371–6386. [Google Scholar] [CrossRef]
- Fernandez-Bachiller, M.I.; Brzozowska, I.; Odolczyk, N.; Zielenkiewicz, U.; Zielenkiewicz, P.; Rademann, J. Mapping protein-protein interactions of the resistance-related bacterial zeta toxin-epsilon antitoxin complex (ε2ζ2) with high affinity peptide ligands using fluorescence polarization. Toxins 2016, 8, 222. [Google Scholar] [CrossRef]
- Rocker, A.; Peschke, M.; Kittila, T.; Sakson, R.; Brieke, C.; Meinhart, A. The ng_zeta 1 toxin of the gonococcal epsilon/zeta toxin/antitoxin system drains precursors for cell wall synthesis. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Lioy, V.S.; Rey, O.; Balsa, D.; Pellicer, T.; Alonso, J.C. A toxin-antitoxin module as a target for antimicrobial development. Plasmid 2010, 63, 31–39. [Google Scholar] [CrossRef]
- Blower, T.R.; Short, F.L.; Rao, F.; Mizuguchi, K.; Pei, X.Y.; Fineran, P.C.; Luisi, B.F.; Salmond, G.P.C. Identification and classification of bacterial type III toxin-antitoxin systems encoded in chromosomal and plasmid genomes. Nucleic Acids Res. 2012, 40, 6158–6173. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology—Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [PubMed]
- Boulange, C.L.; Neves, A.L.; Chilloux, J.; Nicholson, J.K.; Dumas, M.E. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016, 8, 42. [Google Scholar] [PubMed]
- Oke, S.; Martin, A. Insights into the role of the intestinal microbiota in colon cancer. Ther. Adv. Gastroenter. 2017, 10, 417–428. [Google Scholar] [Green Version]
- Meng, C.T.; Bai, C.M.; Brown, T.D.; Hood, L.E.; Tian, Q. Human gut microbiota and gastrointestinal cancer. Genom. Proteom. Bioinf. 2018, 16, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K. Prokaryotic Toxin-Antitoxins; Springer: Heidelberg, Germany; New York, NY, USA, 2013; Volume 8, pp. 1–365. [Google Scholar]
- Albrethsen, J.; Agner, J.; Piersma, S.R.; Hojrup, P.; Pham, T.V.; Weldingh, K.; Jimenez, C.R.; Andersen, P.; Rosenkrands, I. Proteomic profiling of mycobacterium tuberculosis identifies nutrient-starvation-responsive toxin-antitoxin systems. Mol. Cell. Proteom. 2013, 12, 1180–1191. [Google Scholar] [CrossRef]
- Miallau, L.; Jain, P.; Arbing, M.A.; Cascio, D.; Phan, T.; Ahn, C.J.; Chan, S.; Chernishof, I.; Maxson, M.; Chiang, J.; et al. Comparative proteomics identifies the cell-associated lethality of m. Tuberculosis reibe-like toxin-antitoxin complexes. Structure 2013, 21, 627–637. [Google Scholar] [CrossRef]
- Korch, S.B.; Contreras, H.; Clark-Curtiss, J.E. Three mycobacterium tuberculosis rel toxin-antitoxin modules inhibit mycobacterial growth and are expressed in infected human macrophages. J. Bacteriol. 2009, 191, 1618–1630. [Google Scholar]
- Korch, S.B.; Malhotra, V.; Contreras, H.; Clark-Curtiss, J.E. The mycobacterium tuberculosis relbe toxin: Antitoxin genes are stress-responsive modules that regulate growth through translation inhibition. J. Microbiol. 2015, 53, 875. [Google Scholar] [CrossRef]
- De la Cruz, M.A.; Zhao, W.D.; Farenc, C.; Gimenez, G.; Raoult, D.; Cambillau, C.; Gorvel, J.P.; Meresse, S. A toxin-antitoxin module of salmonella promotes virulence in mice. PLoS Pathog. 2013, 9, e1003827. [Google Scholar] [CrossRef]
- Kedzierska, B.; Hayes, F. Emerging roles of toxin-antitoxin modules in bacterial pathogenesis. Molecules 2016, 21, 790. [Google Scholar] [CrossRef] [PubMed]
- Conlon, B.P.; Nakayasu, E.S.; Fleck, L.E.; LaFleur, M.D.; Isabella, V.M.; Coleman, K.; Leonard, S.N.; Smith, R.D.; Adkins, J.N.; Lewis, K. Activated clpp kills persisters and eradicates a chronic biofilm infection. Nature 2013, 503, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.R.; Behiels, E.; Devreese, B. Toxin-antitoxin systems: Their role in persistence, biofilm formation, and pathogenicity. Pathog. Dis. 2014, 70, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Amato, S.M.; Fazen, C.H.; Henry, T.C.; Mok, W.W.K.; Orman, M.A.; Sandvik, E.L.; Volzing, K.G.; Brynildsen, M.P. The role of metabolism in bacterial persistence. Front. Microbiol. 2014, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, E.; Gerdes, K. Molecular mechanisms underlying bacterial persisters. Cell 2014, 157, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K.; Maisonneuve, E. Bacterial persistence and toxin-antitoxin loci. Annu. Rev. Microbiol. 2012, 66, 103–123. [Google Scholar] [CrossRef] [PubMed]
- Engelberg-Kulka, H.; Sat, B.; Reches, M.; Amitai, S.; Hazan, R. Bacterial programmed cell death systems as targets for antibiotics. Trends Microbiol. 2004, 12, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Mutschler, H.; Reinstein, J.; Meinhart, A. Assembly dynamics and stability of the pneumococcal epsilon zeta antitoxin toxin (PezAT) system from streptococcus pneumoniae. J. Biol. Chem. 2010, 285, 21797–21806. [Google Scholar] [CrossRef]
- Kumar, S.; Kolodkin-Gal, I.; Vesper, O.; Alam, N.; Schueler-Furman, O.; Moll, I.; Engelberg-Kulka, H. Escherichia coli quorum-sensing edf, a peptide generated by novel multiple distinct mechanisms and regulated by trans-translation. Mbio 2016, 7. [Google Scholar] [CrossRef]
- Lee, B.; Holkenbrink, C.; Treuner-Lange, A.; Higgs, P.I. Myxococcus xanthus developmental cell fate production: Heterogeneous accumulation of developmental regulatory proteins and reexamination of the role of mazf in developmental lysis. J. Bacteriol. 2012, 194, 3058–3068. [Google Scholar] [CrossRef]
- Boynton, T.O.; McMurry, J.L.; Shimkets, L.J. Characterization of myxococcus xanthus mazf and implications for a new point of regulation. Mol. Microbiol. 2013, 87, 1267–1276. [Google Scholar] [CrossRef]
- Soheili, S.; Ghafourian, S.; Sekawi, Z.; Neela, V.K.; Sadeghifard, N.; Taherikalani, M.; Khosravi, A.; Ramli, R.; Hamat, R.A. The mazef toxin-antitoxin system as an attractive target in clinical isolates of enterococcus faecium and enterococcus faecalis. Drug Des. Dev. Ther. 2015, 9, 2553–2561. [Google Scholar]
- Shimazu, T.; Mirochnitchenko, O.; Phadtare, S.; Inouye, M. Regression of solid tumors by induction of mazf, a bacterial mRNA endoribonuclease. J. Mol. Microbiol. Biotechnol. 2014, 24, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Martinez, L.C.; Vadyvaloo, V. Mechanisms of post-transcriptional gene regulation in bacterial biofilms. Front. Cell Infect. Microbiol. 2014, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Helaine, S.; Cheverton, A.M.; Watson, K.G.; Faure, L.M.; Matthews, S.A.; Holden, D.W. Internalization of salmonella by macrophages induces formation of nonreplicating persisters. Science 2014, 343, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Hershkovitz, I.; Donoghue, H.D.; Minnikin, D.E.; May, H.; Lee, O.Y.C.; Feldman, M.; Galili, E.; Spigelman, M.; Rothschild, B.M.; Bar-Gal, G.K. Tuberculosis origin: The neolithic scenario. Tuberculosis 2015, 95, S122–S126. [Google Scholar] [CrossRef]
- Ashu, E.E.; Jarju, S.; Dione, M.; Mackenzie, G.; Ikumapayi, U.N.; Manjang, A.; Azuine, R.; Antonio, M. Population structure, epidemiology and antibiotic resistance patterns of streptococcus pneumoniae serotype 5: Prior to pcv-13 vaccine introduction in eastern gambia. BMC Infect. Dis. 2016, 16, 33. [Google Scholar] [CrossRef] [PubMed]
- Lobato-Marquez, D.; Moreno-Cordoba, I.; Figueroa, V.; Diaz-Orejas, R.; Garcia-del Portillo, F. Distinct type I and type II toxin-antitoxin modules control salmonella lifestyle inside eukaryotic cells. Sci. Rep. 2015, 5, 9374. [Google Scholar] [CrossRef]
- Zorzini, V.; Buts, L.; Sleutel, M.; Garcia-Pino, A.; Talavera, A.; Haesaerts, S.; De Greve, H.; Cheung, A.; van Nuland, N.A.J.; Loris, R. Structural and biophysical characterization of staphylococcus aureus samazf shows conservation of functional dynamics. Nucleic Acids Res. 2014, 42, 6709–6725. [Google Scholar] [CrossRef]
- Hoffer, E.D.; Miles, S.J.; Dunham, C.M. The structure and function of mycobacterium tuberculosis mazf-mt6 toxin provide insights into conserved features of mazf endonucleases. J. Biol. Chem. 2017, 292, 7718–7726. [Google Scholar] [CrossRef]
- Chen, R.; Tu, J.; Liu, Z.H.; Meng, F.R.; Ma, P.Y.; Ding, Z.S.; Yang, C.W.; Chen, L.; Deng, X.Y.; Xie, W. Structure of the mazf-mt9 toxin, a tRNA-specific endonuclease from mycobacterium tuberculosis. Biochem. Biophys. Res. Commun. 2017, 486, 804–810. [Google Scholar] [CrossRef]
- Min, A.B.; Miallau, L.; Sawaya, M.R.; Habel, J.; Cascio, D.; Eisenberg, D. The crystal structure of the rv0301-rv0300 vapbc-3 toxin-antitoxin complex from M. tuberculosis reveals a Mg2+ ion in the active site and a putative RNA-binding site. Protein Sci. 2012, 21, 1754–1767. [Google Scholar] [CrossRef] [PubMed]
- Miallau, L.; Faller, M.; Chiang, J.; Arbing, M.; Guo, F.; Cascio, D.; Eisenberg, D. Structure and proposed activity of a member of the vapbc family of toxin-antitoxin systems vapbc-5 from mycobacterium tuberculosis. J. Biol. Chem. 2009, 284, 276–283. [Google Scholar] [CrossRef]
- Das, U.; Pogenberg, V.; Subhramanyam, U.K.T.; Wilmanns, M.; Gourinath, S.; Srinivasan, A. Crystal structure of the vapbc-15 complex from mycobacterium tuberculosis reveals a two-metal ion dependent pin-domain ribonuclease and a variable mode of toxin-antitoxin assembly. J. Struct. Biol. 2014, 188, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Deep, A.; Kaundal, S.; Agarwal, S.; Singh, R.; Thakur, K.G. Crystal structure of mycobacterium tuberculosis vapc20 toxin and its interactions with cognate antitoxin, vapb20, suggest a model for toxin-antitoxin assembly. FEBS J. 2017, 284, 4066–4082. [Google Scholar] [CrossRef] [PubMed]
- Jardim, P.; Santos, I.C.D.; Barbosa, J.A.R.G.; de Freitas, S.M.; Valadares, N.F. Crystal structure of vapc21 from mycobacterium tuberculosis at 1.31 angstrom resolution. Biochem. Biophys. Res. Commun. 2016, 478, 1370–1375. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.; Domenech, M.; Moreno-Córdoba, I.; Navarro-Martínez, V.; Nieto, C.; Moscoso, M.; García, E.; Espinosa, M. The streptococcus pneumoniae yefm-yoeb and relbe toxin-antitoxin operons participate in oxidative stress and biofilm formation. Toxins 2018, 10, 378. [Google Scholar] [CrossRef]
- Xie, Y.Z.; Wei, Y.Q.; Shen, Y.; Li, X.B.; Zhou, H.; Tai, C.; Deng, Z.X.; Ou, H.Y. Tadb 2.0: An updated database of bacterial type II toxin-antitoxin loci. Nucleic Acids Res. 2018, 46, D749–D753. [Google Scholar] [CrossRef]
- UniProt Consortium. Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Rose, P.W.; Prlic, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Costanzo, L.D.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The rcsb protein data bank: Integrative view of protein, gene and 3d structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar]
- Williams, J.J.; Hergenrother, P.J. Artificial activation of toxin-antitoxin systems as an antibacterial strategy. Trends Microbiol. 2012, 20, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, S.J.; Poppenberger, B.; Rozhon, W. Toxin-antitoxin systems: Biology, identification, and application. Mob. Genet. Elem. 2013, 3, e26219. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.P.; Mu, Z.X.; Qin, B.; Sun, Y.C.; Cui, S. Structure-based prototype peptides targeting the pseudomonas aeruginosa type VI secretion system effector as a novel antibacterial strategy. Front. Cell. Infect. Microbiol. 2017, 7, 411. [Google Scholar] [CrossRef] [PubMed]
- Khusro, A.; Aarti, C.; Barbabosa-Pliego, A.; Salem, A.Z.M. Neoteric advancement in tb drugs and an overview on the anti-tubercular role of peptides through computational approaches. Microb. Pathog. 2018, 114, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Cook, G.M.; Robson, J.R.; Frampton, R.A.; McKenzie, J.; Przybilski, R.; Fineran, P.C.; Arcus, V.L. Ribonucleases in bacterial toxin-antitoxin systems. BBA—Gene Regul. Mech. 2013, 1829, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Inouye, M. Toxins of prokaryotic toxin-antitoxin systems with sequence-specific endoribonuclease activity. Toxins 2017, 9, 140. [Google Scholar] [CrossRef] [PubMed]
- Kamada, K.; Hanaoka, F.; Burley, S.K. Crystal structure of the maze/mazf complex: Molecular bases of antidote-toxin recognition. Mol. Cell 2003, 11, 875–884. [Google Scholar] [CrossRef]
- Agarwal, S.; Mishra, N.K.; Bhatnagar, S.; Bhatnagar, R. Pemk toxin of bacillus anthracis is a ribonuclease an insight into its active site, structure, and function. J. Biol. Chem. 2010, 285, 7254–7270. [Google Scholar] [CrossRef]
- Chopra, N.; Agarwal, S.; Verma, S.; Bhatnagar, S.; Bhatnagar, R. Modeling of the structure and interactions of the b. Anthracis antitoxin, moxx: Deletion mutant studies highlight its modular structure and repressor function. J. Comput. Aided Mol. Des. 2011, 25, 275–291. [Google Scholar] [CrossRef]
- Pham, T.K.; Kim, D.H.; Lee, B.J.; Kim, Y.W. Truncated and constrained helical analogs of antimicrobial esculentin-2em. Bioorg. Med. Chem. Lett. 2013, 23, 6717–6720. [Google Scholar] [CrossRef]
- Uggerhoj, L.E.; Poulsen, T.J.; Munk, J.K.; Fredborg, M.; Sondergaard, T.E.; Frimodt-Moller, N.; Hansen, P.R.; Wimmer, R. Rational design of alpha-helical antimicrobial peptides: Do’s and don’ts. ChemBioChem 2015, 16, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Fozo, E.M. Srna antitoxins: More than one way to repress a toxin. Toxins 2014, 6, 2310–2335. [Google Scholar] [CrossRef] [PubMed]
- Fineran, P.C.; Blower, T.R.; Foulds, I.J.; Humphreys, D.P.; Lilley, K.S.; Salmond, G.P. The phage abortive infection system, toxin, functions as a protein-RNA toxin-antitoxin pair. Proc. Natl. Acad. Sci. USA 2009, 106, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Short, F.L.; Pei, X.Y.; Blower, T.R.; Ong, S.L.; Fineran, P.C.; Luisi, B.F.; Salmond, G.P. Selectivity and self-assembly in the control of a bacterial toxin by an antitoxic noncoding RNA pseudoknot. Proc. Natl. Acad. Sci. USA 2013, 110, E241–E249. [Google Scholar] [CrossRef] [PubMed]
- Blower, T.R.; Chai, R.; Przybilski, R.; Chindhy, S.; Fang, X.Z.; Kidman, S.E.; Tan, H.; Luisi, B.F.; Fineran, P.C.; Salmond, G.P.C. Evolution of pectobacterium bacteriophage Φm1 to escape two bifunctional type III toxin-antitoxin and abortive infection systems through mutations in a single viral gene. Appl. Environ. Microb. 2017, 83, e03229-16. [Google Scholar] [CrossRef] [PubMed]
- Blower, T.R.; Fineran, P.C.; Johnson, M.J.; Toth, I.K.; Humphreys, D.P.; Salmond, G.P.C. Mutagenesis and functional characterization of the RNA and protein components of the toxin abortive infection and toxin-antitoxin locus of erwinia. J. Bacteriol. 2009, 191, 6029–6039. [Google Scholar] [CrossRef] [PubMed]
- Dy, R.L.; Przybilski, R.; Semeijn, K.; Salmond, G.P.C.; Fineran, P.C. A widespread bacteriophage abortive infection system functions through a type IV toxin-antitoxin mechanism. Nucleic Acids Res. 2014, 42, 4590–4605. [Google Scholar] [CrossRef]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef]
- Koskella, B.; Brockhurst, M.A. Bacteria-phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 2014, 38, 916–931. [Google Scholar] [CrossRef]
- Sengupta, M.; Nielsen, H.J.; Youngren, B.; Austin, S. P1 plasmid segregation: Accurate redistribution by dynamic plasmid pairing and separation. J. Bacteriol. 2010, 192, 1175–1183. [Google Scholar] [CrossRef]
- Samson, J.E.; Spinelli, S.; Cambillau, C.; Moineau, S. Structure and activity of abiq, a lactococcal endoribonuclease belonging to the type III toxinantitoxin system. Mol. Microbiol. 2013, 87, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Samson, J.E.; Belanger, M.; Moineau, S. Effect of the abortive infection mechanism and type III toxin/antitoxin system abiq on the lytic cycle of lactococcus lactis phages. J. Bacteriol. 2013, 195, 3947–3956. [Google Scholar] [CrossRef] [PubMed]
- Blower, T.R.; Evans, T.J.; Przybilski, R.; Fineran, P.C.; Salmond, G.P.C. Viral evasion of a bacterial suicide system by RNA-based molecular mimicry enables infectious altruism. PLoS Genet. 2012, 8, e1003023. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Peterson, D.A.; Gordon, J.I. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 2006, 124, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Littman, D.R. The microbiome in infectious disease and inflammation. Annu. Rev. Immunol. 2012, 30, 759–795. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, L.; McFall-Ngai, M.; Relman, D.A. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 2007, 449, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.C.; Olson, C.A.; Hsiao, E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Maloy, K.J.; Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 2011, 474, 298–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bevins, C.L.; Salzman, N.H. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Microbiol. 2011, 9, 356–368. [Google Scholar] [CrossRef]
- Zhou, M.X.; He, J.; Shen, Y.J.; Zhang, C.; Wang, J.Z.; Chen, Y.W. New frontiers in genetics, gut microbiota, and immunity: A rosetta stone for the pathogenesis of inflammatory bowel disease. Biomed. Res. Int. 2017, 2017, 8201672. [Google Scholar] [CrossRef] [PubMed]
- Kamada, N.; Seo, S.U.; Chen, G.Y.; Nunez, G. Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 2013, 13, 321–335. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Round, J.L.; Kasper, D.L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 2008, 453, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sun, K.J.; Wu, Y.J.; Yang, Y.; Tso, P.; Wu, Z.L. Interactions between intestinal microbiota and host immune response in inflammatory bowel disease. Front. Immunol. 2017, 8, 942. [Google Scholar] [CrossRef] [PubMed]
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system. Nature 2011, 474, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolin, M.J.; Miller, T.L.; Collins, M.D.; Lawson, P.A. Formate-dependent growth and homoacetogenic fermentation by a bacterium from human feces: Description of bryantella formatexigens gen. Nov., sp nov. Appl. Environ. Microbiol. 2003, 69, 6321–6326. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.E.; Faith, J.J.; Bain, J.; Muehlbauer, M.J.; Stevens, R.D.; Newgard, C.B.; Gordon, J.I. Dissecting the in vivo metabolic potential of two human gut acetogens. J. Biol. Chem. 2010, 285, 22082–22090. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Nagai, F.; Morotomi, M. Characterization of phascolarctobacterium succinatutens sp nov., an asaccharolytic, succinate-utilizing bacterium isolated from human feces. Appl. Environ. Microbiol. 2012, 78, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.F.; Guo, X.F.; Zhang, J.C.; Zhang, M.; Ou, Z.H.; Peng, Y.Z. Phascolarctobacterium faecium abundant colonization in human gastrointestinal tract. Exp. Ther. Med. 2017, 14, 3122–3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheah, P.Y.; Bernstein, H. Modification of DNA by bile acids: A possible factor in the etiology of colon cancer. Cancer Lett. 1990, 49, 207–210. [Google Scholar] [CrossRef]
- Reddy, B.S.; Simi, B.; Patel, N.; Aliaga, C.; Rao, C.V. Effect of amount and types of dietary fat on intestinal bacterial 7 alpha-dehydroxylase and phosphatidylinositol-specific phospholipase c and colonic mucosal diacylglycerol kinase and pkc activities during stages of colon tumor promotion. Cancer Res. 1996, 56, 2314–2320. [Google Scholar] [PubMed]
- Kitahara, M.; Takamine, F.; Imamura, T.; Benno, Y. Clostridium hiranonis sp. Nov., a human intestinal bacterium with bile acid 7alpha-dehydroxylating activity. Int. J. Syst. Evol. Microbiol. 2001, 51, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Kassinen, A.; Krogius-Kurikka, L.; Makivuokko, H.; Rinttila, T.; Paulin, L.; Corander, J.; Malinen, E.; Apajalahti, J.; Palva, A. The fecal microbiota of irritable bowel syndrome patients differs significantly from that of healthy subjects. Gastroenterology 2007, 133, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Huttenhower, C.; Kostic, A.D.; Xavier, R.J. Inflammatory bowel disease as a model for translating the microbiome. Immunity 2014, 40, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Kolho, K.L.; Korpela, K.; Jaakkola, T.; Pichai, M.V.A.; Zoetendal, E.G.; Salonen, A.; de Vos, W.M. Fecal microbiota in pediatric inflammatory bowel disease and its relation to inflammation. Am. J. Gastroenterol. 2015, 110, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Zinkernagel, M.S.; Zysset-Burri, D.C.; Keller, I.; Berger, L.E.; Leichtle, A.B.; Largiader, C.R.; Fiedler, G.M.; Wolf, S. Association of the intestinal microbiome with the development of neovascular age-related macular degeneration. Sci. Rep. 2017, 7, 40826. [Google Scholar] [CrossRef]
- Elson, C.O.; Cong, Y. Host-microbiota interactions in inflammatory bowel disease. Gut Microbes 2012, 3, 332–344. [Google Scholar] [CrossRef] [Green Version]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef] [Green Version]
- Amitay, E.L.; Brenner, H. Response to comments on ‘fusobacterium and colorectal cancer: Causal factor or passenger? Results from a large colorectal cancer screening study’. Carcinogenesis 2018, 39, 85. [Google Scholar] [CrossRef]
- Hsieh, Y.Y.; Tung, S.Y.; Pan, H.Y.; Yen, C.W.; Xu, H.W.; Lin, Y.J.; Deng, Y.F.; Hsu, W.T.; Wu, C.S.; Li, C. Increased abundance of clostridium and fusobacterium in gastric microbiota of patients with gastric cancer in taiwan. Sci. Rep. 2018, 8, 158. [Google Scholar] [CrossRef] [PubMed]
- Meehan, C.J.; Beiko, R.G. A phylogenomic view of ecological specialization in the lachnospiraceae, a family of digestive tract-associated bacteria. Genome Biol. Evol. 2014, 6, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Nishida, A.; Fujimoto, T.; Fujii, M.; Shioya, M.; Imaeda, H.; Inatomi, O.; Bamba, S.; Sugimoto, M.; Andoh, A. Reduced abundance of butyrate-producing bacteria species in the fecal microbial community in crohn’s disease. Digestion 2016, 93, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Kato, I. Gut microbiota, inflammation and colorectal cancer. Genes Dis. 2016, 3, 130–143. [Google Scholar] [CrossRef] [Green Version]
- Bultman, S.J.; Jobin, C. Microbial-derived butyrate: An oncometabolite or tumor-suppressive metabolite? Cell Host Microbe 2014, 16, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Gao, Z.; Huang, L.; Qin, H. Gut microbiota and colorectal cancer. Eur. J. Clin. Microbiol. 2017, 36, 757–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De LeBlanc, A.D.; Matar, C.; LeBlanc, N.; Perdigon, G. Effects of milk fermented by lactobacillus helveticus r389 on a murine breast cancer model. Breast Cancer Res. 2005, 7, R477–R486. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, M.W.; Tellez, A.M. Lactobacillus helveticus: The proteolytic system. Front. Microbiol. 2013, 4, 30. [Google Scholar] [CrossRef] [PubMed]
- Elfahri, K.R.; Vasiljevic, T.; Yeager, T.; Donkor, O.N. Anti-colon cancer and antioxidant activities of bovine skim milk fermented by selected lactobacillus helveticus strains. J. Dairy Sci. 2016, 99, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Morotomi, M.; Watanabe, T.; Suegara, N.; Kawai, Y.; Mutai, M. Distribution of indigenous bacteria in the digestive tract of conventional and gnotobiotic rats. Infect. Immun. 1975, 11, 962–968. [Google Scholar] [PubMed]
- Turovskiy, Y.; Noll, K.S.; Chikindas, M.L. The aetiology of bacterial vaginosis. J. Appl. Microbiol. 2011, 110, 1105–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Arango, L.F.; Barrett, H.L.; McIntyre, H.D.; Callaway, L.K.; Morrison, M.; Nitert, M.D.; Grp, S.T. Connections between the gut microbiome and metabolic hormones in early pregnancy in overweight and obese women. Diabetes 2016, 65, 2214–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasai, C.; Sugimoto, K.; Moritani, I.; Tanaka, J.; Oya, Y.; Inoue, H.; Tameda, M.; Shiraki, K.; Ito, M.; Takei, Y.; et al. Comparison of the gut microbiota composition between obese and non-obese individuals in a japanese population, as analyzed by terminal restriction fragment length polymorphism and next-generation sequencing. BMC Gastroenterol. 2015, 15, 100. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riviere, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and butyrate-producing colon bacteria: Importance and strategies for their stimulation in the human gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.D.; Van Domselaar, G.; Bernstein, C.N. The gut microbiota in immune-mediated inflammatory diseases. Front. Microbiol. 2016, 7, 1081. [Google Scholar] [CrossRef]
- Faridani, O.R.; Nikravesh, A.; Pandey, D.P.; Gerdes, K.; Good, L. Competitive inhibition of natural antisense Sok-RNA interactions activates Hok-mediated cell killing in Escherichia coli. Nucleic Acids Res. 2006, 34, 5915–5922. [Google Scholar] [CrossRef]
- Leung, A.; Tsoi, H.; Yu, J. Fusobacterium and Escherichia: Models of colorectal cancer driven by microbiota and the utility of microbiota in colorectal cancer screening. Expert. Rev. Gastroentrol. Hepatol. 2015, 9, 651–657. [Google Scholar] [CrossRef]
- Zuo, T.; Wong, S.H.; Cheung, C.P.; Lam, K.; Lui, R.; Cheung, K.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Wu, J.C.Y.; et al. Gut fungal dysbiosis correlates with reduced efficacy of fecal microbiota transplantation in clostridium difficile infection. Nat. Commun. 2018, 9, 3663. [Google Scholar] [CrossRef]
- Mutschler, H.; Meinhart, A. Epsilon/zeta systems: Their role in resistance, virulence, and their potential for antibiotic development. J. Mol. Med. 2011, 89, 1183–1194. [Google Scholar] [CrossRef]
- Shapiro, S. Speculative strategies for new antibacterials: All roads should not lead to rome. J. Antibiot. 2013, 66, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Koczulla, A.R.; Bals, R. Antimicrobial peptides—Current status and therapeutic potential. Drugs 2003, 63, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, A.; Neundorf, I. Design and application of antimicrobial peptide conjugates. Int. J. Mol. Sci. 2016, 17, 701. [Google Scholar] [CrossRef] [PubMed]
- Brezden, A.; Mohamed, M.F.; Nepal, M.; Harwood, J.S.; Kuriakose, J.; Seleem, M.N.; Chmielewski, J. Dual targeting of intracellular pathogenic bacteria with a cleavable conjugate of kanamycin and an antibacterial cell-penetrating peptide. J. Am. Chem. Soc. 2016, 138, 10945–10949. [Google Scholar] [CrossRef] [PubMed]
- Wadhwani, P.; Heidenreich, N.; Podeyn, B.; Burck, J.; Ulrich, A.S. Antibiotic gold: Tethering of antimicrobial peptides to gold nanoparticles maintains conformational flexibility of peptides and improves trypsin susceptibility. Biomater. Sci. 2017, 5, 817–827. [Google Scholar] [CrossRef]
- Fawell, S.; Seery, J.; Daikh, Y.; Moore, C.; Chen, L.L.; Pepinsky, B.; Barsoum, J. Tat-mediated delivery of heterologous proteins into cells. Proc. Natl. Acad. Sci. USA 1994, 91, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Lewin, M.; Carlesso, N.; Tung, C.H.; Tang, X.W.; Cory, D.; Scadden, D.T.; Weissleder, R. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat. Biotechnol. 2000, 18, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Luong, H.X.; Kim, D.H.; Lee, B.J.; Kim, Y.W. Antimicrobial activity and stability of stapled helices of polybia-mp1. Arch. Pharm. Res. 2017, 40, 1414–1419. [Google Scholar] [CrossRef] [PubMed]
- Luong, H.X.; Kim, D.H.; Mai, N.T.; Lee, B.J.; Kim, Y.W. Mono-substitution effects on antimicrobial activity of stapled heptapeptides. Arch. Pharm. Res. 2017, 40, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Patgiri, A.; Jochim, A.L.; Arora, P.S. A hydrogen bond surrogate approach for stabilization of short peptide sequences in alpha-helical conformation. Acc. Chem. Res. 2008, 41, 1289–1300. [Google Scholar] [CrossRef]
- Perros, M. Infectious disease. A sustainable model for antibiotics. Science 2015, 347, 1062–1064. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garcia, L.; Blasco, L.; Lopez, M.; Bou, G.; Garcia-Contreras, R.; Wood, T.; Tomas, M. Toxin-antitoxin systems in clinical pathogens. Toxins 2016, 8, 227. [Google Scholar] [CrossRef] [PubMed]
- Schuijt, T.J.; van der Poll, T.; de Vos, W.M.; Wiersinga, W.J. The intestinal microbiota and host immune interactions in the critically ill. Trends Microbiol. 2013, 21, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Wahlstrom, A.; Sayin, S.I.; Marschall, H.U.; Backhed, F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Greenhill, C. Metabolism intestinal microbiota affects host physiology. Nat. Rev. Endocrinol. 2017, 13, 64. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, P.; Araujo, J.R.; Di Santo, J.P. A cross-talk between microbiota-derived short-chain fatty acids and the host mucosal immune system regulates intestinal homeostasis and inflammatory bowel disease. Inflamm. Bowel Dis. 2018, 24, 558–572. [Google Scholar] [CrossRef]
- Goldsmith, J.R.; Sartor, R.B. The role of diet on intestinal microbiota metabolism: Downstream impacts on host immune function and health, and therapeutic implications. J. Gastroenterol. 2014, 49, 785–798. [Google Scholar] [CrossRef]
- Samuelson, D.R.; Welsh, D.A.; Shellito, J.E. Regulation of lung immunity and host defense by the intestinal microbiota. Front. Microbiol. 2015, 6, 1085. [Google Scholar] [CrossRef]
- Djuric, Z. Obesity-associated cancer risk: The role of intestinal microbiota in the etiology of the host proinflammatory state. Transl. Res. 2017, 179, 155–167. [Google Scholar] [CrossRef]
- Chowdhury, N.; Kwan, B.W.; Wood, T.K. Persistence increases in the absence of the alarmone guanosine tetraphosphate by reducing cell growth. Sci. Rep. 2016, 6, 20519. [Google Scholar] [CrossRef]





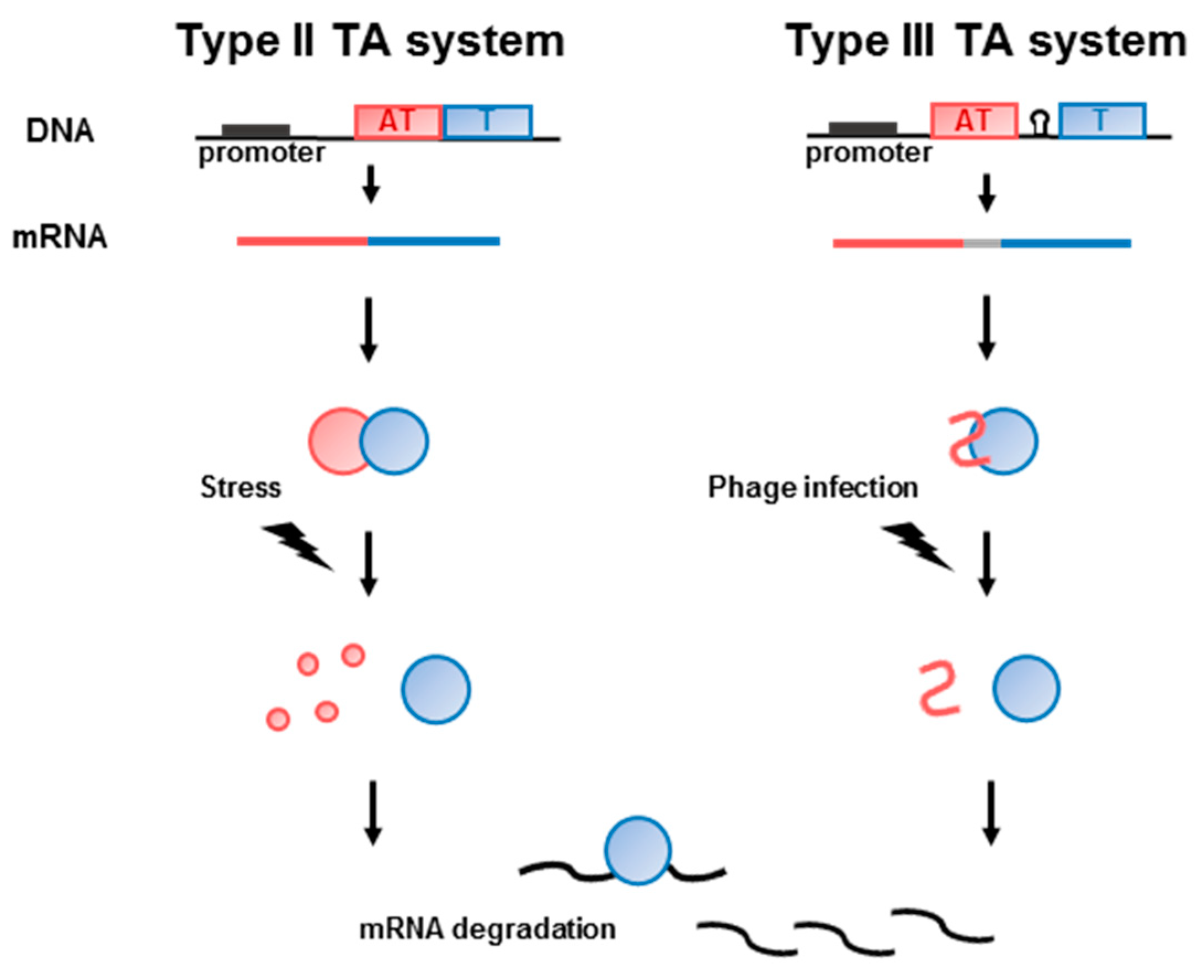{kind=link}
{kind=link}
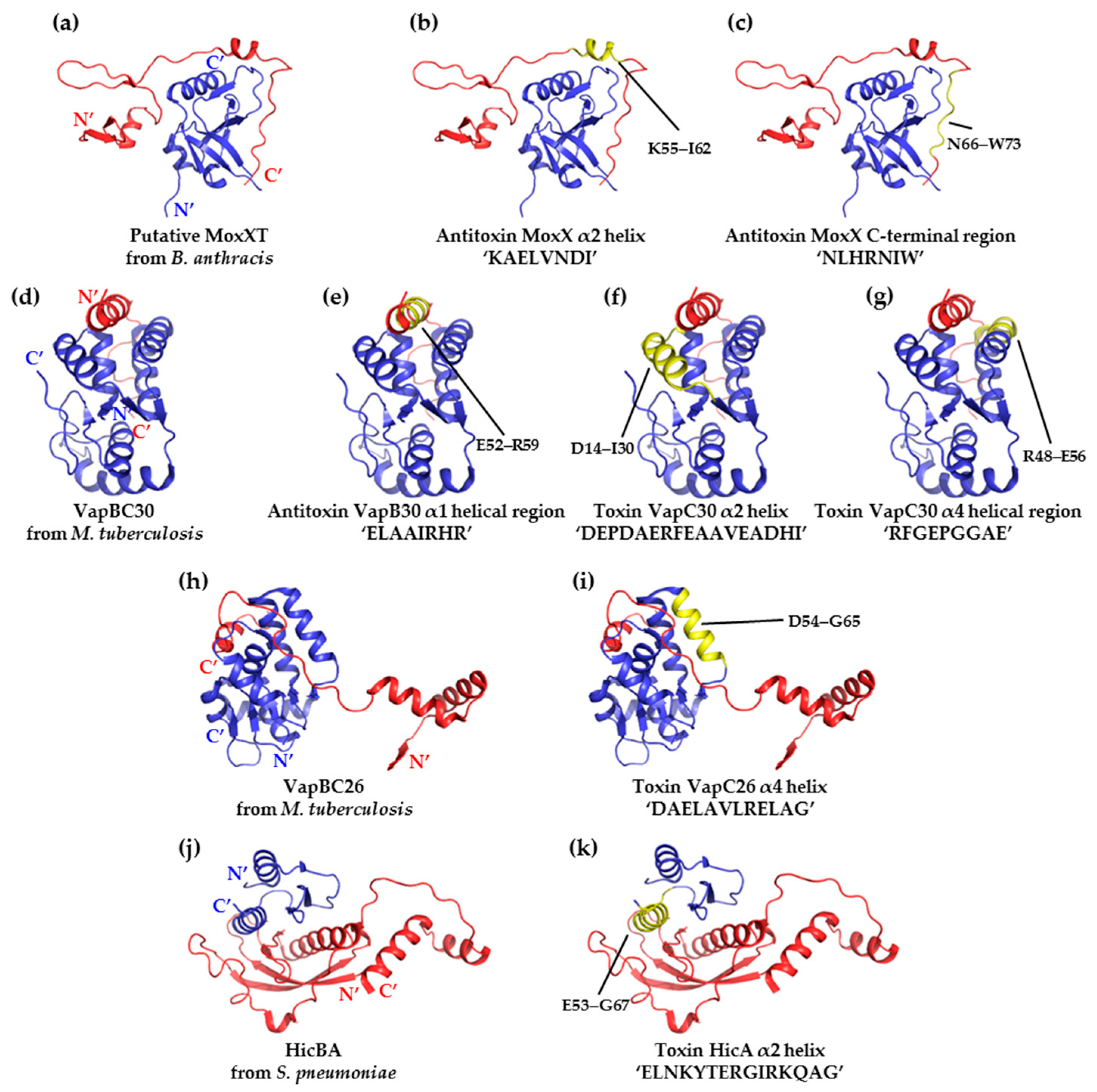{kind=link}
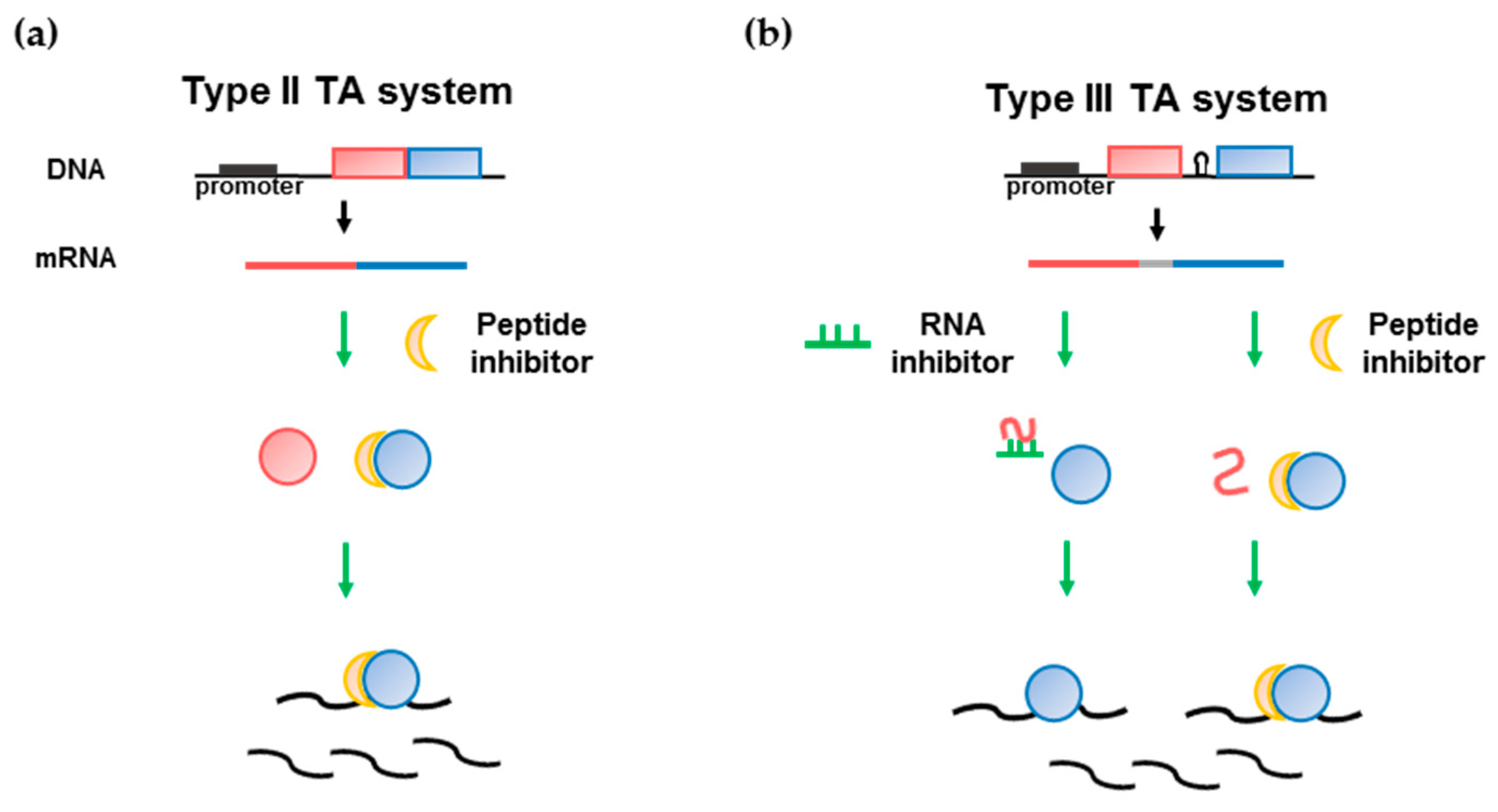{kind=link}
| Pathogenic Bacteria | TA Pair (Antitoxin/Toxin) | Reported Structure | PDB Code | Ref. |
|---|---|---|---|---|
| Staphylococcus aureus | MazE/MazF | Toxin MazF | 4MZM | [70] |
| RelB/RelE (2 distinct loci) | ||||
| Klebsiella pneumoniae | HipB/HipA (2 distinct loci) | |||
| MazE/MazF | ||||
| Phd/Doc | ||||
| RelB/RelE (3 distinct loci) | ||||
| VapB/VapC | ||||
| Pseudomonas aeruginosa | RelB/RelE | |||
| VapB/VapC | ||||
| Mycobacterium tuberculosis | HigA/HigB (2 distinct loci) | |||
| MazE/MazF (9 distinct loci) | Toxin MazF3 | 5CCA | [71] | |
| Complex MazEF4 | 5XE3 | [32] | ||
| Toxin MazF6 | 5HKC | |||
| Toxin MazF7 | 5WYG | [72] | ||
| Toxin MazF9 | 5HJZ | |||
| ParD/ParE (2 distinct loci) | ||||
| RelB/RelE (3 distinct loci) | Complex RelBE2 | 3G5O | ||
| Complex RelBE3 | 3OEI | |||
| VapB/VapC (51 distinct loci) | Complex VapBC2 | 3H87 | [73] | |
| Complex VapBC5 | 3DBO | [74] | ||
| Complex VapBC15 | 4CHG | [75] | ||
| Toxin VapC20 | 5WZF | [76] | ||
| Toxin VapC21 | 5SV2 | [77] | ||
| Complex VapBC26 | 5X3T | [31] | ||
| Complex VapBC30 | 4XGQ | [36] | ||
| Antitoxin VapB45 | 5AF3 | |||
| Streptococcus pneumoniae | HicB/HicA | Complex HicBA | 5YRZ | [37] |
| HigA/HigB | ||||
| RelB/RelE (3 related loci) | [78] | |||
| Phd/Doc | ||||
| Salmonella typhimurium | HigA/HigB | |||
| RelB/RelE (9 distinct loci) | ||||
| Phd/Doc | ||||
| VapB/VapC |
| Target System (PDB Code) | Region Being Mimicked (Residue Range) | Peptide Sequence | % Inhibition |
|---|---|---|---|
| MoxXT (MazEF) from B. anthracis (using structural homolog 1UB4) | Putative α2 helix of the toxin MazF (55–62) | KAELVNDI | 22 |
| Putative C-terminal toxin-binding region of the antitoxin MazE (66–73) | NLHRNIW | 20 | |
| VapBC30 from M. tuberculosis (4XGQ) | α1 helical region of the antitoxin VapB30 (52–59) | ELAAIRHR | 43 |
| α2 helix of the toxin VapC30 (14–30) | DEPDAERFEAAVEADHI | 53 | |
| α4 helical region of the toxin VapC30 (48-56) | RFGEPGGRE | 73 | |
| VapBC26 from M. tuberculosis (5X3T) | α4 helix of the toxin VapC26 (54–65) | DAELAVLRELAG | 82 |
| HicBA from S. pneumoniae (5YRZ) | α2 helix of the toxinHicA (53-67) | ELNKYTERGIRKQAG | 80 |
| Strain | Family | Length T/A (Repeat) | Related Functions or Diseases |
|---|---|---|---|
| Marvinbryantia formatexigens DSM 14469 | toxIN | 172/34 (2.9) | Acetogenesis [117,118] |
| toxIN | 182/38 (2.1) | ||
| cptIN | 161/47 (2) | ||
| cptIN | 66/45 (2) | ||
| Clostridium hiranonis DSM 13275 | cptIN | 157/45 (2.2) | Colorectal cancer Gastric cancer [121,122,123,132] |
| tenpIN | 158/55 (2.1) | ||
| Clostridium nexile DSM 1787 | toxIN | 129/38 (2.2) | |
| Clostridium sp. HGF2 | toxIN | 139/46 (2.1) | |
| cptIN | 161/47 (2.2) | ||
| Coprobacillus sp. 29_1 | toxIN | 163/38 (2.2) | Irritable bowel syndrome [124] |
| Coprococcus catus GD/7 | cptIN | 160/46 (2.2) | Irritable bowel syndrome Obesity [124,143,144] |
| Coprococcus sp. ART55/1 | toxIN | 181/37 (3.4) | |
| Eubacterium rectale ATCC 33656 | cptIN | 162/45 (2.1) | Inflammatory bowel disease Diabetes Macular degeneration Obesity [125,126,127,145,146] |
| cptIN | 158/46 (2.2) | ||
| Eubacterium rectale DSM 17629 | toxIN | 201/38 (2.1) | |
| cptIN | 162/45 (2.1) | ||
| Eubacterium rectale M104/1 | toxIN | 201/38 (2.1) | |
| Eubacterium ventriosum ATCC 27560 | cptIN | 162/46 (2.2) | |
| Fusobacterium sp. 2_1_31 | cptIN | 159/40 (2.9) | Inflammatory bowel disease Colorectal cancer Gastric cancer [128,129,130,131,132] |
| Fusobacterium sp. 3_1_33 | cptIN | 158/41 (3) | |
| tenpIN | 140/41 (3) | ||
| Fusobacterium sp. 3_1_36A2 | tenpIN | 144/53 (2.1) | |
| Fusobacterium sp. 3_1_5R | toxIN | 174/39 (2) | |
| toxIN | 178/38 (3.3) | ||
| toxIN | 189/35 (3.2) | ||
| Fusobacterium sp. 4_1_13 | toxIN | 179/39 (2) | |
| tenpIN | 144/53 (2.1) | ||
| Fusobacterium sp. 7_1 | cptIN | 156/40 (3.1) | |
| Fusobacterium sp. D11 | cptIN | 158/40 (3.1) | |
| Fusobacterium sp. D12 | toxIN | 173/39 (2) | |
| Fusobacterium ulcerans ATCC 49185 | toxIN | 166/35 (3.2) | |
| Lachnospiraceae bacterium 2_1_46FAA | toxIN | 163/38 (3) | Colorectal cancer Crohn’s disease Obesity [133,134,135,136,137,143,144] |
| toxIN | 163/38 (3.2) | ||
| Lachnospiraceae bacterium 4_1_37FAA | toxIN | 163/38 (3.2) | |
| Lachnospiraceae bacterium 5_1_63FAA | cptIN | 162/46 (2.2) | |
| Lachnospiraceae bacterium 8_1_57FAA | toxIN | 163/38 (3.2) | |
| Lachnospiraceae bacterium 9_1_43BFAA | cptIN | 54/45 (2.2) | |
| Lactobacillus helveticus DSM 20075 | toxIN | 124/37 (1.9) | Immune enhancement [44,138,139,140] Antitumor |
| Phascolarctobacterium sp. YIT 12067 | cptIN | 162/46 (2.1) | ATP synthesis [119,120] |
| Roseburia intestinalis M50/1 | toxIN | 146/39 (3.2) | Colorectal cancer Crohn’s disease [133,134,135,136,137,145,146] Diabetes |
| Roseburia intestinalis XB6B4 | toxIN | 166/39 (3.2) | |
| Ruminococcus lactaris ATCC 29176 | cptIN | 162/46 (2.2) | Rheumatoid arthritis, Colorectal cancer, Crohn’s disease, obesity Macular degeneration [125,126,127,133,134,135,136,137,147] |
| Ruminococcus sp. 5_1_39B_FAA | toxIN | 178/36 (2.1) | |
| Ruminococcustorques ATCC 27756 | toxIN | 163/38 (3.2) | |
| Ruminococcus torques L2-14 | cptIN | 162/46 (2.2) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, S.-M.; Kim, D.-H.; Jin, C.; Lee, B.-J. A Systematic Overview of Type II and III Toxin-Antitoxin Systems with a Focus on Druggability. Toxins 2018, 10, 515. https://doi.org/10.3390/toxins10120515
Kang S-M, Kim D-H, Jin C, Lee B-J. A Systematic Overview of Type II and III Toxin-Antitoxin Systems with a Focus on Druggability. Toxins. 2018; 10(12):515. https://doi.org/10.3390/toxins10120515
Chicago/Turabian StyleKang, Sung-Min, Do-Hee Kim, Chenglong Jin, and Bong-Jin Lee. 2018. "A Systematic Overview of Type II and III Toxin-Antitoxin Systems with a Focus on Druggability" Toxins 10, no. 12: 515. https://doi.org/10.3390/toxins10120515
APA StyleKang, S. -M., Kim, D. -H., Jin, C., & Lee, B. -J. (2018). A Systematic Overview of Type II and III Toxin-Antitoxin Systems with a Focus on Druggability. Toxins, 10(12), 515. https://doi.org/10.3390/toxins10120515