1. Introduction
The amnesic shellfish poisoning (ASP) toxin, domoic acid, is produced by some species of the genera
Pseudo-nitzschia and
Nitzschia [
1,
2]. The prevalence of domoic acid and toxic diatoms seems to have increased worldwide [
1]. Domoic acid is a tricarboxylic amino acid that resembles glutamic acid and is a potent glutamate receptor agonist [
3,
4]. Bivalve mollusks are filter feeders, and during harmful algal blooms, they can accumulate toxins in their tissues. This is why they are the primary vectors for causing ASP in humans [
1,
5,
6].
Domoic acid depuration time in bivalves is species-specific and can differ largely from one species to another [
7]. Mussels of the genus
Mytilus [
8,
9,
10,
11] and the oyster
Crassostrea gigas [
9] rapidly eliminate domoic acid, while the king scallop
Pecten maximus [
7] and the razor clam
Siliqua patula [
12] are very slow domoic acid depurators. In mussels and scallops, the digestive gland is the tissue with the highest domoic acid concentration [
7,
10,
13,
14]. Mauriz and Blanco [
15] suggested that the very low depuration rate of
P. maximus could be due to the lack of an efficient transmembrane transporter. Unlike the king scallop, in the queen scallop (
Aequipecten opercularis) the depuration rate is fast [
15].
Domoic acid is excitotoxic in the central nervous system of mammals and other vertebrates [
3,
4], but its putative effects on invertebrates have been less studied. Although it seems that domoic acid-producing organisms are not toxic to shellfish (or at least not highly toxic), they can exert several physiological and sublethal effects on marine bivalves [
16,
17,
18,
19,
20]. Some of these effects include DNA damage in mussels [
16], stress response characterized by shell closure, hemolymph acidosis, hypoxia, an increase in the number and activity of hemocytes in the oyster
C. gigas [
17,
18], reduced larval growth in
P. maximus [
19], and negative impacts on growth rate and survival in juvenile king scallops [
20]. Some authors have suggested that although several harmful algae toxins do not affect the survival of bivalve mollusks, they provoke oxidative stress [
21,
22,
23]. However, the molecular mechanisms that cause oxidative stress are poorly understood, and furthermore, domoic acid causes oxidative stress in the vertebrate central nervous system [
24,
25,
26,
27]. The works cited above [
16,
17,
18,
19,
20,
21] analyzed several physiological and biochemical parameters after exposure to domoic acid but did not study the gene expression patterns in both exposed and non-exposed bivalves.
There are some publications regarding the gene expression changes associated with exposure to domoic acid in vertebrates [
24,
28,
29,
30]. The transcriptome response was dependent on the dose and the exposure duration; among the differentially expressed genes were those involved in transcription (transcription factors), signal transduction, ion transport, generalized stress response, mitochondrial function, inflammatory response, DNA damage, apoptosis, neurological function, and neuroprotection [
24,
28,
29,
30]. In a previous work, we studied (by RNA-seq) the effects of domoic acid-containing
Pseudo-nitzschia on gene expression in the mussel
Mytilus galloprovincialis [
31], and to our knowledge, this is the only published work about the transcriptional effects of domoic acid exposure on mollusks. As stated before, among the bivalves there are large interspecific differences in the domoic acid depuration rate [
7,
8,
9,
10,
11,
12]. It is therefore necessary to carry out gene expression studies on different species.
Understanding the molecular mechanisms of domoic acid uptake and elimination in bivalves and how the toxin (and the toxin-producing species) affects gene expression are two knowledge gaps in this field. The aim of the present work is to contribute to filling these gaps by means of a transcriptomic approach. First, the whole transcriptome of the
A. opercularis digestive gland was de novo assembled, and then, we analyzed by RNA-seq the transcriptional changes after exposure to domoic acid-producing
Pseudo-nitzschia. This approach can provide some clues regarding the biological and molecular processes altered by domoic acid. The transcriptomic approach has been successfully employed to uncover the genetic response of bivalves to diarrhetic shellfish poisoning (DSP) and paralytic shellfish poisoning (PSP) toxins and also to identify the genes putatively involved in detoxification processes [
32,
33,
34,
35,
36].
3. Discussion
There are many studies about the mechanisms of the neurotoxicity of domoic acid in vertebrates (especially mammals), but there is very little knowledge about the putative effects of domoic acid on bivalve mollusks. Several publications showed that domoic acid can exert physiological and sublethal effects on marine bivalves [
16,
17,
18,
19,
20]. Dizer et al. [
16] found that in
Mytilus edulis, DNA damage was significantly increased after the injection of domoic acid and suggested the existence of genotoxic responses in the cells of digestive glands. It is interesting to point out that DNA repair, cellular response to DNA damage stimulus, and cellular response to stress were among the enriched GO terms in the present work for the up-regulated DEGs (
File S4). This could be the transcriptomic response of
A. opercularis to the putative DNA damage provoked by domoic acid. In
C. gigas, domoic acid provoked a generalized stress response [
17] and an increase in the number and activity of hemocytes [
18]. Domoic acid induces oxidative stress in the central nervous system and spinal cord in vertebrates [
24,
25,
26,
27]. Furthermore, harmful algae toxins sometimes provoke oxidative stress in bivalves [
21,
22,
23], and Prego-Faraldo et al. [
40] found that exposure to the toxic dinoflagellate
Prorocentrum lima induces the differential expression of genes coding for antioxidant enzymes. The glutathione S-transferase, thioredoxin, glutaredoxin, and copper/zinc superoxide dismutase Pfam domains were functionally enriched in queen scallops (
File S3), and these genes were, for the most part, up-regulated; these domains are found in proteins involved in protection against reactive oxygen species (ROS).
In a recent work about the effects of environmental stress on gene transcription in oysters, Anderson et al. [
41] proposed a consensus model of sub-cellular stress responses in oysters with the involvement of mitochondria and reactive oxygen species (ROS) production. If the anti-oxidant enzymes and molecular chaperones cannot limit the damage caused by ROS, then the consequences are probably cellular dysfunction and apoptosis [
41]. In vertebrates, domoic acid causes mitochondrial dysfunction as a consequence of oxidative stress [
4,
24,
26]. Hiolski et al. [
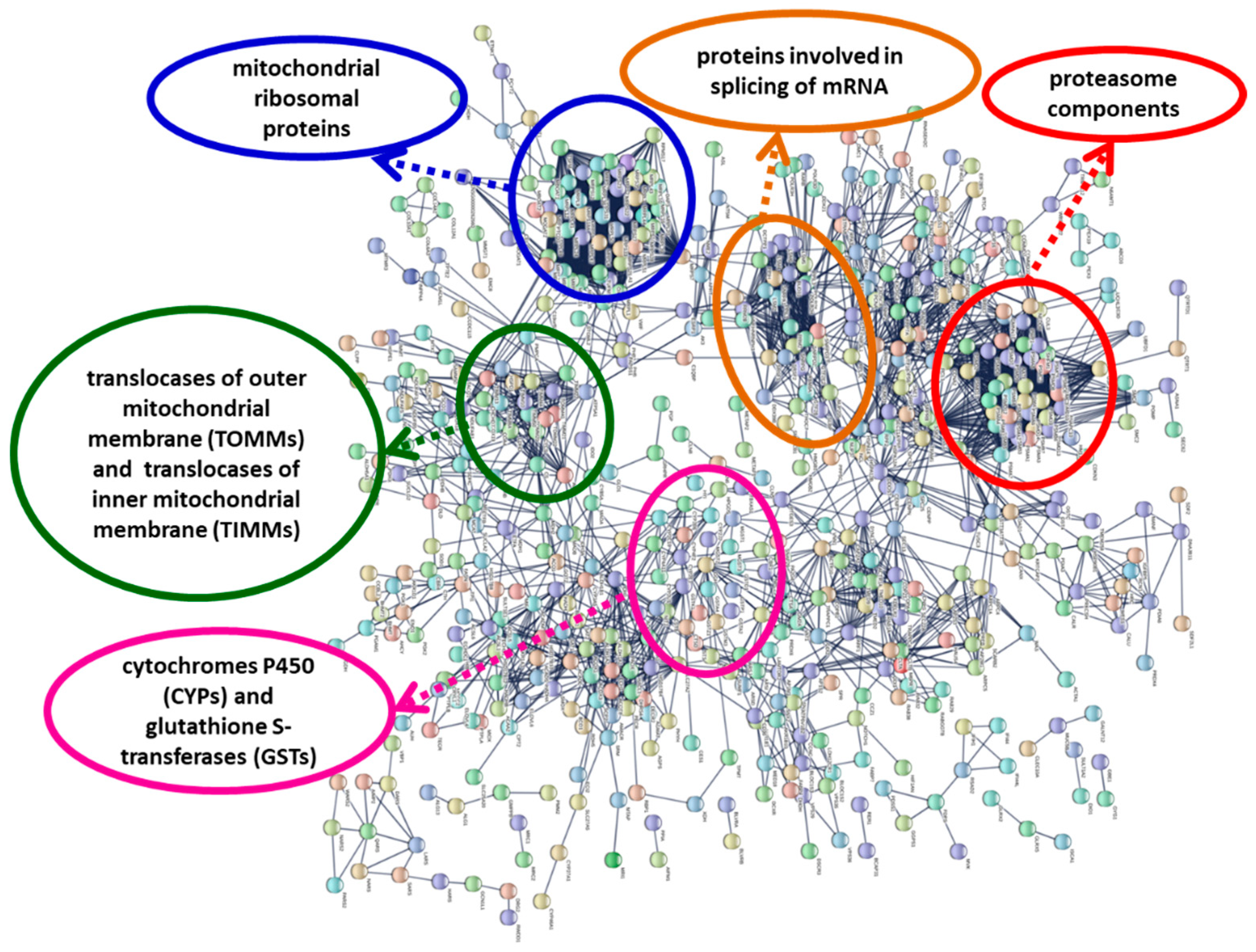
24] suggested the existence of compensatory mitochondrial biogenesis in response to mitochondrial dysfunction. Our results (
Figure 3;
Figure S1; File S2,S6) showed an up-regulation of genes coding for mitochondrial ribosomal proteins and translocases of the outer and inner mitochondrial membrane (proteins involved in mitochondrial biogenesis); in the protein interaction network, these proteins form highly connected protein nodes (
Figure 3;
Figure S1)
Up-regulation of proteasome subunits (
Figure 3;
Figure S1; File S2) is also a possible consequence of oxidative stress [
42], because the proteasome is responsible for the selective degradation of oxidized proteins [
43]. The 26S proteasome is a protease complex, which is responsible for the regulated degradation of proteins in eukaryotic organisms [
42]. The proteasome system can be activated to accomplish the destruction of proteins altered by stress conditions [
44]. The proteasome complex and proteasome core complex were two of the most enriched GO terms in the cellular component category for the up-regulated DEGs (
Table 7), and the proteasome Pfam domain (PF00227) was also enriched (
File S3); the genes coding for proteins with this domain were up-regulated in
Pseudo-nitzschia-exposed scallops (
Files S2 and S3). Proteasome proteins form a group of highly connected nodes in the protein–protein interaction network (
Figure 3,
Figure S1). In
M. galloprovincialis exposed to the toxin okadaic acid, there is an up-regulation of several mRNAs involved in proteasome activity [
45]. Therefore, the results support the hypothesis that exposure to domoic acid-producing
Pseudo-nitzschia causes oxidative stress and the impairment of the mitochondrial function in
A. opercularis and that the transcriptional changes are directed, at least in part, to counteract the stress effects.
The metabolism of xenobiotics (such as toxins) has three phases; phase I (functionalization) and phase II (conjugation) are catalyzed by metabolizing enzymes, while phase III consists of the export from the cell by transmembrane transporter proteins. We found that the Pfam domains of some phase I (cytochromes P450 and aldo-keto reductases) and phase II (glutathione S-transferases and sulfotransferases) drug metabolizing enzymes were functionally enriched, and the genes coding for these enzymes were mostly up-regulated. Cytochromes P450 and glutathione S-transferases constituted a group of highly connected nodes in the protein interaction network (
Figure 3,
Figure S1). Genes of these families were also up-regulated in mussels (
M. galloprovincialis) exposed to domoic acid-containing
Pseudo-nitzschia [
31]. Peña-Llopis et al. [
46] showed that the treatment of scallops (
P. maximus) with N-acetylcysteine increased glutathione S-transferase (GST) activity and enabled the scallops to eliminate domoic acid more efficiently. Therefore, it is possible that glutathione S-transferases play a role in domoic acid detoxification. Li et al. [
35] found an up-regulation of sulfotransferase genes in the kidney of the Zhikong scallop
Chlamys farreri after exposure to paralytic shellfish toxin-producing
Alexandrium minutum. Furthermore, the family of sulfotransferases is significantly expanded in the
C. farreri genome [
35]; the high number of transcripts coding for sulfotransferases in the
A. opercularis transcriptome is indicative of an expansion of this family in the queen scallop.
The molecular mechanisms of domoic acid absorption and excretion in bivalve mollusks are poorly understood [
31]. Mauriz and Blanco [
15] found that in the king scallop
P. maximus, domoic acid is free in the cytosol of the digestive gland and suggested that the low depuration rate of domoic acid in this species could be due to the lack of membrane transporters. Domoic acid is a charged compound and probably needs a transport protein to pass through the plasma membrane, as is the case with glutamic acid [
47,
48]. This putative transmembrane transporter(s) could therefore play an important role in the absorption and/or the excretion of domoic acid. The results of Kimura et al. [
47] suggest that anion exchange transporters are responsible for the transmembrane transport of domoic acid in Caco-2 cell monolayers (which represent the intestinal barrier of mammals); these transporters belong to the solute carrier (
SLC) superfamily. In
A opercularis, we found that transmembrane transport and transmembrane transporter activity were two of the enriched GO terms (
File S4); furthermore, the major facilitator superfamily (MFS) was one of the most significantly enriched Pfam families (
Table 6). Most of the genes belonging to these categories were up-regulated (
Table 6;
Files S2–S4). MFS is a clan of the SLC superfamily [
49], and Hediger et al. [
50] reported that the
SLC gene series included 52 families in the human genome, although it has recently been updated to 65 families [
51]. A total of eight
SLC families (
SLC5,
SLC16,
SLC17,
SLC21,
SLC22,
SLC26,
SLC39, and
SLC49) contain up-regulated genes in
A. opercularis (
File S8) and in
M. galloprovincialis [
31] exposed to domoic acid-containing
Pseudo-nitzschia. The transporter protein(s) putatively involved in the uptake and/or elimination of domoic acid in the digestive gland of bivalve mollusks could be encoded by a gene from one of those families. The families with a higher number of up-regulated genes in both
A. opercularis and
M. galloprovincialis were
SLC16 (the monocarboxylate transporters family) and
SLC22 (organic cation/anion/zwitterion transporters). A total of four members of the human
SLC16 gene family encode monocarboxylate transporters, but the substrates of several members are unknown [
52]. The
SLC22 family [
53] comprises organic cation, zwitterion, and anion transporters (OCTs, OCTNs, and OATs), which participate in the absorption (in the small intestine) and excretion (in the liver and kidney) of xenobiotics and endogenous substances [
53]. Unfortunately, the lack of knowledge about the identity of the domoic acid transmembrane transporter(s) in mammals makes it difficult to identify them in bivalve mollusks. Schultz et al. [
54] suggested that ATP-binding cassette (ABC) transporters are responsible for the absorption of domoic acid in Dungeness crabs, but in
A. opercularis, we found only five up-regulated ABC transporters. A similar result was reported by Pazos et al. [
31] in
M. galloprovincialis (with two up-regulated ABC transporters). This contrasts with the high number of up-regulated
SLC genes found in both bivalves.
Although most of the
SLC genes differentially expressed in
A. opercularis were up-regulated, the
SLC6 family (the sodium- and chloride-dependent neurotransmitter transporter family) is an exception, with 48 down-regulated unigenes (
File S8). Furthermore, neurotransmitter/sodium symporter activity is one of the most enriched GO terms for the down-regulated genes (
Table 8). On the contrary, in
M. galloprovincialis, two genes from this family were up-regulated and none down-regulated [
31]. In
A. opercularis, the number of genes in this family is very high, and this agrees with results from Li et al. [
35], who found that the
SLC6 family is expanded in the
C. farreri (a scallop) genome in relation to other bivalves.
Harmful algae and biotoxins exert different effects on the immune systems of bivalve mollusks [
23,
32,
55]. Immune response and immune system process were two of the most enriched GO terms for the down-regulated DEGs (
File S4), and Pfam families involved in immunological processes were significantly enriched in
A. opercularis (
Table 6;
File S3): the C1q domain-containing proteins, the C-type lectin, the fibrinogen beta and gamma chains C-terminal globular domain, the immunoglobulin domain, and tumor necrosis factors (TNF). Except for the C-type lectins, the genes in these categories were mainly down-regulated (
Table 6;
Files S2 and S3). Differentially expressed genes from these families have been found in several bivalve mollusks after exposure to different biotoxins [
31,
32,
33,
34,
36,
56,
57]. Hégaret et al. [
23] found that some harmful algae provoked a stimulation of immune function of bivalve hemocytes, while others were immunosuppressive. The C1q domain containing proteins and the C-type lectins are particularly abundant in the digestive glands of bivalves [
58,
59]. The C1q domain-containing proteins are indispensable in the innate immune systems of invertebrates [
60] and could be involved in several functions, such as activation of the complement pathway, cell adhesion, pathogen recognition, response to pollutants, and apoptosis [
58,
60,
61]. An expansion of the genes coding for proteins containing the C1q domain was found in several bivalves [
58,
61,
62,
63]. For example, 321 C1q domain-containing proteins are encoded by the
C gigas genome [
62]; this represents approximately 10-fold more than the Ciq proteins encoded by the
Homo sapiens genome [
62]. Some genes coding for C1q domain-containing proteins were down-regulated in
M. galloprovincialis fed with toxigenic strains of
Alexandrium minutum [
34]. The C-type lectins are characterized by a calcium-dependent carbohydrate recognition domain and participate in pathogen recognition and in innate immunity in bivalves [
59], but they can also perform non-immune functions; for example, a role in efficient food particle sorting (food recognition) was found in the oyster
Crassostrea virginica [
64]. There is a high number of genes coding for C-type lectins in bivalve mollusks [
59,
65]. Most of the genes coding for C-type lectins were up-regulated in
A opercularis (
Table 6;
Files S2 and S3) and
M. galloprovincialis [
31] after exposure to domoic acid-producing
Pseudo-nitzschia; this agrees with the up-regulation of C-type lectins in
M. chilensis after exposure to saxitoxin [
33,
56,
57]. On the contrary, these genes were down-regulated in
Argopecten irradians in response to okadaic acid [
32].
One of the most enriched GO terms for the down-regulated DEGs in
A. opercularis was collagen trimer (
Table 8), and collagen triple helix repeat was among the enriched Pfam domains (
Table 6). Furthermore, several collagen components form a group of highly connected protein nodes in the network obtained with the down-regulated genes (
Figure S2). In
M. galloprovincialis, after exposure to domoic acid-containing
Pseudo-nitzschia [
31], some collagen genes (7) were down-regulated, although the number of induced genes was greater (13). Collagens are components of the extracellular matrix characterized by the presence of at least one triple-helical domain [
66]. They are among the most abundant proteins and have mainly a structural function [
66].
Another group of predominantly down-regulated genes (14 up-regulated and 35 down-regulated,
File S2) were those coding for heat shock proteins (HSPs). Half of the induced
HSP genes were mitochondrial forms, and among the repressed ones, the
HSP70 genes predominated. Heat shock proteins are involved in protein folding and can be induced by several types of stress, including high temperature, toxins, pathogens, and hypoxia [
67]. Several publications have reported the increased expression of heat shock protein genes in bivalves after exposure to harmful algae toxins [
32,
57,
67,
68,
69]. Cheng et al. [
67] found an expansion of
Hsp70 (heat shock protein 70 kDa) genes from the
Hspa12 subfamily in
Mizuhopecten yessoensis. Several of these genes were differentially expressed in response to
Alexandrium catenella exposure (most of them were induced, but there were also some
Hsp70 genes down-regulated [
67]). However, Ryan et al. [
29] reported the down-regulation of
Hsp68 (a member of the
Hsp70 family) after domoic acid exposure in mouse brain. Furthermore, the exposure to domoic acid-producing
Pseudo-nitzschia provoked a down-regulation of
HSPs in
M. galloprovincialis [
31]; these results were coincident with those obtained in the present work.
Another result worth highlighting is that some genes coding for glutamate ionotropic receptors, two genes coding for NMDA receptors, and five coding for kainate (KA) receptors, were all down-regulated in the present study (
File S2). The zebrafish
gria2 gene (glutamate ionotropic receptor AMPA 2) was down-regulated after two weeks of low-level domoic acid exposure [
24], and the authors suggested that this down-regulation is a compensatory response to elevated glutamatergic activity [
24]. It is possible that a similar compensatory mechanism takes place in queen scallops after exposure to domoic acid.
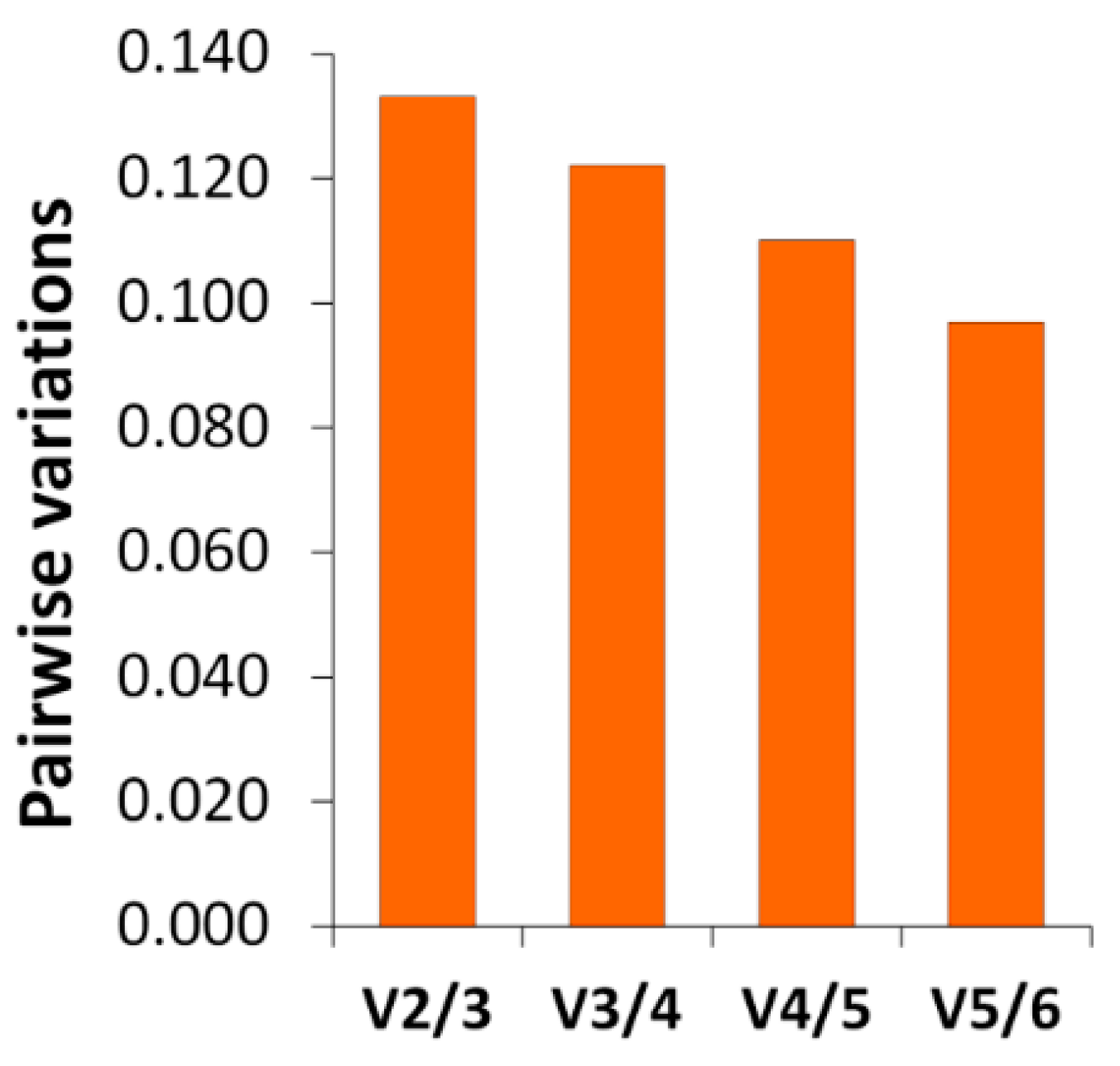
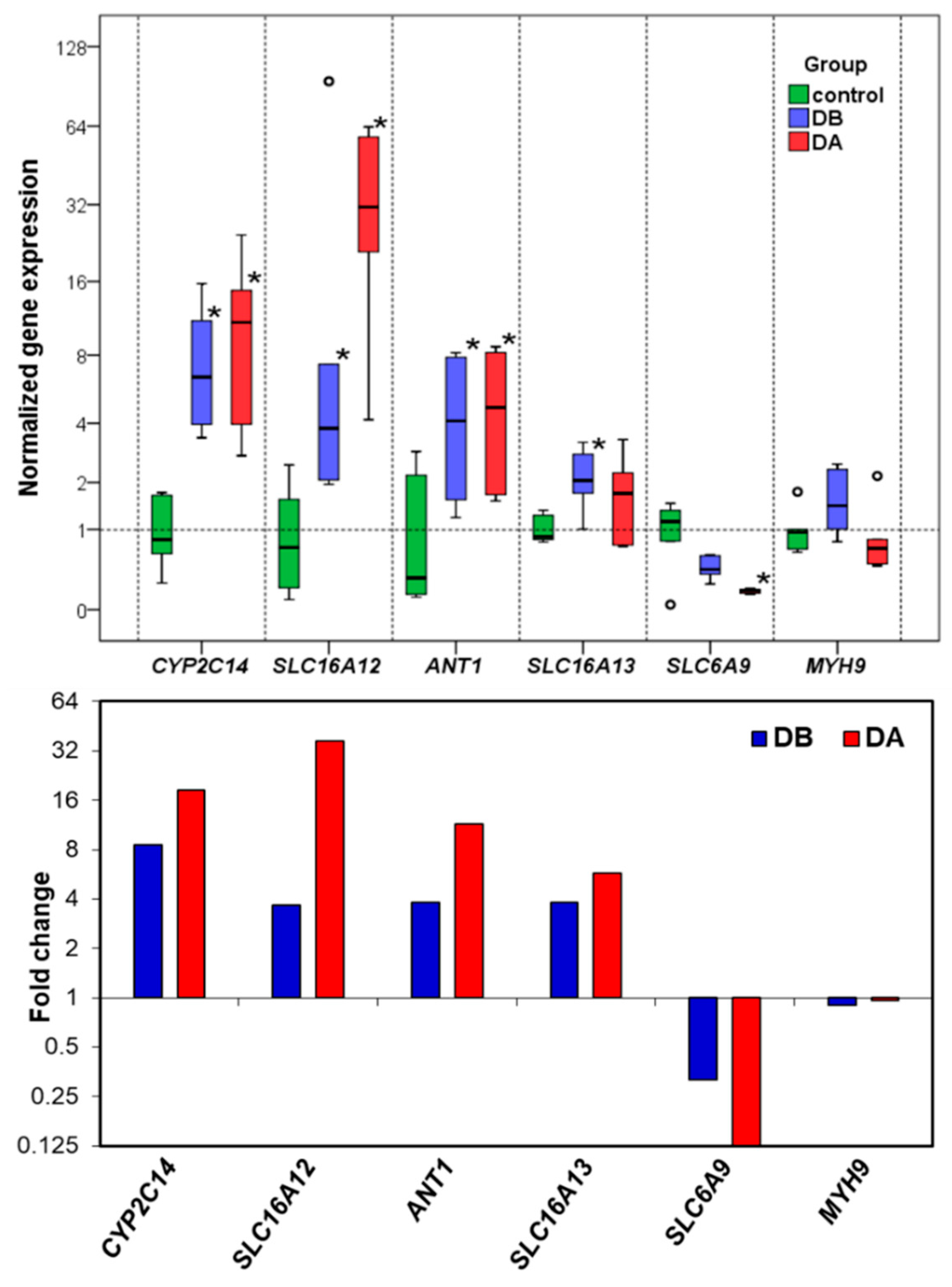
The RT-qPCR results confirm the differential gene expression obtained by RNA-seq. The gene expression changes and expression levels (fold change in relation to control) assessed by the two methods (
Figure 5) were very similar. In the determination of gene expression by means of RT-qPCR, the validation of the reference genes for each experimental situation is very important [
70,
71]. The utilization of RNA-seq expression data allowed us to find more suitable candidate reference genes. Thanks to this, their stability values (
Table 10), calculated by geNorm and NormFinder, were low (which means that the expression was stable). The selected reference genes performed better than some traditional reference genes, such as
18S rRNA,
ACTB, and
EF1A [
70,
71].
5. Materials and Methods
The methods employed were the same as those previously described [
31] except for minor modifications.
5.1. Animals
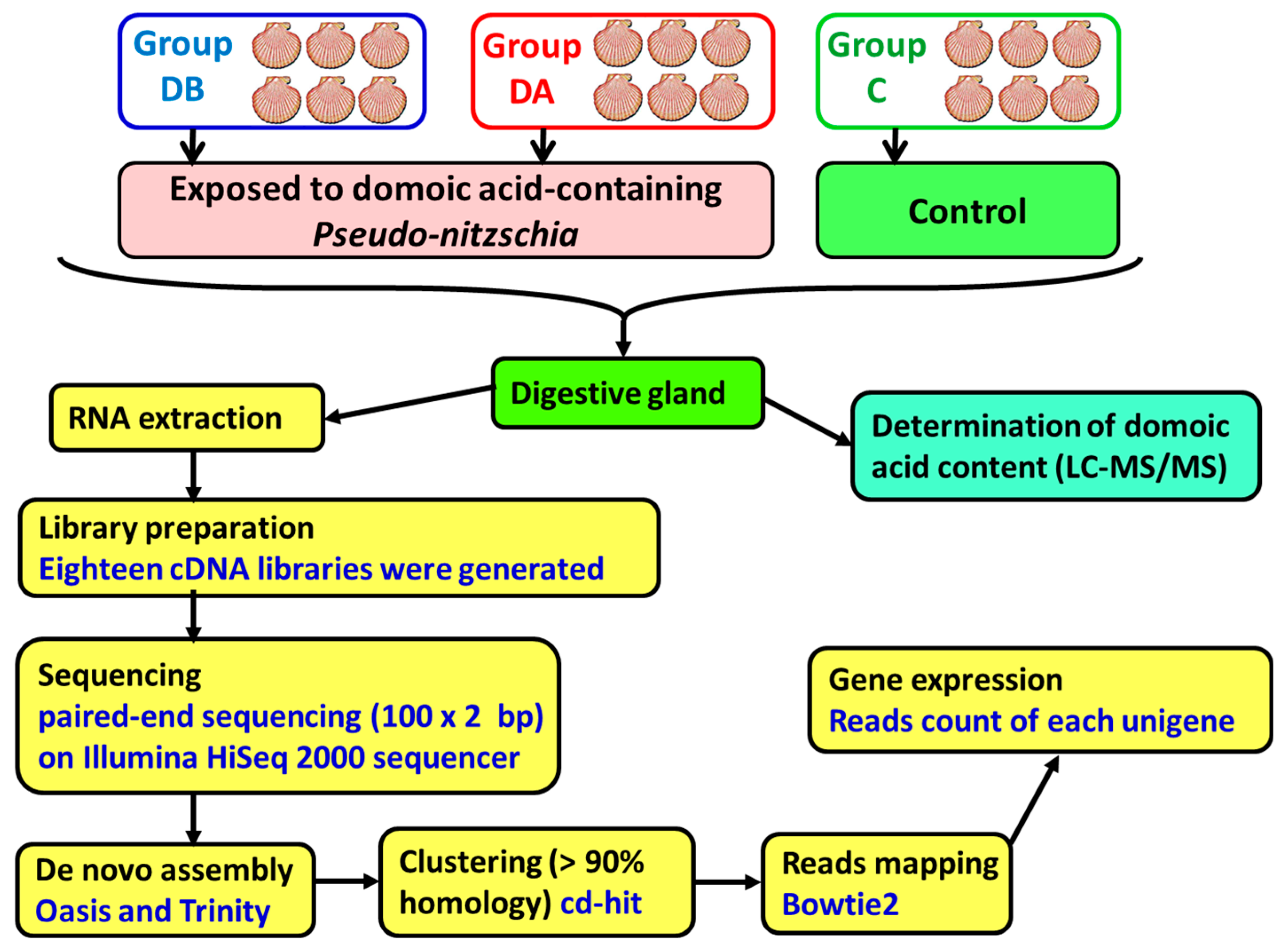
Queen scallops (
A. opercularis) were obtained from a natural bed in the Ría de Arousa in December 2014 and maintained in a 500-L tank, in the Centro de Investigacións Mariñas, (CIMA, Pedras de Corón, Vilanova de Arousa, Spain), with a continuous unfiltered seawater flow (approximate) of 1200 L/h. On April 9, 2015, 2 random samples of the scallops were obtained, and the remaining scallops (control, group C) were maintained in the tank. The scallops in 1 of the samples (group DB) were analyzed to determine their individual content and concentration of domoic acid. The scallops in the other sample (group DA) were placed in culture baskets and transferred to a raft in the culture area Grove C2 in the Ría de Arousa, where a bloom of
Pseudo-nitzschia was taking place. The recorded levels of domoic acid in the mussels from that raft showed a maximum of 22 mg DA/kg on April 9 (data obtained from Intecmar,
www.intecmar.gal [
72]). The scallops were maintained on the raft until April 17, 2015 in order to be exposed to domoic acid-containing
Pseudo-nitzschia. On that date, the scallops (group DA) were brought back to the laboratory to determine their domoic acid content. The scallops from the control group were sampled on May 12, 2015, after the end of the toxic episode caused by
Pseudo-nitzschia. From April 9 to May 12, the main characteristics of the seawater, temperature, salinity, light transmission (index of suspended solids), O
2, and fluorescence (index of phytoplankton abundance) in GROVE and in the area of CIMA were very similar (
Figure S3). As previously explained by Pazos et al. [
31], the experimental approach (animals naturally exposed to domoic acid-producing
Pseudo-nitzschia) was chosen because of the difficulty of supplying toxic
Pseudo-nitzschia under controlled conditions in the laboratory due to the relatively low absorption efficiency of the scallops and to the loss of toxicity of the
Pseudo-nitzschia cultures.
Digestive glands, gills, and the remaining tissues were obtained by dissecting the scallops. Then, 1 part of each digestive gland was used in the determination of the domoic acid content. The second part was stored in RNAlater (ref. AM7021, Ambion, Life Technologies, Carlsbad, CA, USA) at −80°C until RNA extraction.
5.2. Chemicals and Reagents for Toxin Extraction and LC-MS/MS
Methanol for HPLC and formic acid were purchased from RCI Labscan Limited (Bangkok, Thailand) and Sigma-Aldrich (St. Louis, MO, USA), respectively. Ultrapure water was obtained using a Milli-Q Gradient system, coupled with an Elix Advantage 10, both from Millipore (Merck Millipore, Darmstadt, Germany).
5.3. Determination of the Domoic Acid Content
To extract the toxin, each digestive gland was placed in aqueous methanol (50%) in a proportion of 1:2 (w/v) and homogenized with an Ultra-Turrax T25 (IKA, Staufen, Germany). The extract was clarified using centrifugation at 18,000 g at 4 °C for 10 min, retaining a supernatant that was immediately analyzed.
Domoic acid in the obtained extracts was analyzed using LC-MS/MS. The chromatographic separation was carried out using a Thermo Accela chromatographic system (Thermo Fisher Scientific, Waltham, MA, USA), with a high-pressure pump and autosampler. The stationary phase was a solid core Kinetex C18, 50 × 2.1 mm, 2.6 µm particle size, column (Phenomenex, Torrance, CA, USA). An elution gradient, with a flow of 280 µL/min, was used with mobile phase A (formic acid 0.2%) and B (50% MeOH with formic acid 0.2%). The gradient started at 100% A, maintained this condition for 1 min, linearly changed until reaching 55% B in minute 5, was held for 2 min, and then reverted to the initial conditions in order to equilibrate before the next injection. Next, 5 µL of extract, previously filtered through a PES 0.2-µm syringe filter (MFS), were injected.
After the chromatographic separation, domoic acid was detected and quantified by means of a Thermo TSQ Quantum Access MAX triple quadrupole mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA), equipped with a HESI-II electrospray interface, using positive polarization and SRM mode. The transition 312.18 > 266.18 m/z was used to quantify the response and 312.18 > 248.18 was used for confirmation. The spectrometer was operated under the following conditions: spray voltage 3400 V, capillary temperature 270 °C, HESI-II temperature 110 °C, sheet gas (nitrogen) 20 (nominal pressure), auxiliary gas (nitrogen) 10 (nominal pressure), collision energy of 15 V, and collision gas (argon) pressure of 1.5 mTorr.
Concentrations of domoic acid were obtained by comparing the response of the quantification transition in the sample extracts with that of a reference solution obtained from NRC Canada. The quantification limit of the method for tissue analysis is less than 20 ng/mL of extract.
5.4. RNA Extraction
For digestive gland total RNA isolation, the NucleoSpin RNA kit (ref. 740955, Macherey-Nagel, Düren, Germany) was used following the manufacturer’s protocol. Then, RNA was precipitated with 0.5 volumes of Li CL 7.5 M, and the RNA pellet was dissolved in 50 μL of RNA storage solution (ref. AM7000, Ambion, Life Technologies, Carlsbad, CA, USA). To remove DNA contamination, total RNA was treated with DNA-free (ref. AM1907M, Ambion, Life Technologies, Carlsbad, CA, USA). The integrity and quality of the RNA samples were measured using agarose gel electrophoresis, an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and a Nanodrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). The quantity of the total RNA was determined using Qubit 2.0 (Invitrogen, Carlsbad, CA, USA).
5.5. Library Preparation and Sequencing
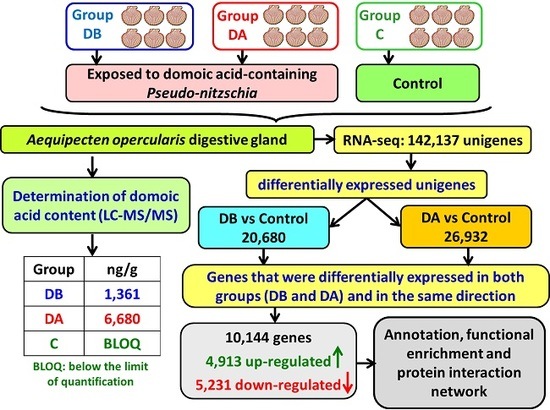
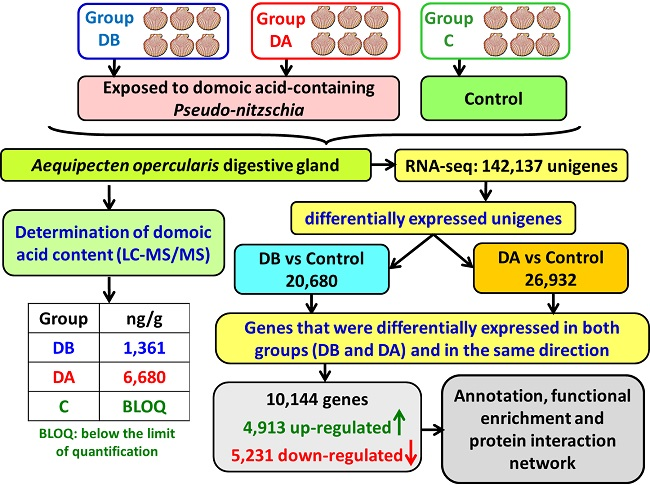
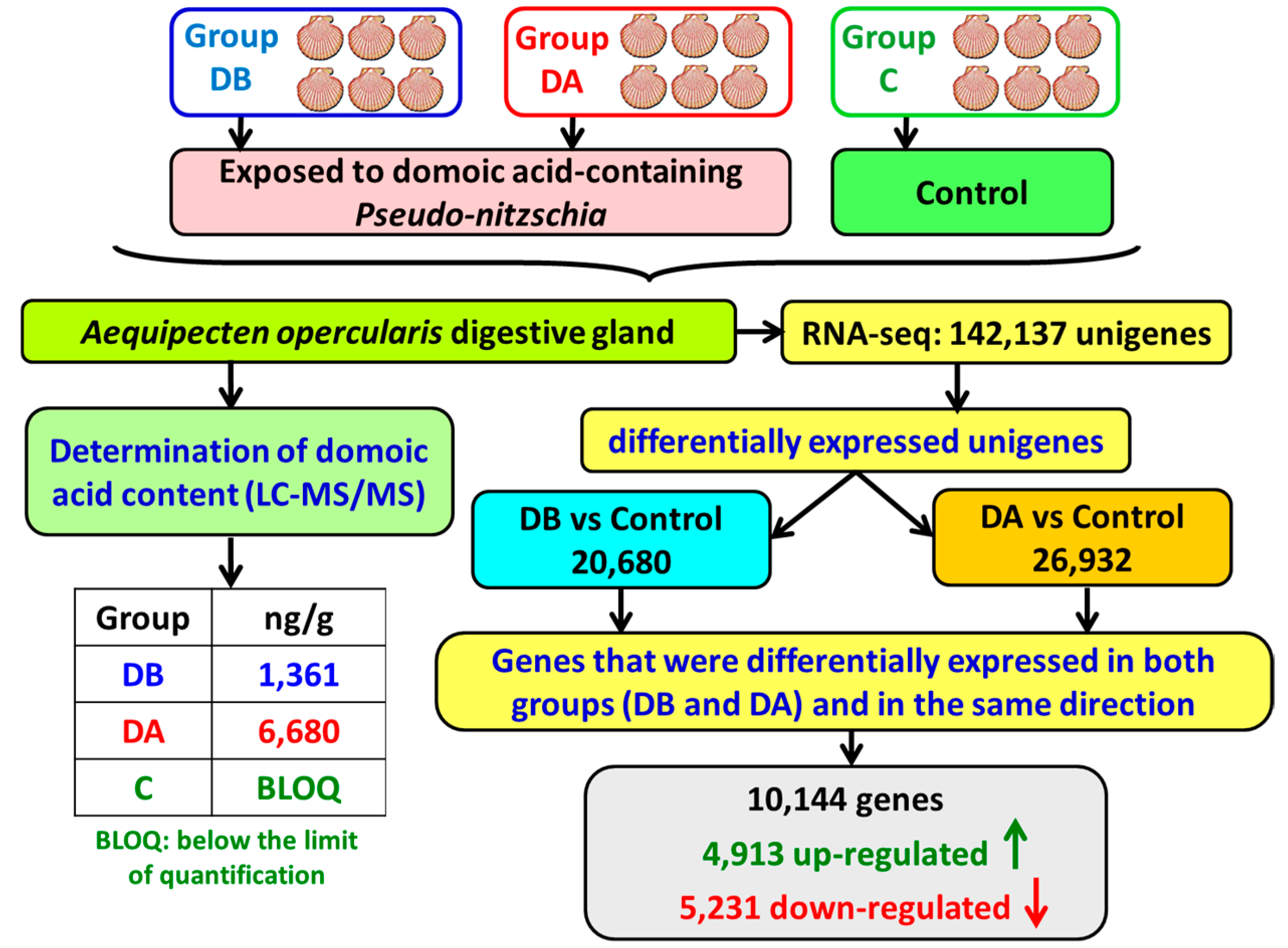
A total of 18 cDNA libraries were generated (
Figure 6) from the digestive gland of the scallops (6 obtained from each group: DB, DA, and control). The poly(A) + mRNA fraction was isolated from total RNA, and cDNA libraries were obtained following Illumina’s recommendations. Briefly, poly(A) + RNA was isolated on poly-T oligoattached magnetic beads and chemically fragmented prior to reverse transcription and cDNA generation. The cDNA fragments then went through an end repair process, the addition of a single ‘A’ base to the 3’ end, and afterwards, the ligation of the adapters. Finally, the products were purified and enriched with PCR to create the indexed final double-stranded cDNA library. The quality of the libraries was analyzed using a Bioanalyzer 2100 high sensitivity assay; the quantity of the libraries was determined by real-time PCR in a LightCycler 480 (Roche Diagnostics, Mannheim, Germany). Prior to cluster generation in cbot (Illumina), an equimolar pooling of the libraries was performed. The pool of the cDNA libraries was sequenced by paired-end sequencing (100 × 2 bp) on an Illumina HiSeq 2000 sequencer (Illumina, San Diego, CA, USA).
5.6. de novo Assembly
Quality control checks of the raw sequencing data were performed with FastQC. The technical adapters were eliminated using Trimgalore software version 0.3.3 (Babraham Bioinformatics, Cambridge, UK) (
http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Additionally, the reads with a mean Phred score >30 were selected. Subsequently, all the samples were combined, and the complexity of the reads was reduced by removing duplicates. Then, a de novo assembly was performed using the programs Oases, version 0.2.09 [
73] and Trinity, version 2.1.1 [
74]. The assembled transcripts were clustered (>90% homology) to reduce redundancy using cd-hit software version 4.6. For each sequence, the potential ORFs were detected using Transdecoder software, version 2.0, with standard parameters.
Each sample was then mapped with Bowtie2, version 2.2.6 [
75] against the reference transcriptome obtained in the previous step. The good quality reads (Mapping Quality ≥20) were selected to increase the resolution of the count expression. Finally, the expression inference was evaluated by means of the counts of properly paired reads in each transcript.
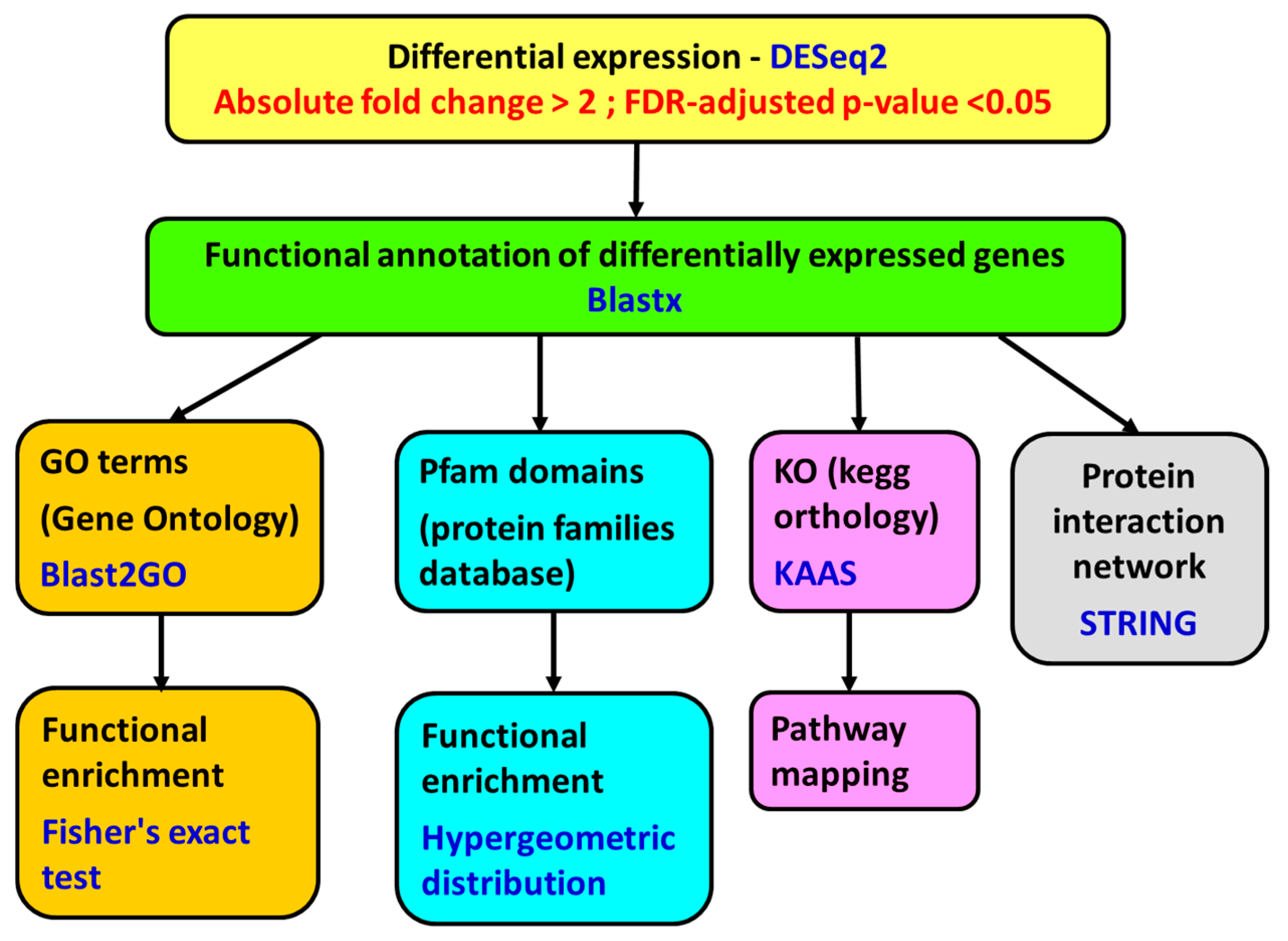
5.7. Differential Expression
The transcriptome expression for each sample was normalized by library size, following the DESeq2 protocols. Considering the whole normalized transcriptome, a study of correlation and Euclidean distance between samples was performed using the statistical software R, version 3.2.3 (
www.r-project.org), for identifying possible samples outliers.
Differential gene expression analysis was performed with DESeq2 algorithm, version 1.8.2 (
http://www.bioconductor.org/packages/devel/bioc/html/DESeq2.html). The genes with a fold change of less than −2 or greater than 2 and a
p-value adjusted using the Benjamini and Hochberg [
76] method for controlling false discovery rate (FDR) <0.05 were considered differentially expressed (
Figure 7).
A filtering step was performed with the DEGs to remove the transcripts from Pseudo-nitzschia; the contigs were blasted against Mizuhopecten yessoensis and Pseudo-nitzschia multistriata genomes (E-value threshold of 10−10, word size 12):
The contigs that had a lower E-value versus Pseudo-nitzschia compared with M. yessoensis were discarded (approximately 0.7% of the sequences).
5.8. Functional Annotation
The genes were annotated using Blastx [
77] against the Uniprot database and Blastn [
77] against the NCBI nucleotide database (
E-value threshold of 10
−2). Then, the annotation was expanded by incorporating information from the species, gene name, and functions using gene ontology and protein structure domains associated with the transcript using InterPro (
https://www.ebi.ac.uk/interpro/). The genes were also annotated with Blast2GO software version 4.1.9 (BioBam Bioinformatics S.L, Valencia, Spain) [
78,
79], using local Blastx 2.4.0+ against a database of
Mizuhopecten yessoensis and
Crassostrea gigas proteins obtained from NCBI (
E-value threshold of 10
−3):
Orthologue assignment and pathway mapping were performed on the KEGG Automatic Annotation Server (KAAS, [
80]) using Blast and the bi-directional best hit (BBH) method (
http://www.genome.jp/tools/kaas/).
5.9. Functional Enrichment
A functional enrichment study was performed using the Pfam [
81] functional information. This study is based on hypergeometric distribution [
82] using the statistical software R version 3.2.3 (
www.r-project.org). The differentially expressed genes were also subjected to GO enrichment analysis with Blast2GO version 4.1.9. (BioBam Bioinformatics S.L, Valencia, Spain) using Fisher’s exact test [
83] (up- and down-regulated genes were analyzed separately). The false discovery rate (FDR) adjusted p-value [
76] was set at a cutoff of 0.05.
5.10. Protein Network Analysis
To search for the protein–protein interactions, network analyses using the String 10.5 algorithm [
84] were performed. The putative human homologues of proteins coded by the up-regulated and the down-regulated genes in the
A. opercularis digestive gland were identified by means of a Blastx search [
85] against the STRING human protein database (9606.protein.sequences.v10.fa), with an E-value threshold of 10
−5. The top Blastx search results were used as input in the String program. The up-regulated and the down-regulated genes were analyzed separately.
5.11. Real Time RT-qPCR Validation
cDNA was synthesized from 0.5 μg of total RNA with the iScript™cDNA Synthesis kit (ref. 170-8891, BioRad, Hercules, CA, USA) in a 20-µL reaction volume, and the conditions were 5 min at 25 °C, 30 min at 42 °C, and 5 min at 85 °C.
For the relative quantification of gene expression by means of RT-qPCR, a normalization step must be performed using internal reference genes, whose expression levels are stable [
38,
86,
87,
88]. Suitable reference genes should be selected for each experimental condition to ensure their stable expression [
70,
89].
A total of 6 reference gene candidates (
Table 9),
VAMP7,
RPS4,
MYH9,
EIF4EBP2,
DNAJ, and
RAP1B and 5 target genes (
Table 9),
CYP2C14,
SLC16A12,
ANT1,
SLC16A13, and
SLC6A9, were used in the gene expression study. The candidate reference genes were selected for their stable expression based on the RNA-seq data. Oligonucleotide primers were designed with OligoAnalyzer 3.1 (
http://eu.idtdna.com/analyzer/Applications/OligoAnalyzer/; Integrated DNA Technologies, Leuven, Belgium) from the sequences in
Table 9 and were synthesized by Integrated DNA Technologies (Leuven, Belgium). The primer sequences and amplicon lengths are listed in
Table 9. The specificity of the primers was confirmed by the presence of a single peak in the melting curve and by the presence of a single band of the expected size when PCR products were run in a 2% agarose gel. The PCR amplification efficiency (E) of each transcript was determined by means of Real-Time PCR Miner software (Version 4.0;
http://www.miner.ewindup.info/ [
90]). The mean amplification efficiency (E) of each amplicon (
Table 9) was used in the calculation of gene expression.
Real-time qPCR analysis was conducted in technical duplicates and 6 biological replicates, in 96-well reaction plates on an iCycler iQ® Real-Time System (Bio-Rad, Hercules, CA, USA, 2003). The PCR final volume was 20 μL, containing 4 μL of 1:5 diluted cDNA (20 ng of cDNA), 10 μL of SsoFast EvaGreen Supermix (ref. 172-5201, Bio-Rad, Hercules, CA, USA), 400 nM of forward and reverse primers, and 4.4 μL of PCR-grade water. The cycling conditions were as follows: 30 s at 95 °C (initial template denaturation) and 40 cycles of 5 s at 95 °C (denaturation) followed by 10 s at 60 °C (annealing and elongation) and 10 s at 75 °C for fluorescence measurement. At the end of each run, a melting curve was carried out: 95 °C for 20 s and 60 °C for 20 s followed by an increase in temperature from 60 to 100 °C (with temperature increases in steps of 0.5 °C every 10 s). Baseline values were automatically determined for all the plates using Bio-Rad iCycler iQ software V3.1 (IQ™ Real-Time PCR Detection System). The threshold value was set manually at 100 RFU (relative fluorescence units) to calculate the Cq values. Non-reverse transcriptase controls and non-template controls (NTC) were also included in each run.
The gene expression was normalized to reference genes that had stable expression levels [
38,
86,
87,
88]. The gene expression stability of candidate reference genes was analyzed using 3 Microsoft Excel-based software applications, geNorm V3.5 [
38], NormFinder V0.953 [
86], and BestKeeper V1 [
88]. The non-normalized expression (Q) was calculated using the equation Q = (1 + E)
–Cq. Then, the expression was normalized by dividing it by the normalization factor (the geometric mean of the non-normalized expression of the selected reference genes) [
89].
The statistical analyses were performed with the IBM SPSS Statistics 24.0 package (IBM SPSS, Chicago, IL, USA). The data were tested for normality (Shapiro–Wilk test) and for homogeneity of variance (Levene’s test). The gene expression was log-transformed (base 2) to meet the requirements of normality and homogeneity of variances. The expression of target genes in domoic acid-exposed scallops (groups DB and DA) in relation to the control group was compared using ANOVA and post hoc Dunnett’s t test. p <0.05 was considered statistically significant.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}