The Toxin-Antitoxin Systems of the Opportunistic Pathogen Stenotrophomonas maltophilia of Environmental and Clinical Origin


Abstract
:1. Introduction
2. Results
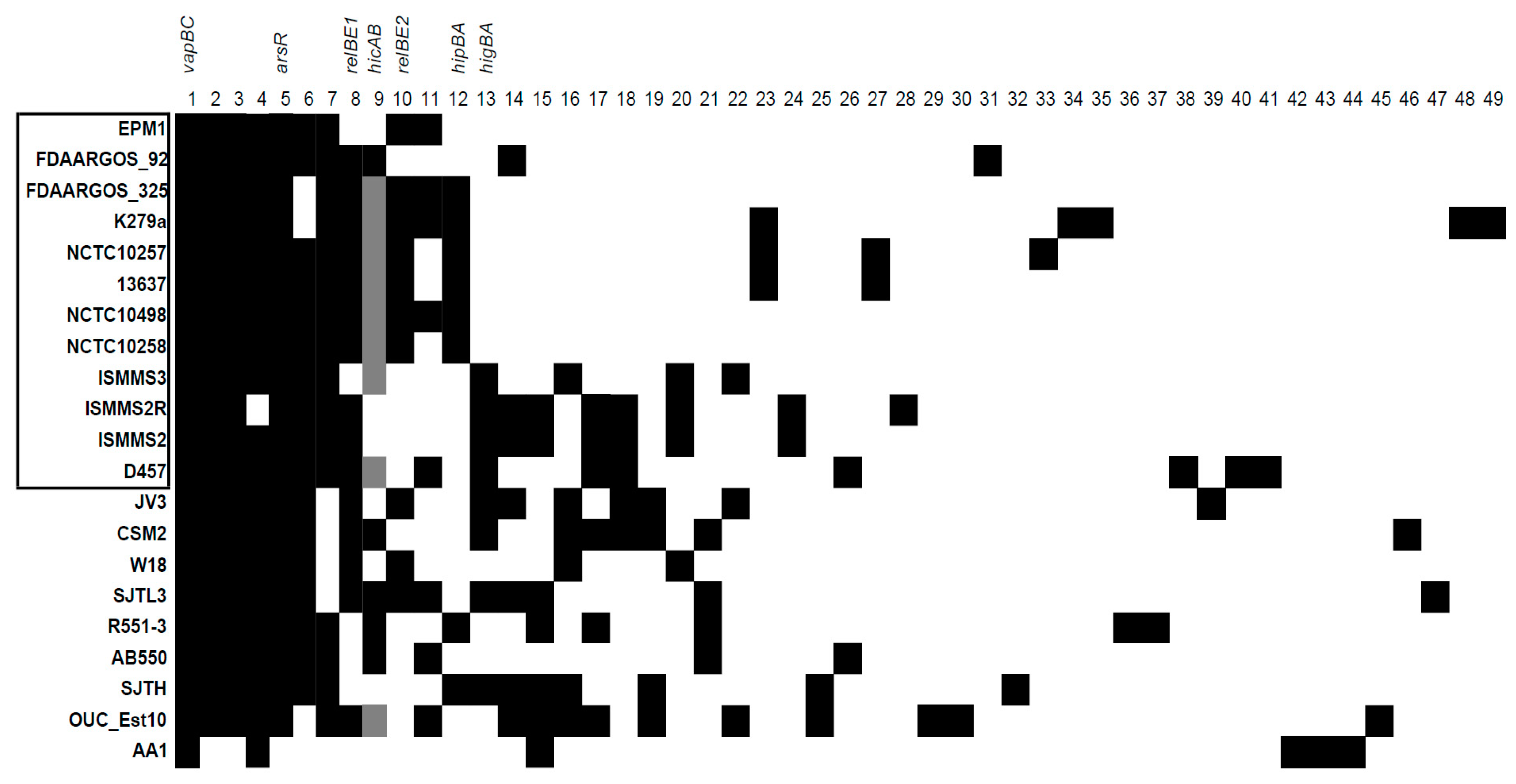
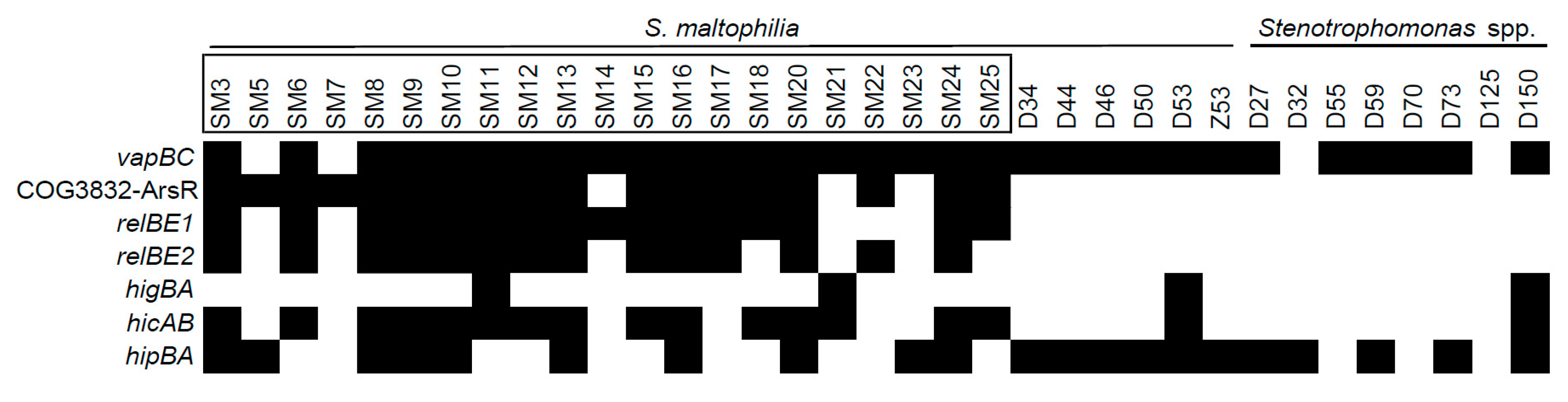
2.1. Detection of S. maltophilia TA Systems in Genomes
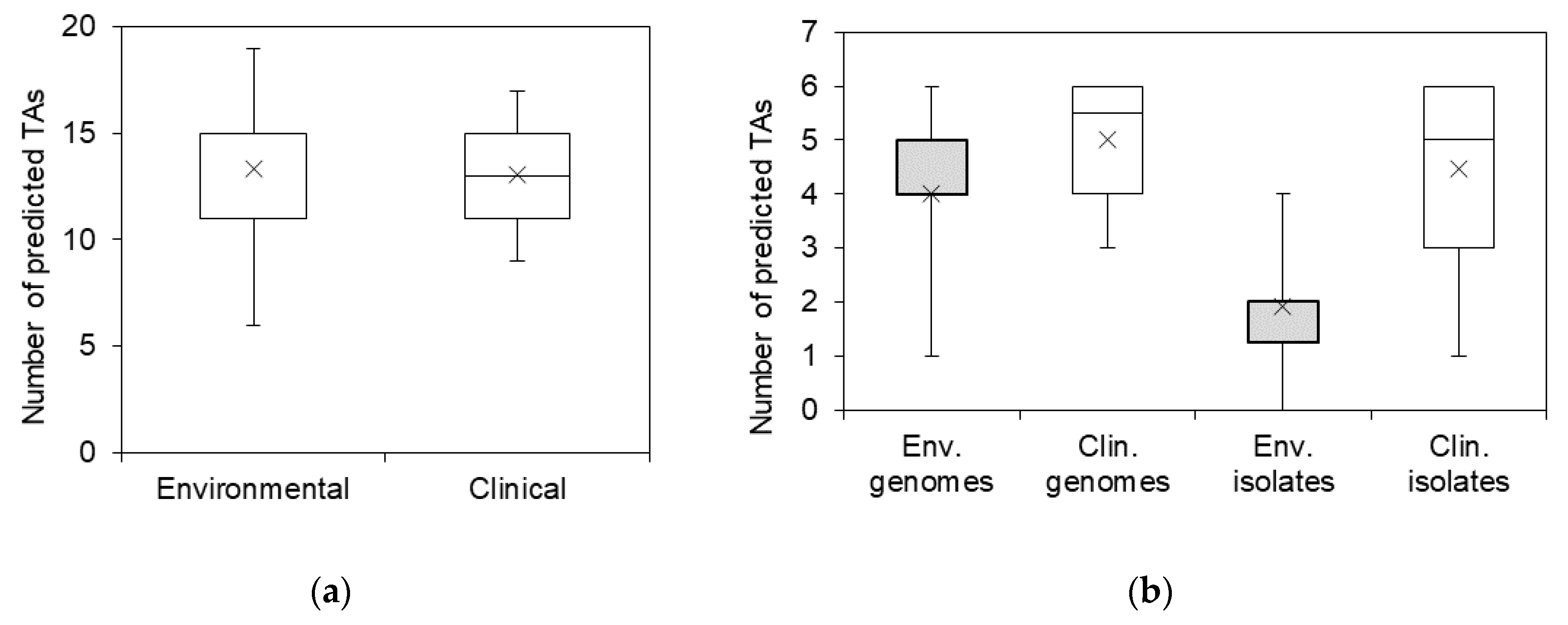
2.2. The Prevalence of TA Systems in Stenotrophomonas spp. of Clinical and Environmental Origin
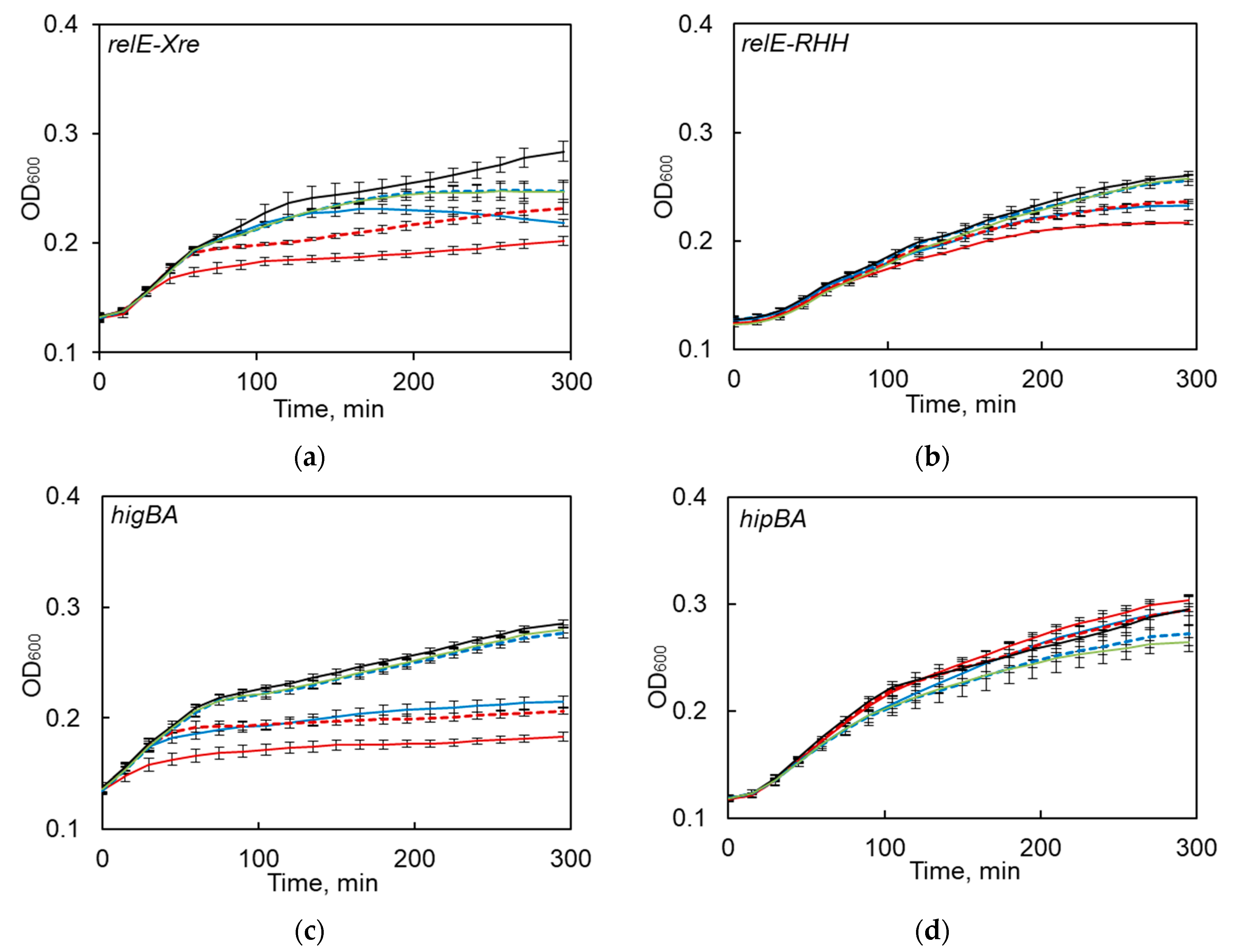
2.3. Functionality of S. maltophilia TA Systems
2.4. Characterization of Stenotrophomonas spp. Isolates
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Bioinformatic Assays
5.2. The Bacteria Used in the Study and Growth Conditions
5.3. Detection of TA Systems in Stenotrophomonas
5.4. Cloning of TA System Genes
5.5. Kill-Rescue Assay
5.6. Random Amplified Polymorphic DNA (RAPD) Assay
5.7. The Evaluation of Antibiotic Resistance
5.8. Biofilm Formation Assay
5.9. Pellicle Formation Assay
5.10. Extraction of Capsular Polysaccharides
5.11. Serum Resistance
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brooke, J.S. Stenotrophomonas maltophilia: An Emerging Global Opportunistic Pathogen. Clin. Microbiol. Rev. 2012, 25, 2–41. [Google Scholar] [CrossRef] [Green Version]
- Ryan, R.P.; Monchy, S.; Cardinale, M.; Taghavi, S.; Crossman, L.; Avison, M.B.; Berg, G.; van der Lelie, D.; Dow, J.M. The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat. Rev. Microbiol. 2009, 7, 514–525. [Google Scholar] [CrossRef]
- Falagas, M.E.; Kastoris, A.C.; Vouloumanou, E.K.; Rafailidis, P.I.; Kapaskelis, A.M.; Dimopoulos, G. Attributable mortality of Stenotrophomonas maltophilia infections: A systematic review of the literature. Future Microbiol. 2009, 4, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- WHO | World Health Organization. Available online: https://www.who.int/drugresistance/AMR_Importance/en/ (accessed on 28 July 2020).
- Pompilio, A.; Savini, V.; Fiscarelli, E.; Gherardi, G.; Di Bonaventura, G. Clonal Diversity, Biofilm Formation, and Antimicrobial Resistance among Stenotrophomonas maltophilia Strains from Cystic Fibrosis and Non-Cystic Fibrosis Patients. Antibiotics 2020, 9, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifonova, A.; Strateva, T. Stenotrophomonas maltophilia—A low-grade pathogen with numerous virulence factors. Infect. Dis. 2019, 51, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Adamek, M.; Overhage, J.; Bathe, S.; Winter, J.; Fischer, R.; Schwartz, T. Genotyping of Environmental and Clinical Stenotrophomonas maltophilia Isolates and their Pathogenic Potential. PLoS ONE 2011, 6, e27615. [Google Scholar] [CrossRef] [Green Version]
- Bostanghadiri, N.; Ghalavand, Z.; Fallah, F.; Yadegar, A.; Ardebili, A.; Tarashi, S.; Pournajaf, A.; Mardaneh, J.; Shams, S.; Hashemi, A. Characterization of Phenotypic and Genotypic Diversity of Stenotrophomonas maltophilia Strains Isolated From Selected Hospitals in Iran. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef]
- Pompilio, A.; Pomponio, S.; Crocetta, V.; Gherardi, G.; Verginelli, F.; Fiscarelli, E.; Dicuonzo, G.; Savini, V.; D’Antonio, D.; Di Bonaventura, G. Phenotypic and genotypic characterization of Stenotrophomonas maltophiliaisolates from patients with cystic fibrosis: Genome diversity, biofilm formation, and virulence. BMC Microbiol. 2011, 11, 159. [Google Scholar] [CrossRef] [Green Version]
- Madi, H.; Lukić, J.; Vasiljević, Z.; Biočanin, M.; Kojić, M.; Jovčić, B.; Lozo, J. Genotypic and Phenotypic Characterization of Stenotrophomonas maltophilia Strains from a Pediatric Tertiary Care Hospital in Serbia. PLoS ONE 2016, 11, e0165660. [Google Scholar] [CrossRef]
- Valdezate, S.; Vindel, A.; Martin-Davila, P.; Del Saz, B.S.; Baquero, F.; Canton, R. High Genetic Diversity among Stenotrophomonas maltophilia Strains Despite Their Originating at a Single Hospital. J. Clin. Microbiol. 2004, 42, 693–699. [Google Scholar] [CrossRef] [Green Version]
- Lira, F.; Berg, G.; Martínez, J.L. Double-Face Meets the Bacterial World: The Opportunistic Pathogen Stenotrophomonas maltophilia. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Dzidic, S.; Bedeković, V. Horizontal gene transfer-emerging multidrug resistance in hospital bacteria. Acta Pharmacol. Sin. 2003, 24, 519–526. [Google Scholar] [PubMed]
- Brown, S.P.; Cornforth, D.M.; Mideo, N. Evolution of virulence in opportunistic pathogens: Generalism, plasticity, and control. Trends Microbiol. 2012, 20, 336–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, A.; Brodersen, D.E.; Mitarai, N.; Gerdes, K. Toxins, Targets, and Triggers: An Overview of Toxin-Antitoxin Biology. Mol. Cell 2018, 70, 768–784. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-Y.; Lee, B.-J. Structure, Biology, and Therapeutic Application of Toxin-Antitoxin Systems in Pathogenic Bacteria. Toxins 2016, 8, 305. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Park, J.-H.; Inouye, M. Toxin-Antitoxin Systems in Bacteria and Archaea. Annu. Rev. Genet. 2011, 45, 61–79. [Google Scholar] [CrossRef] [Green Version]
- Lobato-Márquez, D.; Díaz-Orejas, R.; García-del Portillo, F. Toxin-antitoxins and bacterial virulence. FEMS Microbiol. Rev. 2016, 40, 592–609. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, H.; Hay, A.J.; Zhong, Z.; Zhu, J.; Kan, B. Functional RelBE-Family Toxin-Antitoxin Pairs Affect Biofilm Maturation and Intestine Colonization in Vibrio cholerae. PLoS ONE 2015, 10, e0135696. [Google Scholar] [CrossRef] [Green Version]
- Wood, T.L.; Wood, T.K. The HigB/HigA toxin/antitoxin system of Pseudomonas aeruginosa influences the virulence factors pyochelin, pyocyanin, and biofilm formation. Microbiol. Open 2016, 5, 499–511. [Google Scholar] [CrossRef]
- Leplae, R.; Geeraerts, D.; Hallez, R.; Guglielmini, J.; Drèze, P.; Van Melderen, L. Diversity of bacterial type II toxin–antitoxin systems: A comprehensive search and functional analysis of novel families. Nucleic Acids Res. 2011, 39, 5513–5525. [Google Scholar] [CrossRef] [Green Version]
- Georgiades, K.; Raoult, D. Genomes of the Most Dangerous Epidemic Bacteria Have a Virulence Repertoire Characterized by Fewer Genes but More Toxin-Antitoxin Modules. PLoS ONE 2011, 6, e17962. [Google Scholar] [CrossRef] [PubMed]
- Sala, A.; Bordes, P.; Genevaux, P. Multiple Toxin-Antitoxin Systems in Mycobacterium tuberculosis. Toxins 2014, 6, 1002–1020. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Wei, Y.; Shen, Y.; Li, X.; Zhou, H.; Tai, C.; Deng, Z.; Ou, H.-Y. TADB 2.0: An updated database of bacterial type II toxin–antitoxin loci. Nucleic Acids Res. 2018, 46, D749–D753. [Google Scholar] [CrossRef] [PubMed]
- Sevin, E.W.; Barloy-Hubler, F. RASTA-Bacteria: A web-based tool for identifying toxin-antitoxin loci in prokaryotes. Genome Biol. 2007, 8, R155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akarsu, H.; Bordes, P.; Mansour, M.; Bigot, D.-J.; Genevaux, P.; Falquet, L. TASmania: A bacterial Toxin-Antitoxin Systems database. PLoS Comput. Biol. 2019, 15, e1006946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butt, A.; Müller, C.; Harmer, N.; Titball, R.W. Identification of type II toxin–antitoxin modules in Burkholderia pseudomallei. FEMS Microbiol. Lett. 2013, 338, 86–94. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhang, N.; Cao, M.; Ren, S.; Zeng, T.; Qin, M.; Zhao, X.; Yuan, F.; Chen, H.; Bei, W. Identification of Three Type II Toxin-Antitoxin Systems in Streptococcus suis Serotype 2. Toxins 2018, 10, 467. [Google Scholar] [CrossRef] [Green Version]
- Coussens, N.P.; Daines, D.A. Wake me when it’s over—Bacterial toxin-antitoxin proteins and induced dormancy. Exp. Biol. Med. 2016, 241, 1332–1342. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct 2009, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, M.B. Antibiotic resistance in the opportunistic pathogen Stenotrophomonas maltophilia. Front. Microbiol. 2015, 6, 658. [Google Scholar] [CrossRef] [Green Version]
- Bjarnsholt, T. The role of bacterial biofilms in chronic infections. Apmis. Suppl. 2013, 1–51. [Google Scholar] [CrossRef] [PubMed]
- Di Bonaventura, G.; Spedicato, I.; D’Antonio, D.; Robuffo, I.; Piccolomini, R. Biofilm formation by Stenotrophomonas maltophilia: Modulation by quinolones, trimethoprim-sulfamethoxazole, and ceftazidime. Antimicrob. Agents Chemother. 2004, 48, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armitano, J.; Méjean, V.; Jourlin-Castelli, C. Gram-negative bacteria can also form pellicles. Environ. Microbiol. Rep. 2014, 6, 534–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cress, B.F.; Englaender, J.A.; He, W.; Kasper, D.; Linhardt, R.J.; Koffas, M.A.G. Masquerading microbial pathogens: Capsular polysaccharides mimic host-tissue molecules. FEMS Microbiol. Rev. 2014, 38, 660–697. [Google Scholar] [CrossRef] [Green Version]
- Niu, T.; Guo, L.; Luo, Q.; Zhou, K.; Yu, W.; Chen, Y.; Huang, C.; Xiao, Y. Wza gene knockout decreases Acinetobacter baumannii virulence and affects Wzy-dependent capsular polysaccharide synthesis. Virulence 2020, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Geisinger, E.; Isberg, R.R. Antibiotic modulation of capsular exopolysaccharide and virulence in Acinetobacter baumannii. PLoS Pathog. 2015, 11, e1004691. [Google Scholar] [CrossRef] [Green Version]
- Skerniškytė, J.; Krasauskas, R.; Péchoux, C.; Kulakauskas, S.; Armalytė, J.; Sužiedėlienė, E. Surface-Related Features and Virulence Among Acinetobacter baumannii Clinical Isolates Belonging to International Clones I and II. Front. Microbiol. 2018, 9, 3116. [Google Scholar] [CrossRef] [Green Version]
- Van Melderen, L.; De Bast, M.S. Bacterial Toxin–Antitoxin Systems: More Than Selfish Entities? PLoS Genet. 2009, 5, e1000437. [Google Scholar] [CrossRef]
- Rowe-Magnus, D.A. Comparative Analysis of Superintegrons: Engineering Extensive Genetic Diversity in the Vibrionaceae. Genome Res. 2003, 13, 428–442. [Google Scholar] [CrossRef] [Green Version]
- Szekeres, S.; Dauti, M.; Wilde, C.; Mazel, D.; Rowe-Magnus, D.A. Chromosomal toxin-antitoxin loci can diminish large-scale genome reductions in the absence of selection: Chromosomal addiction loci minimize gene loss. Mol. Microbiol. 2007, 63, 1588–1605. [Google Scholar] [CrossRef]
- Gotfredsen, M.; Gerdes, K. The Escherichia coli relBE genes belong to a new toxin-antitoxin gene family. Mol. Microbiol. 1998, 29, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Schureck, M.A.; Repack, A.; Miles, S.J.; Marquez, J.; Dunham, C.M. Mechanism of endonuclease cleavage by the HigB toxin. Nucleic Acids Res. 2016, 44, 7944–7953. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Grishin, N.V.; Koonin, E.V. The HicAB cassette, a putative novel, RNA-targeting toxin-antitoxin system in archaea and bacteria. Bioinformatics 2006, 22, 2581–2584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, L.; Zhang, J.; Inouye, M. Characterization of ChpBK, an mRNA interferase from Escherichia coli. J. Biol. Chem. 2005, 280, 26080–26088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winther, K.S.; Gerdes, K. Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc. Natl. Acad. Sci. USA 2011, 108, 7403–7407. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, P.; Arora, G.; Singh, M.; Kidwai, S.; Narayan, O.P.; Singh, R. MazF ribonucleases promote Mycobacterium tuberculosis drug tolerance and virulence in guinea pigs. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wen, W.; Liu, B.; Xue, L.; Zhu, Z.; Niu, L.; Sun, B. Autoregulation and Virulence Control by the Toxin-Antitoxin System SavRS in Staphylococcus aureus. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [Green Version]
- Ramage, H.R.; Connolly, L.E.; Cox, J.S. Comprehensive Functional Analysis of Mycobacterium tuberculosis Toxin-Antitoxin Systems: Implications for Pathogenesis, Stress Responses, and Evolution. PLoS Genet. 2009, 5. [Google Scholar] [CrossRef] [Green Version]
- Frampton, R.; Aggio, R.B.M.; Villas-Bôas, S.G.; Arcus, V.L.; Cook, G.M. Toxin-Antitoxin Systems of Mycobacterium smegmatis Are Essential for Cell Survival. J. Biol. Chem. 2012, 287, 5340–5356. [Google Scholar] [CrossRef] [Green Version]
- Lopes, A.P.Y.; Lopes, L.M.; Fraga, T.R.; Chura-Chambi, R.M.; Sanson, A.L.; Cheng, E.; Nakajima, E.; Morganti, L.; Martins, E.A.L. VapC from the Leptospiral VapBC Toxin-Antitoxin Module Displays Ribonuclease Activity on the Initiator tRNA. PLoS ONE 2014, 9, e101678. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.T.; Espinosa, M.; Yeo, C.C. Keeping the Wolves at Bay: Antitoxins of Prokaryotic Type II Toxin-Antitoxin Systems. Front. Mol. Biosci. 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Strateva, T.; Trifonova, A.; Savov, E.; Dimov, S. An update on the antimicrobial susceptibility and molecular epidemiology of Stenotrophomonas maltophilia in Bulgaria: A 5-year study (2011–2016). Infect. Dis. 2019, 51, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Travassos, L.H.; Pinheiro, M.N.; Coelho, F.S.; Sampaio, J.L.M.; Merquior, V.L.C.; Marques, E.A. Phenotypic properties, drug susceptibility and genetic relatedness of Stenotrophomonas maltophilia clinical strains from seven hospitals in Rio de Janeiro, Brazil. J. Appl. Microbiol. 2004, 96, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.; Weigel, W.; Sztukowska, M.; Demuth, D.R. Identification and functional characterization of type II toxin/antitoxin systems in Aggregatibacter actinomycetemcomitans. Mol. Oral Microbiol. 2018, 33, 224–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bliven, K.A.; Maurelli, A.T. Antivirulence Genes: Insights into Pathogen Evolution through Gene Loss. Infect. Immun. 2012, 80, 4061–4070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima-Mendez, G.; Alvarenga, D.O.; Ross, K.; Hallet, B.; Melderen, L.V.; Varani, A.M.; Chandler, M. Toxin-Antitoxin Gene Pairs Found in Tn3 Family Transposons Appear To Be an Integral Part of the Transposition Module. mBio 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.-F.; Chang, X.; Ye, Y.; Wang, Z.-X.; Shao, Y.-B.; Shi, W.; Li, X.; Li, J.-B. Stenotrophomonas maltophilia resistance to trimethoprim/sulfamethoxazole mediated by acquisition of sul and dfrA genes in a plasmid-mediated class 1 integron. Int. J. Antimicrob. Agents 2011, 37, 230–234. [Google Scholar] [CrossRef]
- Walsh, T.R.; Macgowan, A.P.; Bennett, P.M. Sequence Analysis and Enzyme Kinetics of the L2 Serine β-Lactamase from Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 1997, 41, 5. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Liu, W.; Cui, Q.; Niu, W.; Li, H.; Zhao, X.; Wei, X.; Wang, X.; Huang, S.; Dong, D.; et al. Prevalence and detection of Stenotrophomonas maltophilia carrying metallo-Î2-lactamase blaL1 in Beijing, China. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Youenou, B.; Favre-Bonté, S.; Bodilis, J.; Brothier, E.; Dubost, A.; Muller, D.; Nazaret, S. Comparative Genomics of Environmental and Clinical Stenotrophomonas maltophilia Strains with Different Antibiotic Resistance Profiles. Genome Biol. Evol. 2015, 7, 2484–2505. [Google Scholar] [CrossRef] [Green Version]
- Deredjian, A.; Alliot, N.; Blanchard, L.; Brothier, E.; Anane, M.; Cambier, P.; Jolivet, C.; Khelil, M.N.; Nazaret, S.; Saby, N.; et al. Occurrence of Stenotrophomonas maltophilia in agricultural soils and antibiotic resistance properties. Res. Microbiol. 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Berg, G.; Eberl, L.; Hartmann, A. The rhizosphere as a reservoir for opportunistic human pathogenic bacteria. Environ. Microbiol. 2005, 7, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Muder, R.R.; Harris, A.P.; Muller, S.; Edmond, M.; Chow, J.W.; Papadakis, K.; Wagener, M.W.; Bodey, G.P.; Steckelberg, J.M. Bacteremia due to Stenotrophomonas (Xanthomonas) maltophilia: A prospective, multicenter study of 91 episodes. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 1996, 22, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.A.; Mehta, N.J. Stenotrophomonas maltophilia endocarditis: A systematic review. Angiology 2002, 53, 49–55. [Google Scholar] [CrossRef]
- Horio, N.; Horiguchi, M.; Murakami, K.; Yamamoto, E.; Miyake, Y. Stenotrophomonas maltophilia endophthalmitis after intraocular lens implantation. Graefes Arch. Clin. Exp. Ophthalmol. Albrecht Von Graefes Arch. Klin. Exp. Ophthalmol. 2000, 238, 299–301. [Google Scholar] [CrossRef]
- de Vidipó, L.A.; de Marques, E.A.; Puchelle, E.; Plotkowski, M.-C. Stenotrophomonas maltophilia Interaction with Human Epithelial Respiratory Cells In Vitro. Microbiol. Immunol. 2001, 45, 563–569. [Google Scholar] [CrossRef]
- De Oliveira-Garcia, D.; Dall’Agnol, M.; Rosales, M.; Azzuz, A.C.G.S.; Alcántara, N.; Martinez, M.B.; Girón, J.A. Fimbriae and adherence of Stenotrophomonas maltophilia to epithelial cells and to abiotic surfaces. Cell. Microbiol. 2003, 5, 625–636. [Google Scholar] [CrossRef]
- Zhuo, C.; Zhao, Q.; Xiao, S. The Impact of spgM, rpfF, rmlA Gene Distribution on Biofilm Formation in Stenotrophomonas maltophilia. PLoS ONE 2014, 9, e108409. [Google Scholar] [CrossRef]
- Branda, S.S.; Vik, Å.; Friedman, L.; Kolter, R. Biofilms: The matrix revisited. Trends Microbiol. 2005, 13, 20–26. [Google Scholar] [CrossRef]
- Yamamoto, K.; Arai, H.; Ishii, M.; Igarashi, Y. Trade-off between oxygen and iron acquisition in bacterial cells at the air–liquid interface. FEMS Microbiol. Ecol. 2011, 77, 83–94. [Google Scholar] [CrossRef]
- Campa, M.; Bendinelli, M.; Friedman, H. Pseudomonas Aeruginosa as an Opportunistic Pathogen; Springer Science & Business Media: New York, NY, USA, 2012; ISBN 978-1-4615-3036-7. [Google Scholar]
- Tipton, K.A.; Dimitrova, D.; Rather, P.N. Phase-Variable Control of Multiple Phenotypes in Acinetobacter baumannii Strain AB5075. J. Bacteriol. 2015, 197, 2593–2599. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.Y.; Tipton, K.A.; Farokhyfar, M.; Burd, E.M.; Weiss, D.S.; Rather, P.N. A high-frequency phenotypic switch links bacterial virulence and environmental survival in Acinetobacter baumannii. Nat. Microbiol. 2018, 3, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Boulnois, G.J.; Roberts, I.S. Genetics of Capsular Polysaccharide Production in Bacteria. In Proceedings of the Bacterial Capsules; Jann, K., Jann, B., Eds.; Springer: Berlin/Heidelberg, Germany, 1990; pp. 1–18. [Google Scholar]
- McKay, G.A.; Woods, D.E.; MacDonald, K.L.; Poole, K. Role of phosphoglucomutase of Stenotrophomonas maltophilia in lipopolysaccharide biosynthesis, virulence, and antibiotic resistance. Infect. Immun. 2003, 71, 3068–3075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 2014, 1079, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.; Morita, M.; Nishimura, Y.; Sugino, Y. A rapid and highly efficient method for preparation of competent Escherichia coli cells. Nucleic Acids Res. 1990, 18, 6169. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Dong, H.; Huang, Y.-P. Highly efficient transformation of Stenotrophomonas maltophilia S21, an environmental isolate from soil, by electroporation. J. Microbiol. Methods 2014, 107, 92–97. [Google Scholar] [CrossRef]
- Yanisch-Perron, C.; Vieira, J.; Messing, J. Improved M13 phage cloning vectors and host strains: Nucleotide sequences of the M13mpl8 and pUC19 vectors. Gene 1985, 33, 103–119. [Google Scholar] [CrossRef]
- Motiejūnaitė, R.; Armalytė, J.; Markuckas, A.; Sužiedėlienė, E. Escherichia coli dinJ-yafQ genes act as a toxin-antitoxin module. FEMS Microbiol. Lett. 2007, 268, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Guzman, L.M.; Belin, D.; Carson, M.J.; Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995, 177, 4121–4130. [Google Scholar] [CrossRef] [Green Version]
- Petkevičius, V.; Vaitekūnas, J.; Stankevičiūtė, J.; Gasparavičiūtė, R.; Meškys, R. Catabolism of 2-Hydroxypyridine by Burkholderia sp. Strain MAK1: A 2-Hydroxypyridine 5-Monooxygenase Encoded by hpdABCDE Catalyzes the First Step of Biodegradation. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armalytė, J.; Skerniškytė, J.; Bakienė, E.; Krasauskas, R.; Šiugždinienė, R.; Kareivienė, V.; Kerzienė, S.; Klimienė, I.; Sužiedėlienė, E.; Ružauskas, M. Microbial Diversity and Antimicrobial Resistance Profile in Microbiota From Soils of Conventional and Organic Farming Systems. Front. Microbiol. 2019, 10, 892. [Google Scholar] [CrossRef] [PubMed]
- Jurėnaitė, M.; Markuckas, A.; Sužiedėlienė, E. Identification and Characterization of Type II Toxin-Antitoxin Systems in the Opportunistic Pathogen Acinetobacter baumannii. J. Bacteriol. 2013, 195, 3165–3172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasauskas, R.; Skerniškytė, J.; Armalytė, J.; Sužiedėlienė, E. The role of Acinetobacter baumannii response regulator BfmR in pellicle formation and competitiveness via contact-dependent inhibition system. BMC Microbiol. 2019, 19, 241. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Predicted TA Family | Protein Domains (T-A) 1 | Found in Genomes (n = 21) | Conservativity of Operon (%) 2 | Prevalence in Other Bacteria 3 | Remarks |
|---|---|---|---|---|---|
| vapBC | PIN-AbrB | 21 | 84 | Stenotrophomonas spp., Xantomonas spp. | |
| - | COG3832- ArsR | 20 | 90 | Stenotrophomonas spp. and other genera | |
| relBE | RelE-Xre | 15 | 73 | Stenotrophomonas spp. | T before A |
| relBE | RelE-RHH | 10 | 92 | Stenotrophomonas spp. | |
| relBE (higBA) | HigB-Xre | 8 | 78 | Stenotrophomonas spp., Pseudomonas spp. | T before A |
| hicAB | HicA-HicB | 14 | 79 | Stenotrophomonas spp. | T before A |
| hipBA | HipA-Xre | 8 | 70 | Stenotrophomonas spp. (T common to Burkholderia spp.) | T is 440 a.a. |
| Strain/Plasmid | Description | Reference |
|---|---|---|
| E. coli JM107 | endA1 glnV44 thi-1 relA1 gyrA96 Δ(lac-proAB) [F’ traD36 proAB+ lacIq lacZΔM15] hsdR17(RK− mK+) λ− | [81] |
| E. coli BW25113 F’ | proA+B+lacIqΔlacZ)M15 zzf::mini-Tn10 (KanR) | [82] |
| pBAD30 | Expression plasmid | [83] |
| pUHE 25-2(cat) | Expression plasmid | [82] |
| pBAD1_gfp | gfp gene cloned into pBAD-MCS-1 vector | [84] |
| Isolate | Genus/Species | Origin | Source 1 | Isolation Year |
|---|---|---|---|---|
| D27 | S. maltophilia/rhizophila | Environmental | Soil (Conventional wheat) | 2016 |
| D32 | Stenotrophomonas sp. | Environmental | Soil (Conventional wheat) | 2016 |
| D34 | S. maltophilia | Environmental | Soil (Conventional wheat) | 2016 |
| D44 | S. maltophilia | Environmental | Soil (Conventional wheat) | 2016 |
| D46 | S. maltophilia | Environmental | Soil (Conventional wheat) | 2016 |
| D50 | S. maltophilia | Environmental | Soil (Conventional wheat) | 2016 |
| D53 | S. maltophilia | Environmental | Soil (Conventional wheat) | 2016 |
| D55 | Stenotrophomonas sp. | Environmental | Soil (Organic rapeseed) | 2016 |
| D59 | Stenotrophomonas sp. | Environmental | Soil (Organic rapeseed) | 2016 |
| D70 | S. rhizophila | Environmental | Soil (Organic rapeseed) | 2016 |
| D73 | Stenotrophomonas sp. | Environmental | Soil (Organic rapeseed) | 2016 |
| D125 | Stenotrophomonas sp. | Environmental | Soil (Organic maize) | 2016 |
| D150 | Stenotrophomonas sp. | Environmental | Soil (Conventional maize) | 2016 |
| Z53 | S. maltophilia | Environmental | Fish (Carp) | 2016 |
| SM3 | S. maltophilia | Clinical | NCI | 2017 |
| SM5 | S. maltophilia | Clinical | VCCH LM | 2018 |
| SM6 | S. maltophilia | Clinical | VCCH LM | 2018 |
| SM7 | S. maltophilia | Clinical | VCCH LM | 2018 |
| SM8 | S. maltophilia | Clinical | NCI | 2019 |
| SM9 | S. maltophilia | Clinical | NCI | 2019 |
| SM10 | S. maltophilia | Clinical | VCCH LM | 2019 |
| SM11 | S. maltophilia | Clinical | VUH SK PD | 2019 |
| SM12 | S. maltophilia | Clinical | VUH SK PD | 2019 |
| SM13 | S. maltophilia | Clinical | VUH SK PD | 2019 |
| SM14 | S. maltophilia | Clinical | VCCH LM | 2019 |
| SM15 | S.maltophilia | Clinical | KCPHC | 2019 |
| SM16 | S. maltophilia | Clinical | VCCH LM | 2019 |
| SM17 | S. maltophilia | Clinical | NCI | 2019 |
| SM18 | S. maltophilia | Clinical | NCI | 2019 |
| SM20 | S. maltophilia | Clinical | NCI | 2019 |
| SM21 | S. maltophilia | Clinical | NCI | 2019 |
| SM22 | S. maltophilia | Clinical | NCI | 2019 |
| SM23 | S. maltophilia | Clinical | NCI | 2019 |
| SM24 | S. maltophilia | Clinical | NCI | 2019 |
| SM25 | S. maltophilia | Clinical | NCI | 2019 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klimkaitė, L.; Armalytė, J.; Skerniškytė, J.; Sužiedėlienė, E. The Toxin-Antitoxin Systems of the Opportunistic Pathogen Stenotrophomonas maltophilia of Environmental and Clinical Origin. Toxins 2020, 12, 635. https://doi.org/10.3390/toxins12100635
Klimkaitė L, Armalytė J, Skerniškytė J, Sužiedėlienė E. The Toxin-Antitoxin Systems of the Opportunistic Pathogen Stenotrophomonas maltophilia of Environmental and Clinical Origin. Toxins. 2020; 12(10):635. https://doi.org/10.3390/toxins12100635
Chicago/Turabian StyleKlimkaitė, Laurita, Julija Armalytė, Jūratė Skerniškytė, and Edita Sužiedėlienė. 2020. "The Toxin-Antitoxin Systems of the Opportunistic Pathogen Stenotrophomonas maltophilia of Environmental and Clinical Origin" Toxins 12, no. 10: 635. https://doi.org/10.3390/toxins12100635
APA StyleKlimkaitė, L., Armalytė, J., Skerniškytė, J., & Sužiedėlienė, E. (2020). The Toxin-Antitoxin Systems of the Opportunistic Pathogen Stenotrophomonas maltophilia of Environmental and Clinical Origin. Toxins, 12(10), 635. https://doi.org/10.3390/toxins12100635