Targeting Type II Toxin–Antitoxin Systems as Antibacterial Strategies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
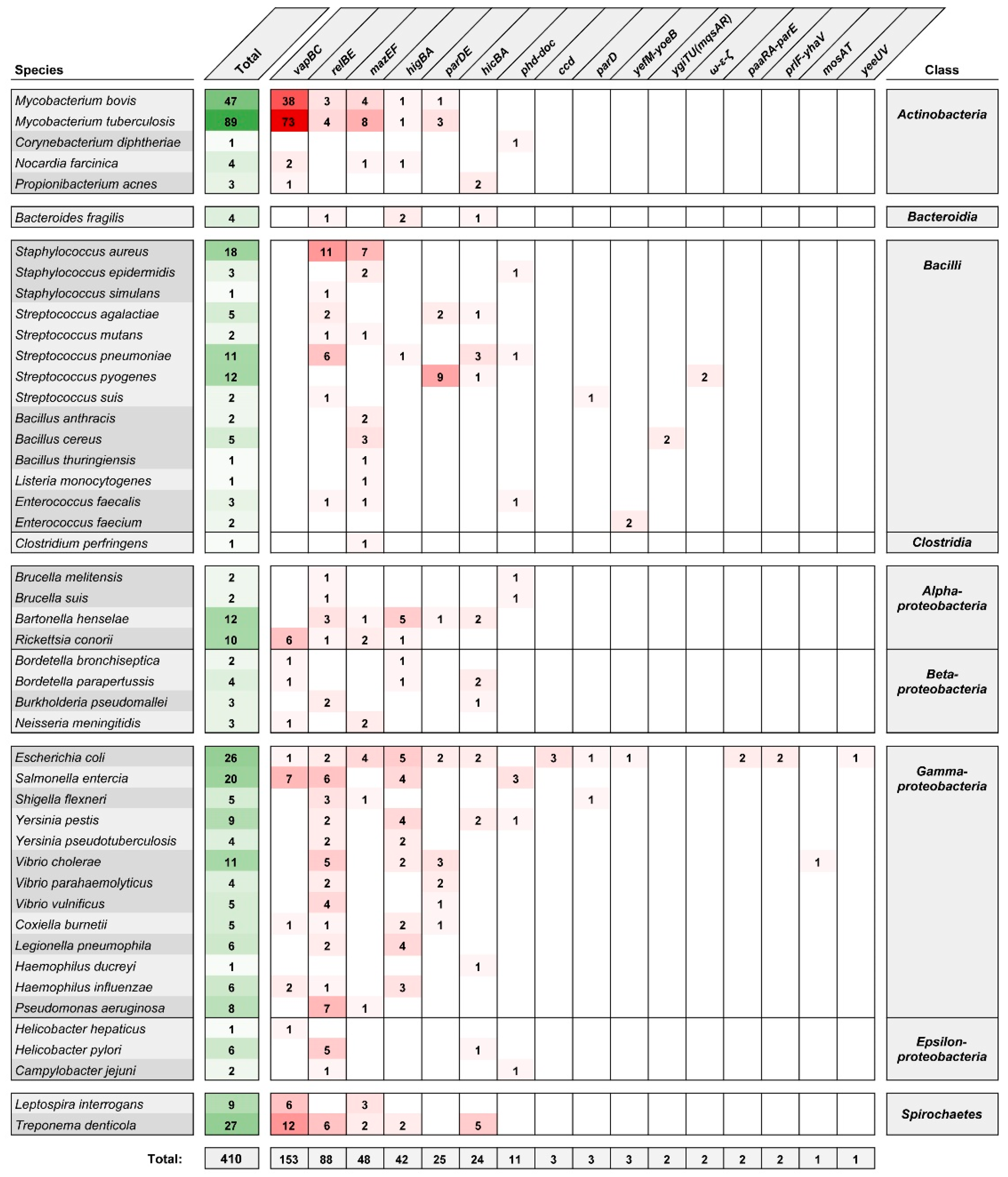
2. Type II TA Systems in Pathogenic Bacteria
3. Strategies for the Artificial Activation of Toxin–Antitoxin Systems
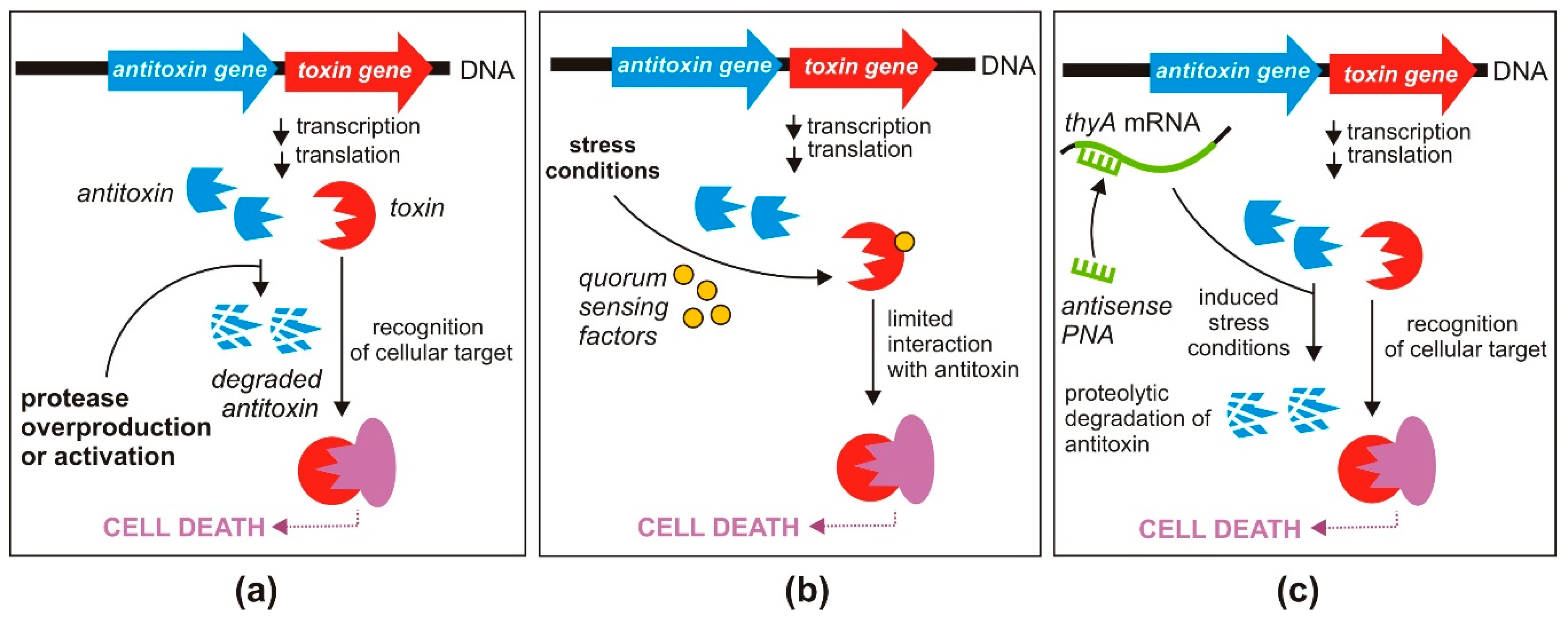
3.1. Direct Activation of TA Systems
3.1.1. Disruption or Preventing the Formation of TA Complexes
3.1.2. Inhibition of Antitoxin Translation
3.2. Indirect Activation of TA Systems
3.2.1. Enhanced Expression of Proteases Degrading Antitoxins
3.2.2. Triggering of TA Systems by Quorum-Sensing Factors
3.2.3. Induction of the Stringent Response
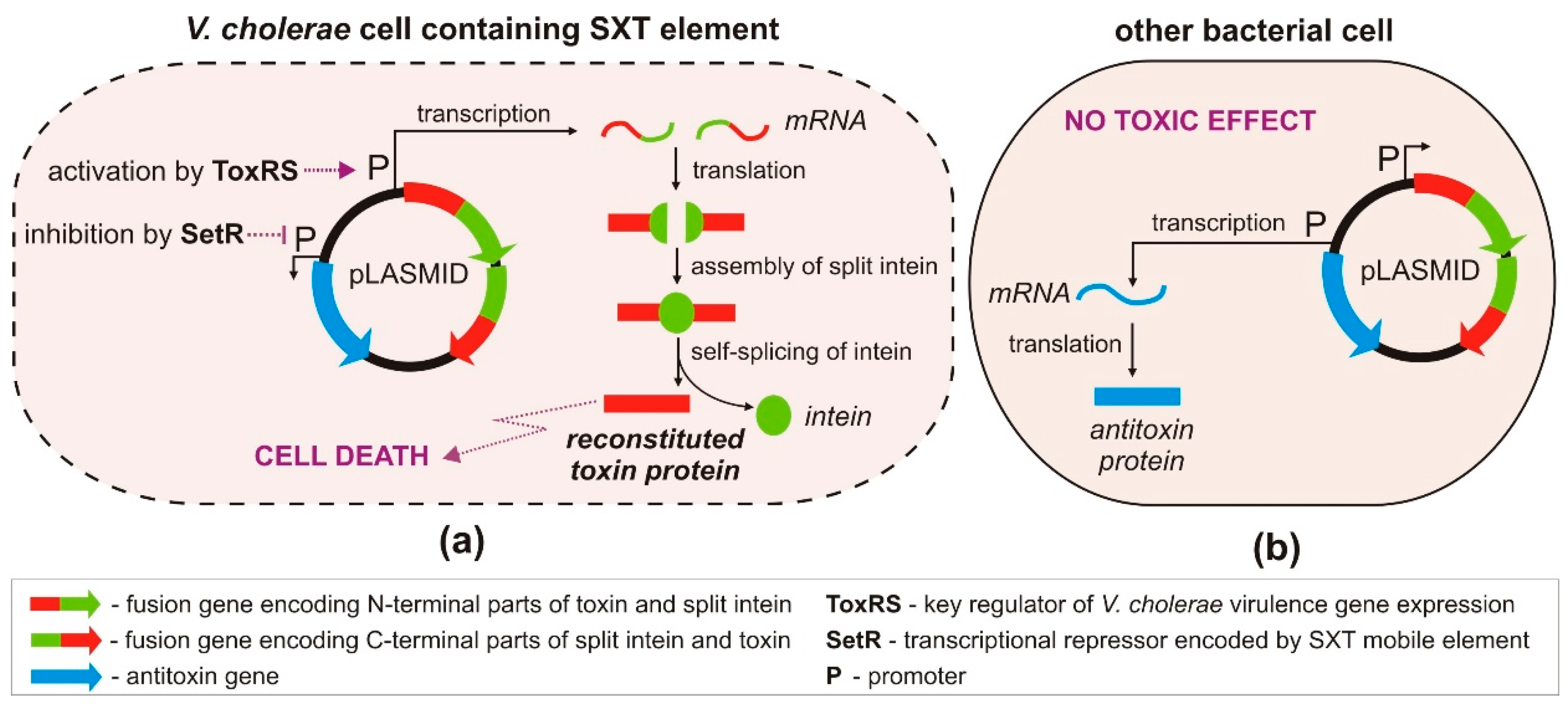
3.3. Engineered TA Systems in a Targeted Killing Strategy
4. Toxicity to Eukaryotic Cells
5. Role of Type II TA Modules in Biofilm Formation and Bacterial Persistence
6. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Ventola, C.L. The antibiotic resistance crisis: Causes and threats. Pharm. Ther. J. 2015, 40, 277–283. [Google Scholar]
- Gerdes, K.; Nielsen, A.; Thorsted, P.; Wagner, E.G.H. Mechanism of killer gene activation. Antisense RNA-dependent RNase III cleavage ensures rapid turn-over of the stable Hok, SrnB and PndA effector messenger RNAs. J. Mol. Biol. 1992, 226, 637–649. [Google Scholar] [CrossRef]
- Fraikin, N.; Goormaghtigh, F.; Van Melderen, L. Type II toxin-antitoxin systems: Evolution and revolutions. J. Bacteriol. 2020, 202, e00763-19. [Google Scholar] [CrossRef] [Green Version]
- Page, R.; Peti, W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 2016, 12, 208–214. [Google Scholar] [CrossRef]
- Coussens, N.P.; Daines, D.A. Wake me when it’s over-Bacterial toxin-antitoxin proteins and induced dormancy. Exp. Biol. Med. 2016, 241, 1332–1342. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Inouye, M. Regulation of growth and death in Escherichia coli by toxin–antitoxin systems. Nat. Rev. Microbiol. 2011, 9, 779–790. [Google Scholar] [CrossRef]
- Piscotta, F.J.; Jeffrey, P.D.; James Link, A. ParST is a widespread toxin–antitoxin module that targets nucleotide metabolism. Proc. Natl. Acad. Sci. USA 2019, 116, 826–834. [Google Scholar] [CrossRef] [Green Version]
- Unterholzner, S.J.; Poppenberger, B.; Rozhon, W. Toxin–antitoxin systems. Biology, identification, and application. Mob. Genet. Elem. 2013, 3, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Keren, I.; Shah, D.; Spoering, A.; Kaldalu, N.; Lewis, K. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 2004, 186, 8172–8180. [Google Scholar] [CrossRef] [Green Version]
- Robson, J.; McKenzie, J.L.; Cursons, R.; Cook, G.M.; Arcus, V.L. The vapBC operon from Mycobacterium smegmatis is an autoregulated toxin-antitoxin module that controls growth via inhibition of translation. J. Mol. Biol. 2009, 390, 353–367. [Google Scholar] [CrossRef]
- Dziewit, L.; Jazurek, M.; Drewniak, L.; Baj, J.; Bartosik, D. The SXT conjugative element and linear prophage N15 encode toxin-antitoxin-stabilizing systems homologous to the tad-ata module of the Paracoccus aminophilus plasmid pAMI2. J. Bacteriol. 2007, 189, 1983–1997. [Google Scholar] [CrossRef] [Green Version]
- Pandey, D.P.; Gerdes, K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005, 33, 966–976. [Google Scholar] [CrossRef]
- Christensen, S.K.; Mikkelsen, M.; Pedersen, K.; Gerdes, K. RelE, a global inhibitor of translation, is activated during nutritional stress. Proc. Natl. Acad. Sci. USA 2001, 98, 14328–14333. [Google Scholar] [CrossRef] [Green Version]
- Aizenman, E.; Engelberg-Kulka, H.; Glaser, G. An Escherichia coli chromosomal “addiction module” regulated by 3′,5′-bispyrophosphate: A model for programmed bacterial cell death. Proc. Natl. Acad. Sci. USA 1996, 93, 6059–6063. [Google Scholar] [CrossRef] [Green Version]
- Wen, Y.; Behiels, E.; Devreese, B. Toxin-antitoxin systems: Their role in persistence, biofilm formation, and pathogenicity. Pathog. Dis. 2014, 70, 240–249. [Google Scholar] [CrossRef]
- Kȩdzierska, B.; Lian, L.Y.; Hayes, F. Toxin-antitoxin regulation: Bimodal interaction of YefM-YoeB with paired DNA palindromes exerts transcriptional autorepression. Nucleic Acids Res. 2007, 35, 325–339. [Google Scholar] [CrossRef]
- Williams, J.; Hergenrother, P. Artificial activation of toxin-antitoxin systems as an antibacterial strategy. Trends Microbiol. 2014, 20, 291–298. [Google Scholar] [CrossRef] [Green Version]
- Harms, A.; Brodersen, D.E.; Mitarai, N.; Gerdes, K. Toxins, targets, and triggers: An overview of toxin-antitoxin biology. Mol. Cell 2018, 70, 768–784. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.Y.; Lee, B.J. Structure, biology, and therapeutic application of toxin-antitoxin systems in pathogenic bacteria. Toxins (Basel) 2016, 8, 305. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.T.; Balsa, D.; Espinosa, M. One cannot rule them all: Are bacterial toxins-antitoxins druggable? FEMS Microbiol. Rev. 2015, 39, 522–540. [Google Scholar] [CrossRef] [Green Version]
- Bravo, A.; Ruiz-Cruz, S.; Alkorta, I.; Espinosa, M. When humans met superbugs: Strategies to tackle bacterial resistances to antibiotics. Biomol. Concepts 2018, 9, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Carlos, L.; Barbosa, B.; Sânder, A.; Cangussu, R.; Garrido, S.S.; Marchetto, R. Toxin-antitoxin systems and its biotechnological applications. Afr. J. Biotechnol. 2014, 13, 11–17. [Google Scholar]
- Shao, Y.; Harrison, E.M.; Bi, D.; Tai, C.; He, X.; Ou, H.Y.; Rajakumar, K.; Deng, Z. TADB: A web-based resource for type 2 toxin-antitoxin loci in bacteria and archaea. Nucleic Acids Res. 2011, 39, D606–D611. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Wei, Y.; Shen, Y.; Li, X.; Zhou, H.; Tai, C.; Deng, Z.; Ou, H.Y. TADB 2.0: An updated database of bacterial type II toxin-antitoxin loci. Nucleic Acids Res. 2018, 46, D749–D753. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K.; Christensen, S.K.; Løbner-Olesen, A. Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 2005, 3, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Van Melderen, L.; De Bast, M.S. Bacterial toxin-antitoxin systems: More than selfish entities? PLoS Genet. 2009, 5, e1000437. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Comprehensive comparative-genomic analysis of Type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct 2009, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Garcia, L.; Blasco, L.; Lopez, M.; Bou, G.; Garcia-Contreras, R.; Wood, T.; Tomas, M. Toxin-antitoxin systems in clinical pathogens. Toxins (Basel) 2016, 8, 227. [Google Scholar] [CrossRef] [Green Version]
- Lobato-Márquez, D.; Díaz-Orejas, R.; García-del Portillo, F. Toxin-antitoxins and bacterial virulence. FEMS Microbiol. Rev. 2016, 40, 592–609. [Google Scholar] [CrossRef] [Green Version]
- Kędzierska, B.; Hayes, F. Emerging roles of toxin-antitoxin modules in bacterial pathogenesis. Molecules 2016, 21, 790. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.E.; Walsh, T.R. Toxin-antitoxin systems and their role in disseminating and maintaining antimicrobial resistance. FEMS Microbiol. Rev. 2017, 41, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.M.; Kim, D.H.; Jin, C.; Lee, B.J. A systematic overview of type II and III toxin-antitoxin systems with a focus on druggability. Toxins (Basel) 2018, 10, 515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slayden, R.A.; Dawson, C.C.; Cummings, J.E. Toxin-antitoxin systems and regulatory mechanisms in Mycobacterium tuberculosis. Pathog. Dis. 2018, 76, fty039. [Google Scholar] [CrossRef] [PubMed]
- Korch, S.B.; Malhotra, V.; Contreras, H.; Clark-Curtiss, J.E. The Mycobacterium tuberculosis relBE toxin:antitoxin genes are stress-responsive modules that regulate growth through translation inhibition. J. Microbiol. 2015, 53, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Venkataraman, B.; Vasudevan, M.; Gopinath Bankar, K. Co-expression network analysis of toxin-antitoxin loci in Mycobacterium tuberculosis reveals key modulators of cellular stress. Sci. Rep. 2017, 7, 5868. [Google Scholar] [CrossRef] [PubMed]
- Thakur, Z.; Saini, V.; Arya, P.; Kumar, A.; Mehta, P.K. Computational insights into promoter architecture of toxin-antitoxin systems of Mycobacterium tuberculosis. Gene 2018, 641, 161–171. [Google Scholar] [CrossRef]
- Yang, M.; Gao, C.; Wang, Y.; Zhang, H.; He, Z.G. Characterization of the interaction and cross-regulation of three Mycobacterium tuberculosis RelBE modules. PLoS ONE 2010, 5, e10672. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Tiwari, P.; Deep, A.; Kidwai, S.; Gupta, S.; Thakur, K.G.; Singh, R. System-wide analysis unravels the differential regulation and in vivo essentiality of virulence-associated proteins B and C toxin-antitoxin systems of Mycobacterium tuberculosis. J. Infect. Dis. 2018, 217, 1809–1820. [Google Scholar] [CrossRef]
- Zhu, L.; Sharp, J.D.; Kobayashi, H.; Woychik, N.A.; Inouye, M. Noncognate Mycobacterium tuberculosis toxin-antitoxins can physically and functionally interact. J. Biol. Chem. 2010, 285, 39732–39738. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, M.V.; Dawson, C.C.; Crew, R.; England, K.; Slayden, R.A. MazF6 toxin of Mycobacterium tuberculosis demonstrates antitoxin specificity and is coupled to regulation of cell growth by a Soj-like protein. BMC Microbiol. 2013, 13, 240. [Google Scholar] [CrossRef] [Green Version]
- Tandon, H.; Melarkode Vattekatte, A.; Srinivasan, N.; Sandhya, S. Molecular and structural basis of cross-reactivity in M. tuberculosis toxin–antitoxin systems. Toxins (Basel) 2020, 12, 481. [Google Scholar] [CrossRef] [PubMed]
- Solano-Gutierrez, J.S.; Pino, C.; Robledo, J. Toxin-antitoxin systems shows variability among Mycobacterium tuberculosis lineages. FEMS Microbiol. Lett. 2019, 366, fny276. [Google Scholar] [CrossRef] [PubMed]
- Tandon, H.; Sharma, A.; Wadhwa, S.; Varadarajan, R.; Singh, R.; Srinivasan, N.; Sandhya, S. Bioinformatic and mutational studies of related toxin–antitoxin pairs in Mycobacterium tuberculosis predict and identify key functional residues. J. Biol. Chem. 2019, 294, 9048–9063. [Google Scholar] [CrossRef] [Green Version]
- Schuster, C.F.; Bertram, R. Toxin-antitoxin systems of Staphylococcus aureus. Toxins (Basel) 2016, 8, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukowski, M.; Hyz, K.; Janczak, M.; Hydzik, M.; Dubin, G.; Wladyka, B. Identification of novel mazEF/pemIK family toxin-antitoxin loci and their distribution in the Staphylococcus genus. Sci. Rep. 2017, 7, 13462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, G.; Zhu, Q.; Sun, B. Bioinformatics and functional assessment of toxin-antitoxin systems in Staphylococcus aureus. Toxins (Basel) 2018, 10, 473. [Google Scholar] [CrossRef] [Green Version]
- Kato, F.; Yoshizumi, S.; Yamaguchi, Y.; Inouye, M. Genome-wide screening for identification of novel toxin-antitoxin systems in Staphylococcus aureus. Appl. Environ. Microbiol. 2019, 85, e00915-19. [Google Scholar] [CrossRef]
- Sierra, R.; Viollier, P.; Renzoni, A. Linking toxin-antitoxin systems with phenotypes: A Staphylococcus aureus viewpoint. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 742–751. [Google Scholar] [CrossRef]
- Horesh, G.; Fino, C.; Harms, A.; Dorman, M.J.; Parts, L.; Gerdes, K.; Heinz, E.; Thomson, N.R. Type II and type IV toxin-antitoxin systems show different evolutionary patterns in the global Klebsiella pneumoniae population. Nucleic Acids Res. 2020, 48, 4357–4370. [Google Scholar] [CrossRef] [Green Version]
- Akarsu, H.; Bordes, P.; Mansour, M.; Bigot, D.J.; Genevaux, P.; Falquet, L. TASmania: A bacterial toxin-antitoxin systems database. PLoS Comput. Biol. 2019, 15, e1006946. [Google Scholar] [CrossRef] [Green Version]
- Agüero, J.A.; Akarsu, H.; Aguilar-Bultet, L.; Oevermann, A.; Falquet, L. Large-scale comparison of toxin and antitoxins in listeria monocytogenes. Toxins (Basel) 2020, 12, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, S. Speculative strategies for new antibacterials: All roads should not lead to Rome. J. Antibiot. (Tokyo) 2013, 66, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Germain, E.; Castro-Roa, D.; Zenkin, N.; Gerdes, K. Molecular mechanism of bacterial persistence by HipA. Mol. Cell 2013, 52, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-P.; Wang, Q.; Quan, S.-W.; Yu, X.-Q.; Wang, Y.; Guo, D.-D.; Peng, L.; Feng, H.-Y.; Yong-Xing, H. Type II toxin–antitoxin system in bacteria: Activation, function, and mode of action. Biophys. Rep. 2020, 6, 68–79. [Google Scholar] [CrossRef]
- Ramisetty, B.C.M.; Santhosh, R.S. Endoribonuclease type II toxin-antitoxin systems: Functional or selfish? Microbiology 2017, 163, 931–939. [Google Scholar] [CrossRef]
- Lioy, V.S.; Rey, O.; Balsa, D.; Pellicer, T.; Alonso, J.C. A toxin-antitoxin module as a target for antimicrobial development. Plasmid 2010, 63, 31–39. [Google Scholar] [CrossRef]
- Agarwal, S.; Agarwal, S.; Bhatnagar, R. Identification and characterization of a novel toxin-antitoxin module from Bacillus anthracis. FEBS Lett. 2007, 581, 1727–1734. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Mishra, N.K.; Bhatnagar, S.; Bhatnagar, R. PemK toxin of Bacillus anthracis is a ribonuclease: An insight into its active site, structure, and function. J. Biol. Chem. 2010, 285, 7254–7270. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; Kumar, S.; Gupta, V.P.; Gourinath, S.; Bhatnagar, S.; Bhatnagar, R. Structural basis of Bacillus anthracis MoxXT disruption and the modulation of MoxT ribonuclease activity by rationally designed peptides. J. Biomol. Struct. Dyn. 2015, 33, 606–624. [Google Scholar] [CrossRef]
- Chopra, N.; Agarwal, S.; Verma, S.; Bhatnagar, S.; Bhatnagar, R. Modeling of the structure and interactions of the B. anthracis antitoxin, MoxX: Deletion mutant studies highlight its modular structure and repressor function. J. Comput. Aided. Mol. Des. 2011, 25, 275–291. [Google Scholar] [CrossRef]
- Lee, I.G.; Lee, S.J.; Chae, S.; Lee, K.Y.; Kim, J.H.; Lee, B.J. Structural and functional studies of the Mycobacterium tuberculosis VapBC30 toxin-antitoxin system: Implications for the design of novel antimicrobial peptides. Nucleic Acids Res. 2015, 43, 7624–7637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, W.T.; Moreno-Córdoba, I.; Yeo, C.C.; Espinosa, M. Toxin-antitoxin genes of the Gram-positive pathogen Streptococcus pneumoniae: So few and yet so many. Microbiol. Mol. Biol. Rev. 2012, 76, 773–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Równicki, M.; Pieńko, T.; Czarnecki, J.; Kolanowska, M.; Bartosik, D.; Trylska, J. Artificial activation of Escherichia coli mazEF and hipBA toxin–antitoxin systems by antisense peptide nucleic acids as an antibacterial strategy. Front. Microbiol. 2018, 9, 2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Good, L.; Awasthi, S.K.; Dryselius, R.; Larsson, O.; Nielsen, P.E. Bactericidal antisense effects of peptide-PNA conjugates. Nat. Biotechnol. 2001, 19, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowska, M.; Równicki, M.; Mieczkowski, A.; Miszkiewicz, J.; Trylska, J. Antibacterial peptide nucleic acids—Facts and perspectives. Molecules 2020, 25, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, S.K.; Maenhaut-Michel, G.; Mine, N.; Gottesman, S.; Gerdes, K.; Van Melderen, L. Overproduction of the Lon protease triggers inhibition of translation in Escherichia coli: Involvement of the yefM-yoeB toxin-antitoxin system. Mol. Microbiol. 2004, 51, 1705–1717. [Google Scholar] [CrossRef]
- Brzozowska, I.; Zielenkiewicz, U. The ClpXP protease is responsible for the degradation of the Epsilon antidote to the Zeta toxin of the streptococcal pSM19035 plasmid. J. Biol. Chem. 2014, 289, 7514–7523. [Google Scholar] [CrossRef] [Green Version]
- Van Melderen, L.; Thi, M.H.D.; Lecchi, P.; Gottesman, S.; Couturier, M.; Maurizi, M.R. ATP-dependent degradation of CcdA by Lon protease. Effects of secondary structure and heterologous subunit interactions. J. Biol. Chem. 1996, 271, 27730–27738. [Google Scholar] [CrossRef] [Green Version]
- Muthuramalingam, M.; White, J.C.; Bourne, C.R. Toxin-antitoxin modules are pliable switches activated by multiple protease pathways. Toxins (Basel) 2016, 8, 214. [Google Scholar] [CrossRef]
- Goff, S.A.; Goldberg, A.L. An increased content of protease La, the lon gene product, increases protein degradation and blocks growth in Escherichia coli. J. Biol. Chem. 1987, 262, 4508–4515. [Google Scholar]
- Brötz-Oesterhelt, H.; Beyer, D.; Kroll, H.P.; Endermann, R.; Ladel, C.; Schroeder, W.; Hinzen, B.; Raddatz, S.; Paulsen, H.; Henninger, K.; et al. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat. Med. 2005, 11, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Engelberg-Kulka, H. Quorum sensing peptides mediating interspecies bacterial cell death as a novel class of antimicrobial agents. Curr. Opin. Microbiol. 2014, 21, 22–27. [Google Scholar] [CrossRef]
- Kumar, S.; Kolodkin-Gal, I.; Engelberg-Kulka, H. Novel quorum-sensing peptides mediating interspecies bacterial cell death. MBio 2013, 4, e00314-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelberg-Kulka, H.; Hazan, R.; Amitai, S. mazEF: A chromosomal toxin-antitoxin module that triggers programmed cell death in bacteria. J. Cell Sci. 2005, 118, 4327–4332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigam, A.; Kumar, S.; Engelberg-Kulka, H. Quorum sensing extracellular death peptides enhance the endoribonucleolytic activities of mycobacterium tuberculosis MazF toxins. MBio 2018, 9, e00685-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramisetty, B.C.M.; Raj, S.; Ghosh, D. Escherichia coli MazEF toxin-antitoxin system does not mediate programmed cell death. J. Basic Microbiol. 2016, 56, 1398–1402. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Tamber, S.; Memmi, G.; Donegan, N.P.; Cheung, A.L. Overexpression of MazF in Staphylococcus aureus induces bacteriostasis by selectively targeting mRNAs for cleavage. J. Bacteriol. 2009, 191, 2051–2059. [Google Scholar] [CrossRef] [Green Version]
- Ronneau, S.; Helaine, S. Clarifying the link between toxin–antitoxin modules and bacterial persistence. J. Mol. Biol. 2019, 431, 3462–3471. [Google Scholar] [CrossRef]
- Jimmy, S.; Saha, C.K.; Kurata, T.; Stavropoulos, C.; Oliveira, S.R.A.; Koh, A.; Cepauskas, A.; Takada, H.; Rejman, D.; Tenson, T.; et al. A widespread toxin–antitoxin system exploiting growth control via alarmone signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 10500–10510. [Google Scholar] [CrossRef]
- Török, I.; Kari, C. Accumulation of ppGpp in a relA mutant of Escherichia coli during amino acid starvation. J. Biol. Chem. 1980, 255, 3838–3840. [Google Scholar]
- Ramisetty, B.C.M.; Natarajan, B.; Santhosh, R.S. MazEF-mediated programmed cell death in bacteria: “What is this?”. Crit. Rev. Microbiol. 2015, 41, 89–100. [Google Scholar] [CrossRef] [PubMed]
- López-Igual, R.; Bernal-Bayard, J.; Rodríguez-Patón, A.; Ghigo, J.M.; Mazel, D. Engineered toxin–intein antimicrobials can selectively target and kill antibiotic-resistant bacteria in mixed populations. Nat. Biotechnol. 2019, 37, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Collin, F.; Karkare, S.; Maxwell, A. Exploiting bacterial DNA gyrase as a drug target: Current state and perspectives. Appl. Microbiol. Biotechnol. 2011, 92, 479–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, P.; Kézdy, K.E.; Van Melderen, L.; Steyaert, J.; Wyns, L.; Pato, M.L.; Higgins, P.N.; Couturier, M. The F plasmid CcdB protein induces efficient ATP-dependent DNA cleavage by gyrase. J. Mol. Biol. 1993, 234, 534–541. [Google Scholar] [CrossRef]
- Childers, B.M.; Klose, K.E. Regulation of virulence in Vibrio cholerae: The ToxR regulon. Future Microbiol. 2007, 2, 335–344. [Google Scholar] [CrossRef]
- Beaber, J.W.; Hochhut, B.; Waldor, M.K. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature 2004, 427, 72–74. [Google Scholar] [CrossRef]
- Shimazu, T.; Mirochnitchenko, O.; Phadtare, S.; Inouye, M. Regression of solid tumors by induction of MazF, a bacterial mRNA endoribonuclease. J. Mol. Microbiol. Biotechnol. 2014, 24, 228–233. [Google Scholar] [CrossRef]
- Chono, H.; Saito, N.; Tsuda, H.; Shibata, H.; Ageyama, N.; Terao, K.; Yasutomi, Y.; Mineno, J.; Kato, I. In vivo safety and persistence of endoribonuclease gene-transduced CD4+ t cells in cynomolgus macaques for HIV-1 gene therapy model. PLoS ONE 2011, 6, e23585. [Google Scholar] [CrossRef]
- Chono, H.; Matsumoto, K.; Tsuda, H.; Saito, N.; Lee, K.; Kim, S.; Shibata, H.; Ageyama, N.; Terao, K.; Yasutomi, Y.; et al. Acquisition of HIV-1 resistance in T lymphocytes using an ACA-specific E. coli mRNA interferase. Hum. Gene Ther. 2011, 22, 35–43. [Google Scholar] [CrossRef]
- Lewis, K. Persister cells. Annu. Rev. Microbiol. 2010, 64, 357–372. [Google Scholar] [CrossRef]
- Lewis, K. Persister cells and the riddle of biofilm survival. Biochemistry 2005, 70, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Multidrug tolerance of biofilms and persister cells. Curr. Top. Microbiol. Immunol. 2008, 322, 107–131. [Google Scholar] [PubMed]
- Wood, T.K.; Knabel, S.J.; Kwan, B.W. Bacterial persister cell formation and dormancy. Appl. Environ. Microbiol. 2013, 79, 7116–7121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebeaux, D.; Ghigo, J.-M.; Beloin, C. Biofilm-related infections: Bridging the gap between clinical management and fundamental aspects of recalcitrance toward antibiotics. Microbiol. Mol. Biol. Rev. 2014, 78, 510–543. [Google Scholar] [CrossRef] [Green Version]
- Soo, V.W.C.; Wood, T.K. Antitoxin MqsA represses curli formation through the master biofilm regulator CsgD. Sci. Rep. 2013, 3, 3186. [Google Scholar] [CrossRef] [Green Version]
- González Barrios, A.F.; Zuo, R.; Hashimoto, Y.; Yang, L.; Bentley, W.E.; Wood, T.K. Autoinducer 2 controls biofilm formation in Escherichia coli through a novel motility quorum-sensing regulator (MqsR, B3022). J. Bacteriol. 2006, 188, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Kasari, V.; Kurg, K.; Margus, T.; Tenson, T.; Kaldalu, N. The Escherichia coli mqsR and ygiT genes encode a new toxin-antitoxin pair. J. Bacteriol. 2010, 192, 2908–2919. [Google Scholar] [CrossRef] [Green Version]
- Fraikin, N.; Rousseau, C.J.; Goeders, N.; Van Melderen, L. Reassessing the role of the type II MqsRA toxin-antitoxin system in stress response and biofilm formation: MqsA is transcriptionally uncoupled from mqsR. MBio 2019, 10, e02678-19. [Google Scholar] [CrossRef] [Green Version]
- Ramisetty, B.C.M.; Ghosh, D.; Chowdhury, M.R.; Santhosh, R.S. Corrigendum: What is the link between stringent response, endoribonuclease encoding type II toxin-antitoxin systems and persistence? Front. Microbiol. 2017, 8, 458. [Google Scholar] [CrossRef]
- Moyed, H.S.; Bertrand, K.P. hipA, a newly recognized gene of Escherichia coli K-12 that affects frequency of persistence after inhibition of murein synthesis. J. Bacteriol. 1983, 155, 768–775. [Google Scholar] [CrossRef] [Green Version]
- Van Melderen, L.; Wood, T.K. Commentary: What is the link between stringent response, endoribonuclease encoding type II toxin-antitoxin systems and persistence? Front. Microbiol. 2017, 14, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, Y.; Gandt, A.B.; Rowe, S.E.; Deisinger, J.P.; Conlon, B.P.; Lewis, K. ATP-Dependent persister formation in Escherichia coli. MBio 2017, 8, e02267-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramisetty, B.C.M.; Ghosh, D.; Chowdhury, M.R.; Santhosh, R.S. What is the link between stringent response, endoribonuclease encoding type II toxin-antitoxin systems and persistence? Front. Microbiol. 2016, 7, 1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, A.; Maisonneuve, E.; Gerdes, K. Mechanisms of bacterial persistence during stress and antibiotic exposure. Science 2016, 16, aaf4268. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, E.; Castro-Camargo, M.; Gerdes, K. Retraction notice to: (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 2018, 172, 1135. [Google Scholar] [CrossRef] [Green Version]
- Goormaghtigh, F.; Fraikin, N.; Putrinš, M.; Hallaert, T.; Hauryliuk, V.; Garcia-Pino, A.; Sjödin, A.; Kasvandik, S.; Udekwu, K.; Tenson, T.; et al. Reassessing the role of type II toxin-antitoxin systems in formation of escherichia coli type II persister cells. MBio 2018, 9, e00640-18. [Google Scholar] [CrossRef] [Green Version]
- Goeders, N.; Van Melderen, L. Toxin-antitoxin systems as multilevel interaction systems. Toxins (Basel) 2013, 6, 304–324. [Google Scholar] [CrossRef] [Green Version]
- Van Melderen, L. Toxin-antitoxin systems: Why so many, what for? Curr. Opin. Microbiol. 2010, 13, 781–785. [Google Scholar] [CrossRef]
- Pacios, O.; Blasco, L.; Bleriot, I.; Fernandez-Garcia, L.; Ambroa, A.; López, M.; Bou, G.; Cantón, R.; Garcia-Contreras, R.; Wood, T.K.; et al. (p)ppGpp and its role in bacterial persistence: New challenges. Antimicrob. Agents Chemother. 2020. [Google Scholar] [CrossRef]
- Tripathi, A.; Dewan, P.C.; Siddique, S.A.; Varadarajan, R. MazF-induced growth inhibition and persister generation in Escherichia coli. J. Biol. Chem. 2014, 289, 4191–4205. [Google Scholar] [CrossRef] [Green Version]
- Harrison, J.J.; Wade, W.D.; Akierman, S.; Vacchi-Suzzi, C.; Stremick, C.A.; Turner, R.J.; Ceri, H. The chromosomal toxin gene yafQ is a determinant of multidrug tolerance for Escherichia coli growing in a biofilm. Antimicrob. Agents Chemother. 2009, 53, 2253–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norton, J.P.; Mulvey, M.A. Toxin-Antitoxin Systems Are Important for Niche-Specific Colonization and Stress Resistance of Uropathogenic Escherichia coli. PLoS Pathog. 2012, 8, e1002954. [Google Scholar] [CrossRef] [PubMed]
- Slattery, A.; Victorsen, A.H.; Brown, A.; Hillman, K.; Phillips, G.J. Isolation of highly persistent mutants of Salmonella enterica serovar typhimurium reveals a new toxin-antitoxin module. J. Bacteriol. 2013, 195, 647–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helaine, S.; Cheverton, A.M.; Watson, K.G.; Faure, L.M.; Matthews, S.A.; Holden, D.W. Internalization of salmonella by macrophages induces formation of nonreplicating persisters. Science 2014, 343, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Cheverton, A.M.; Gollan, B.; Przydacz, M.; Wong, C.T.; Mylona, A.; Hare, S.A.; Helaine, S. A Salmonella toxin promotes persister formation through acetylation of tRNA. Mol. Cell 2016, 63, 86–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rycroft, J.A.; Gollan, B.; Grabe, G.J.; Hall, A.; Cheverton, A.M.; Larrouy-Maumus, G.; Hare, S.A.; Helaine, S. Activity of acetyltransferase toxins involved in Salmonella persister formation during macrophage infection. Nat. Commun. 2018, 9, 1993. [Google Scholar] [CrossRef]
- Fisher, R.A.; Gollan, B.; Helaine, S. Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 2017, 15, 453–464. [Google Scholar] [CrossRef]
- Wilmaerts, D.; Dewachter, L.; De Loose, P.-J.; Bollen, C.; Verstraeten, N.; Michiels, J. HokB Monomerization and Membrane Repolarization Control Persister Awakening. Mol. Cell 2019, 75, 1031–1042. [Google Scholar] [CrossRef]
- Dörr, T.; Vulic, M.; Lewis, K. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 2010, 8, e1000317. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.Y.; Soo, V.W.C.; Islam, S.; McAnulty, M.J.; Benedik, M.J.; Wood, T.K. Toxin GhoT of the GhoT/GhoS toxin/antitoxin system damages the cell membrane to reduce adenosine triphosphate and to reduce growth under stress. Environ. Microbiol. 2014, 16, 1741–1754. [Google Scholar] [CrossRef]
- Wang, X.; Lord, D.M.; Hong, S.H.; Peti, W.; Benedik, M.J.; Page, R.; Wood, T.K. Type II toxin/antitoxin MqsR/MqsA controls type V toxin/antitoxin GhoT/GhoS. Environ. Microbiol. 2013, 15, 1734–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Defraine, V.; Fauvart, M.; Michiels, J. Fighting bacterial persistence: Current and emerging anti-persister strategies and therapeutics. Drug Resist. Updates 2018, 38, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Conlon, B.P.; Rowe, S.E.; Gandt, A.B.; Nuxoll, A.S.; Donegan, N.P.; Zalis, E.A.; Clair, G.; Adkins, J.N.; Cheung, A.L.; Lewis, K. Persister formation in Staphylococcus aureus is associated with ATP depletion. Nat. Microbiol. 2016, 1, 16051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Yin, N.; Liu, H.; Pei, J.; Lai, L. Novel inhibitors of toxin HipA reduce multidrug tolerant persisters. ACS Med. Chem. Lett. 2016, 7, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Solecki, O.; Mosbah, A.; Baudy Floc’h, M.; Felden, B. Converting a Staphylococcus aureus toxin into effective cyclic pseudopeptide antibiotics. Chem. Biol. 2015, 22, 329–335. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Równicki, M.; Lasek, R.; Trylska, J.; Bartosik, D. Targeting Type II Toxin–Antitoxin Systems as Antibacterial Strategies. Toxins 2020, 12, 568. https://doi.org/10.3390/toxins12090568
Równicki M, Lasek R, Trylska J, Bartosik D. Targeting Type II Toxin–Antitoxin Systems as Antibacterial Strategies. Toxins. 2020; 12(9):568. https://doi.org/10.3390/toxins12090568
Chicago/Turabian StyleRównicki, Marcin, Robert Lasek, Joanna Trylska, and Dariusz Bartosik. 2020. "Targeting Type II Toxin–Antitoxin Systems as Antibacterial Strategies" Toxins 12, no. 9: 568. https://doi.org/10.3390/toxins12090568
APA StyleRównicki, M., Lasek, R., Trylska, J., & Bartosik, D. (2020). Targeting Type II Toxin–Antitoxin Systems as Antibacterial Strategies. Toxins, 12(9), 568. https://doi.org/10.3390/toxins12090568