In Silico Modeling of Spirolides and Gymnodimines: Determination of S Configuration at Butenolide Ring Carbon C-4


Abstract
:
1. Introduction
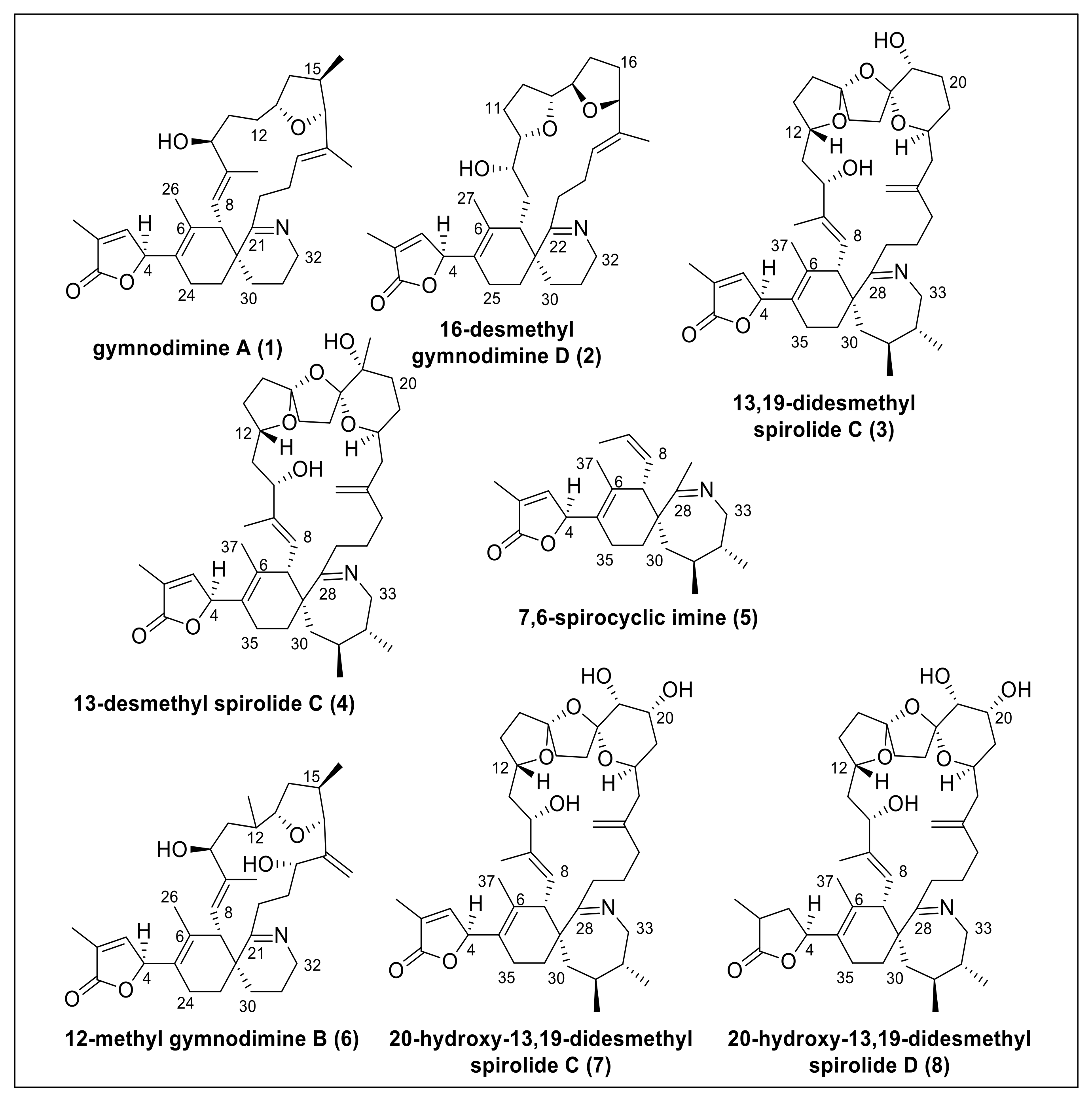
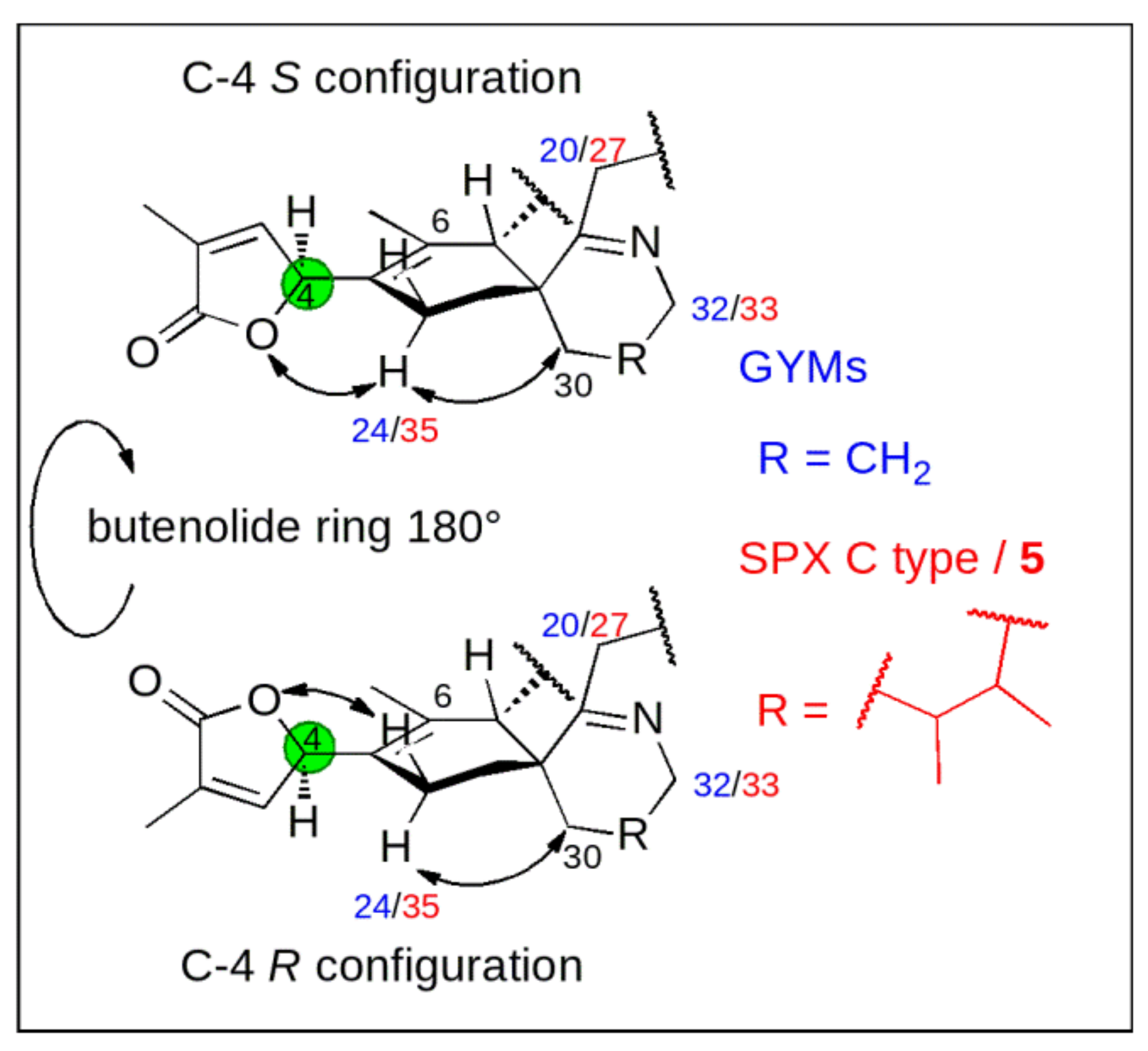
Conceptual Idea
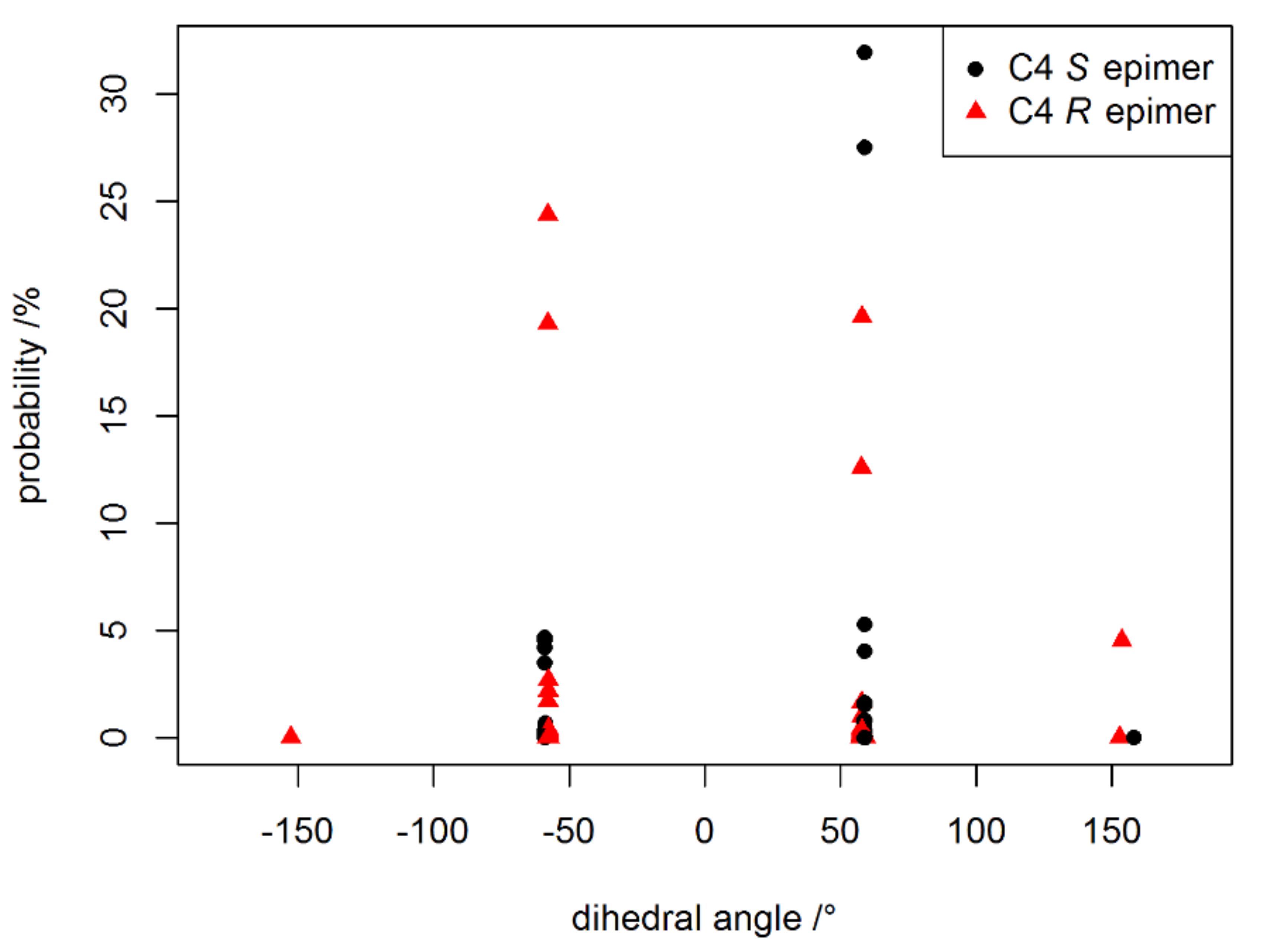
2. Results and Discussion
2.1. Simulation of Shielding Tensors
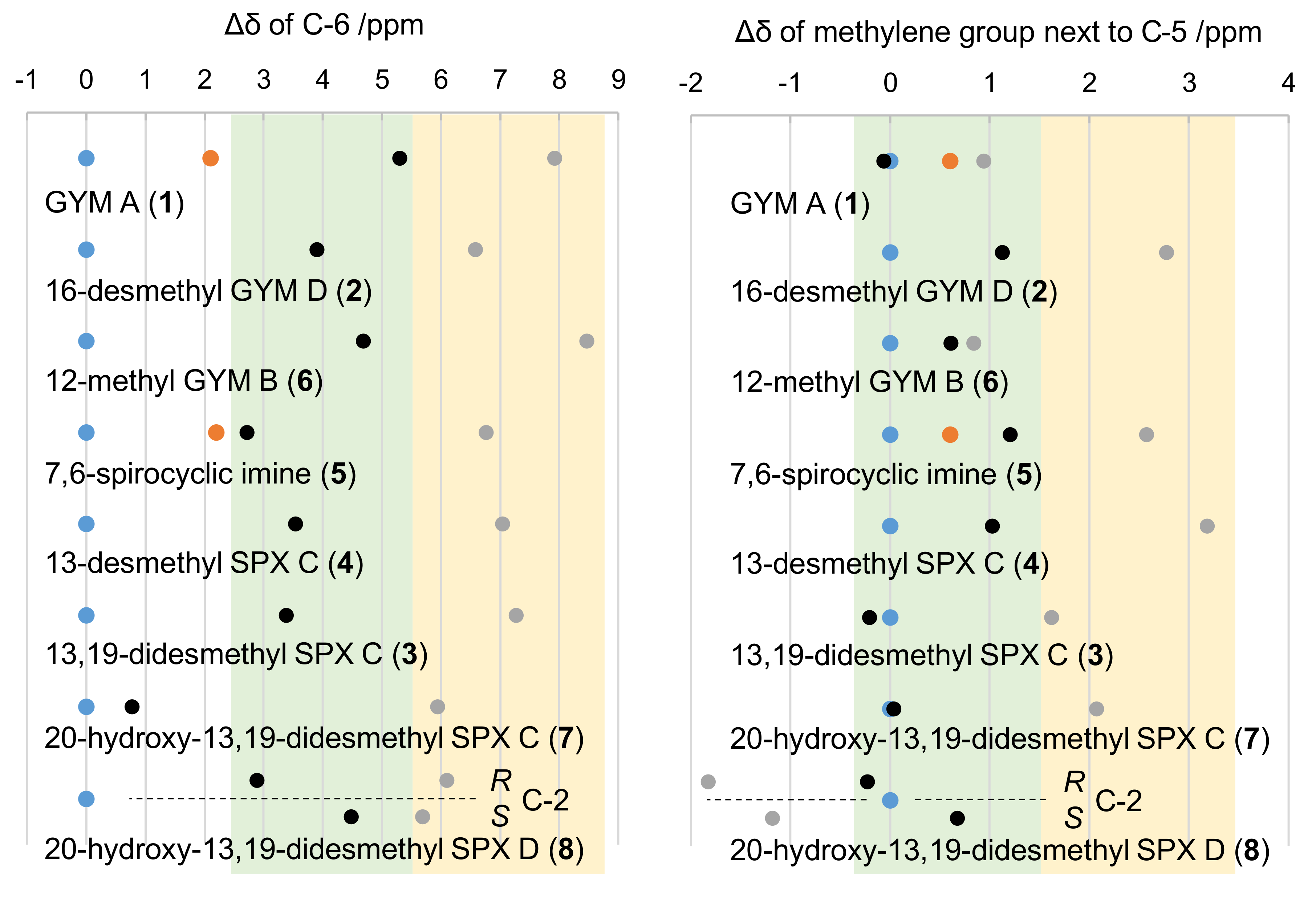
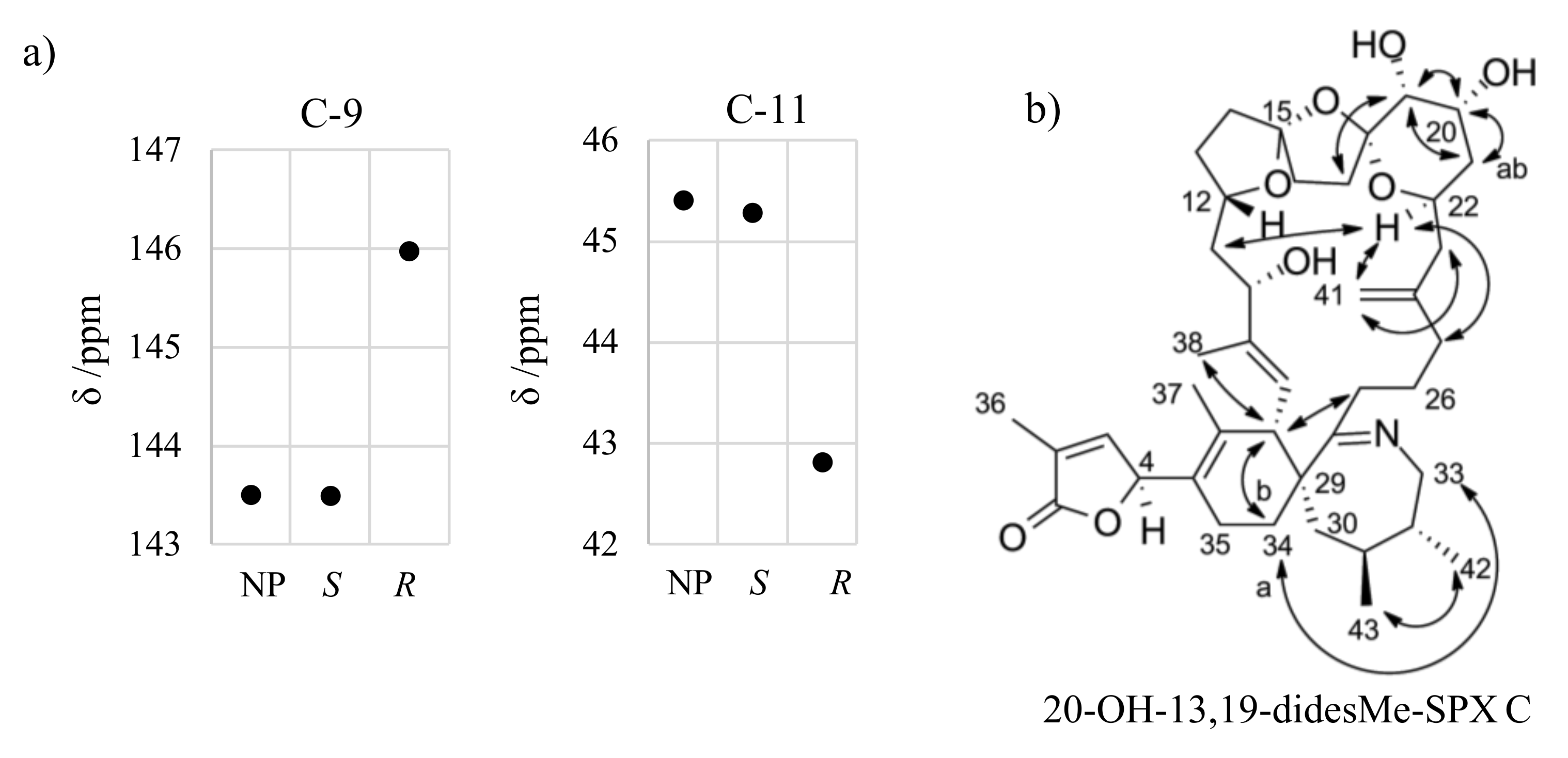
2.2. Individual Carbon Atoms
2.3. DP4+ Probability
2.4. Re-Examination of C-4 in 3
3. Conclusions
4. Materials and Methods
4.1. Parameter Setup
4.2. Conformational Search
4.3. Determination of Shielding Tensors and NMR Chemical Shifts
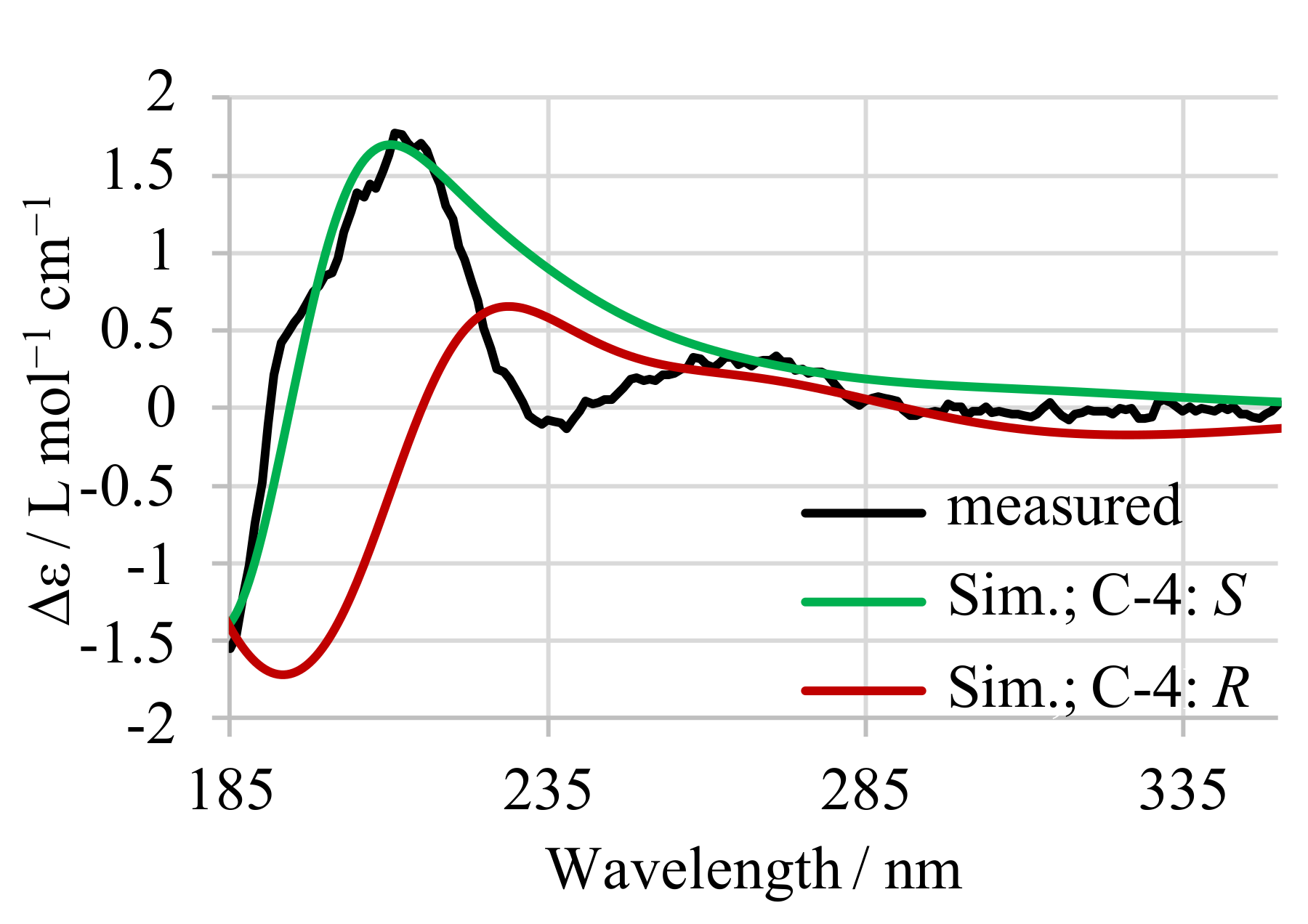
4.4. Circular Dichroism Spectroscopy
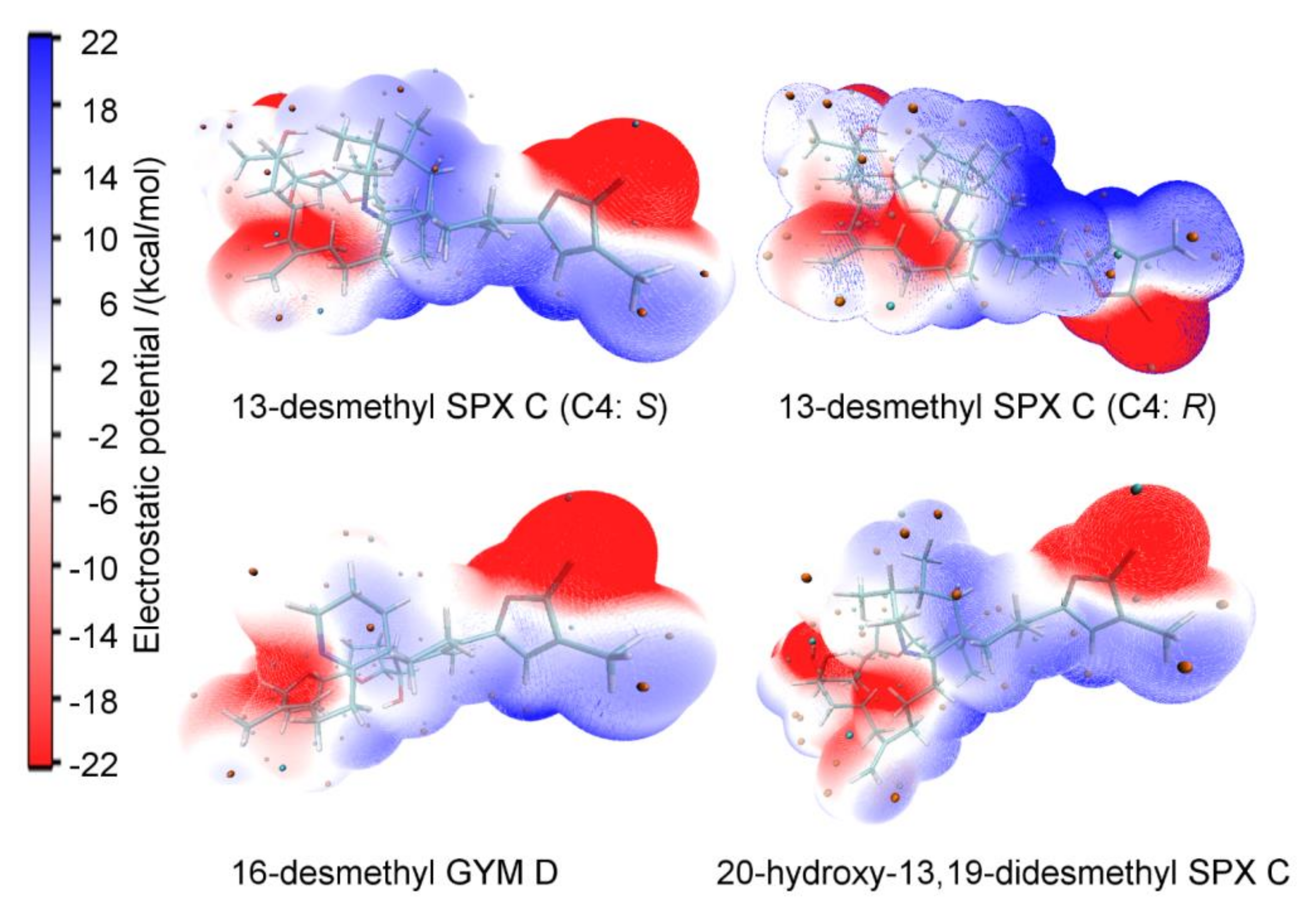
4.5. Electrostatic Potential Surface
4.6. Reference NMR Spectra
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. CD Spectroscopy of 13-desmethyl SPX C

Appendix B. Reanalysis of 13,19-didesmethyl SPX C

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family/Compound | Dihedral Angle α | H-3–H-37/Å | H-4–H-37/Å | H-4–H-35b/Å | Probability/% |
|---|---|---|---|---|---|
| C4: R | |||||
| 13,19-didesmethyl SPX C: anti | 168° | 3.3 | 3.8 | 2.5 | 0.0 |
| 13,19-didesmethyl SPX C: gauche− | −53° | 2.5 | 2.1 | 3.3 | 50.0 |
| 13,19-didesmethyl SPX C: gauche+ | 43° | 4.7 | 2.3 | 3.7 | 50.0 |
| 13,19-didesmethyl SPX C: averaged | 3.7 ± 0.5 | 1.9 ± 0.4 | 3.5 ± 0.4 | ||
| C4: S | |||||
| 13,19-didesmethyl SPX C: anti | −176° | 3.4 | 3.7 | 2.2 | 0.0 |
| 13,19-didesmethyl SPX C: gauche+ | 39° | 2.3 | 2.3 | 3.7 | 58.0 |
| 13,19-didesmethyl SPX C: gauche− | −41° | 4.2 | 2.4 | 3.7 | 42.0 |
| 13,19-didesmethyl SPX C: averaged | 2.6 ± 0.7 | 2.2 ± 0.3 | 3.3 ± 0.2 | ||
| measured NOE strength | weak | strong | very weak | ||

Appendix C. Stereochemistry of 20-Hydroxy-13,19-didesmethyl SPX C

Appendix D. Electrostatic Potential Surface of SPXs and GYMs

References
- Stivala, C.E.; Benoit, E.; Aráoz, R.; Servent, D.; Novikov, A.; Molgó, J.; Zakarian, A. Synthesis and biology of cyclic imine toxins, an emerging class of potent, globally distributed marine toxins. Nat. Prod. Rep. 2015, 32, 411–435. [Google Scholar] [CrossRef] [PubMed]
- Otero, A.; Chapela, M.-J.; Atanassova, M.; Vieites, J.M.; Cabado, A.G. Cyclic Imines: Chemistry and Mechanism of Action: A Review. Chem. Res. Toxicol. 2011, 24, 1817–1829. [Google Scholar] [CrossRef] [PubMed]
- Kharrat, R.; Servent, D.; Girard, E.; Ouanounou, G.; Amar, M.; Marrouchi, R.; Benoit, E.; Molgó, J. The marine phycotoxin gymnodimine targets muscular and neuronal nicotinic acetylcholine receptor subtypes with high affinity. J. Neurochem. 2008, 107, 952–963. [Google Scholar] [CrossRef]
- Hu, T.; Curtis, J.M.; Walter, J.A.; Wright, J.L.C. Characterization of biologically inactive spirolides E and F: Identification of the spirolide pharmacophore. Tetrahedron Lett. 1996, 37, 7671–7674. [Google Scholar] [CrossRef]
- Boente-Juncal, A.; Méndez, A.G.; Vale, C.; Vieytes, M.R.; Botana, L.M. In Vitro Effects of Chronic Spirolide Treatment on Human Neuronal Stem Cell Differentiation and Cholinergic System Development. ACS Chem. Neurosci. 2018, 9, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- Alonso, E.; Otero, P.; Vale, C.; Alfonso, A.; Antelo, A.; Gimenez-Llort, L.; Chabaud, L.; Guillou, C.; Botana, L.M. Benefit of 13-desmethyl Spirolide C Treatment in Triple Transgenic Mouse Model of Alzheimer Disease: Beta-Amyloid and Neuronal Markers Improvement. Curr. Alzheimer Res. 2013, 10, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Alonso, E.; Vale, C.; Vieytes, M.R.; Laferla, F.M.; Giménez-Llort, L.; Botana, L.M. 13-Desmethyl spirolide-C is neuroprotective and reduces intracellular Aβ and hyperphosphorylated tau in vitro. Neurochem. Int. 2011, 59, 1056–1065. [Google Scholar] [CrossRef] [PubMed]
- Ciminiello, P.; Catalanotti, B.; Dell’Aversano, C.; Fattorusso, C.; Fattorusso, E.; Forino, M.; Grauso, L.; Leo, A.; Tartaglione, L. Full relative stereochemistry assignment and conformational analysis of 13,19-didesmethyl spirolide C via NMR- and molecular modeling-based techniques. A step towards understanding spirolide’s mechanism of action. Org. Biomol. Chem. 2009, 7, 3674. [Google Scholar] [CrossRef]
- Van Wagoner, R.M.; Misner, I.; Tomas, C.R.; Wright, J.L.C. Occurrence of 12-methylgymnodimine in a spirolide-producing dinoflagellate Alexandrium peruvianum and the biogenetic implications. Tetrahedron Lett. 2011, 52, 4243–4246. [Google Scholar] [CrossRef]
- Minamino, K.; Murata, M.; Tsuchikawa, H. Synthesis of 7,6-Spirocyclic Imine with Butenolide Ring Provides Evidence for the Relative Configuration of Marine Toxin 13-desMe Spirolide C. Org. Lett. 2019, 21, 8970–8975. [Google Scholar] [CrossRef]
- Bourne, Y.; Radic, Z.; Araoz, R.; Talley, T.T.; Benoit, E.; Servent, D.; Taylor, P.; Molgo, J.; Marchot, P. Structural determinants in phycotoxins and AChBP conferring high affinity binding and nicotinic AChR antagonism. Proc. Natl. Acad. Sci. USA 2010, 107, 6076–6081. [Google Scholar] [CrossRef] [Green Version]
- Stewart, M.; Blunt, J.W.; Munro, M.H.; Robinson, W.T.; Hannah, D.J. The absolute stereochemistry of the New Zealand shellfish toxin gymnodimine. Tetrahedron Lett. 1997, 38, 4889–4890. [Google Scholar] [CrossRef]
- Kong, K.; Moussa, Z.; Lee, C.; Romo, D. Total Synthesis of the Spirocyclic Imine Marine Toxin (−)-Gymnodimine and an Unnatural C4-Epimer. J. Am. Chem. Soc. 2011, 133, 19844–19856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zurhelle, C.; Nieva, J.; Tillmann, U.; Harder, T.; Krock, B.; Tebben, J. Identification of Novel Gymnodimines and Spirolides from the Marine Dinoflagellate Alexandrium ostenfeldii. Mar. Drugs 2018, 16, 446. [Google Scholar] [CrossRef] [Green Version]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An Improved Probability for the Stereochemical Assignment of Isomeric Compounds using Quantum Chemical Calculations of NMR Shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef]
- Bifulco, G.; Dambruoso, P.; Gomez-Paloma, L.; Riccio, R. Determination of Relative Configuration in Organic Compounds by NMR Spectroscopy and Computational Methods. Chem. Rev. 2007, 107, 3744–3779. [Google Scholar] [CrossRef]
- Bühl, M.; Von Ragué Schleyer, P. Application and Evaluation of ab Initio Chemical Shift Calculations for Boranes and Carboranes. How reliable are “accurate” Experimental Structures? J. Am. Chem. Soc. 1992, 114, 477–491. [Google Scholar] [CrossRef]
- Onak, T.; Diaz, M.; Barfield, M. Ab Initio/NMR Studies on Per-B-F and Per-B-Cl Derivatives of C4B2H6 and C2B4H6: “Classical” vs. “Carborane-Cage” Structures. Correlation of 11B, 19F, and 35Cl NMR Data among Related Fluoroboron and Chloroboron Compounds. J. Am. Chem. Soc. 1995, 117, 1403–1410. [Google Scholar] [CrossRef]
- Dokalik, A.; Kalchhauser, H.; Mikenda, W.; Schweng, G. NMR spectra of nitrogen-containing compounds. Correlations between experimental and GIAO calculated data. Magn. Reson. Chem. 1999, 37, 895–902. [Google Scholar] [CrossRef]
- Zuschneid, T.; Fischer, H.; Handel, T.; Albert, K.; Häfelinger, G. Experimental gas phase 1H NMR spectra and basis set dependence of ab initio GIAO MO calculations of 1H and 13C NMR absolute shieldings and chemical shifts of small hydrocarbons. Z. Naturforsch. Sect. B J. Chem. Sci. 2004, 59, 1153–1176. [Google Scholar] [CrossRef]
- Miles, C.O.; Wilkins, A.L.; Stirling, D.J.; MacKenzie, A.L. New Analogue of Gymnodimine from a Gymnodinium Species. J. Agric. Food Chem. 2000, 48, 1373–1376. [Google Scholar] [CrossRef] [PubMed]
- Miles, C.O.; Wilkins, A.L.; Stirling, D.J.; MacKenzie, A.L. Gymnodimine C, an Isomer of Gymnodimine B, from Karenia selliformis. J. Agric. Food Chem. 2003, 51, 4838–4840. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Burton, I.W.; Cembella, A.D.; Curtis, J.M.; Quilliam, M.A.; Walter, J.A.; Wright, J.L.C. Characterization of Spirolides A, C, and 13-Desmethyl C, New Marine Toxins Isolated from Toxic Plankton and Contaminated Shellfish. J. Nat. Prod. 2001, 64, 308–312. [Google Scholar] [CrossRef]
- Strangman, W.; Anttila, M.; Tomas, C.; Wright, J. (5S)-5-[(4aR,8aS,9E,11S,13R,14S,16R,17R,19S)-11,19-Dihydroxy-8,10,13,16-tetramethyl-18-methylidene-3,4,5,6,8a,11,12,13,14,15,16,17,18,19,20,21-hexadecahydro-2H-14,17-epoxybenzo[2,3]cyclohexadeca[1,2-b]pyridine-7-yl]-3-methylfuran-2(5H)-one (12-Methylgymnodimine B). Molbank 2016, 2016, M896. [Google Scholar] [CrossRef] [Green Version]
- Pierens, G.K. 1H and 13C NMR scaling factors for the calculation of chemical shifts in commonly used solvents using density functional theory. J. Comput. Chem. 2014, 35, 1388–1394. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A fast force field generation tool for small organic molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Kirkpatrick, S.; Gelatt, C.D.; Vecchi, M.P. Optimization by Simulated Annealing. Science 1983, 220, 671–680. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. AM1: A New General Purpose Quantum Mechanical Molecular Model1. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Gordon, M.S.; Schmidt, M.W. Advances in electronic structure theory. In Theory and Applications of Computational Chemistry; Elsevier: Amsterdam, The Netherlands, 2005; pp. 1167–1189. ISBN 9780444517197. [Google Scholar]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism I. A gauge-invariant LCAO method for N.M.R. Chemical shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-C.; Ferreira, D.; Ding, Y. Determination of Absolute Configuration of Natural Products: Theoretical Calculation of Electronic Circular Dichroism as a Tool. Curr. Org. Chem. 2010, 14, 1678–1697. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Gupta, V.P. Electron Density Analysis and Electrostatic Potential. In Principles and Applications of Quantum Chemistry; Elsevier: Amsterdam, The Netherlands, 2016; pp. 195–214. ISBN 9780128034781. [Google Scholar]



| Level of Theory | 13C | Level of Theory | 13C | ||
|---|---|---|---|---|---|
| Optimization | Shielding Tensors | RMSE | Optimization | Shielding Tensors | RMSE |
| B3LYP,CPCM | B3LYP,CPCM | 2.02 | none | B3LYP,CPCM | 3.09 |
| B3LYP | B3LYP | 2.00 | none | TPSSh, CPCM | 3.20 |
| B3LYP | PBE0 | 2.40 | none | B3LYP | 2.86 |
| B3LYP | RHF | 3.89 | none | RHF | 4.88 |
| RHF | RHF | 4.10 | |||
| DFT | MP2 | |||
|---|---|---|---|---|
| RMSEall | RMSEC-6,C-24 | RMSEall | RMSEC-6,C-24 | |
| S epimer | 2.909 | 3.751 | 3.478 | 1.957 |
| R epimer | 3.030 | 5.637 | 3.284 | 3.404 |
| C-4 Configuration | S | R | ||
|---|---|---|---|---|
| GYM A (1) | 99.49% | 0.51% | ||
| 12-methyl GYM B (6) | 93.27% | 6,73% | ||
| 16-desmethyl GYM D (2) | 100.00% | 0.00% | ||
| 7,6-spirocyclic imine (5) | 96.21% | 3.79% | ||
| 13-desmethyl SPX C (4) | 100.00% | 0.00% | ||
| 13,19-didesmethyl SPX C (3) | 99.99% | 0.01% | ||
| 20-hydroxy-13,19-didesmethyl SPX C (7) | 100.00% | 0.00% | ||
| C-2 Configuration | S | R | S | R |
| 20-hydroxy-13,19-didesmethyl SPX D (8) | 0.01% | 99.99% | 0.00% | 0.00% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zurhelle, C.; Harder, T.; Tillmann, U.; Tebben, J. In Silico Modeling of Spirolides and Gymnodimines: Determination of S Configuration at Butenolide Ring Carbon C-4. Toxins 2020, 12, 685. https://doi.org/10.3390/toxins12110685
Zurhelle C, Harder T, Tillmann U, Tebben J. In Silico Modeling of Spirolides and Gymnodimines: Determination of S Configuration at Butenolide Ring Carbon C-4. Toxins. 2020; 12(11):685. https://doi.org/10.3390/toxins12110685
Chicago/Turabian StyleZurhelle, Christian, Tilmann Harder, Urban Tillmann, and Jan Tebben. 2020. "In Silico Modeling of Spirolides and Gymnodimines: Determination of S Configuration at Butenolide Ring Carbon C-4" Toxins 12, no. 11: 685. https://doi.org/10.3390/toxins12110685
APA StyleZurhelle, C., Harder, T., Tillmann, U., & Tebben, J. (2020). In Silico Modeling of Spirolides and Gymnodimines: Determination of S Configuration at Butenolide Ring Carbon C-4. Toxins, 12(11), 685. https://doi.org/10.3390/toxins12110685