Scorpion Toxins and Ion Channels: Potential Applications in Cancer Therapy

 , , ,
, , , 
Abstract
:1. Apoptosis
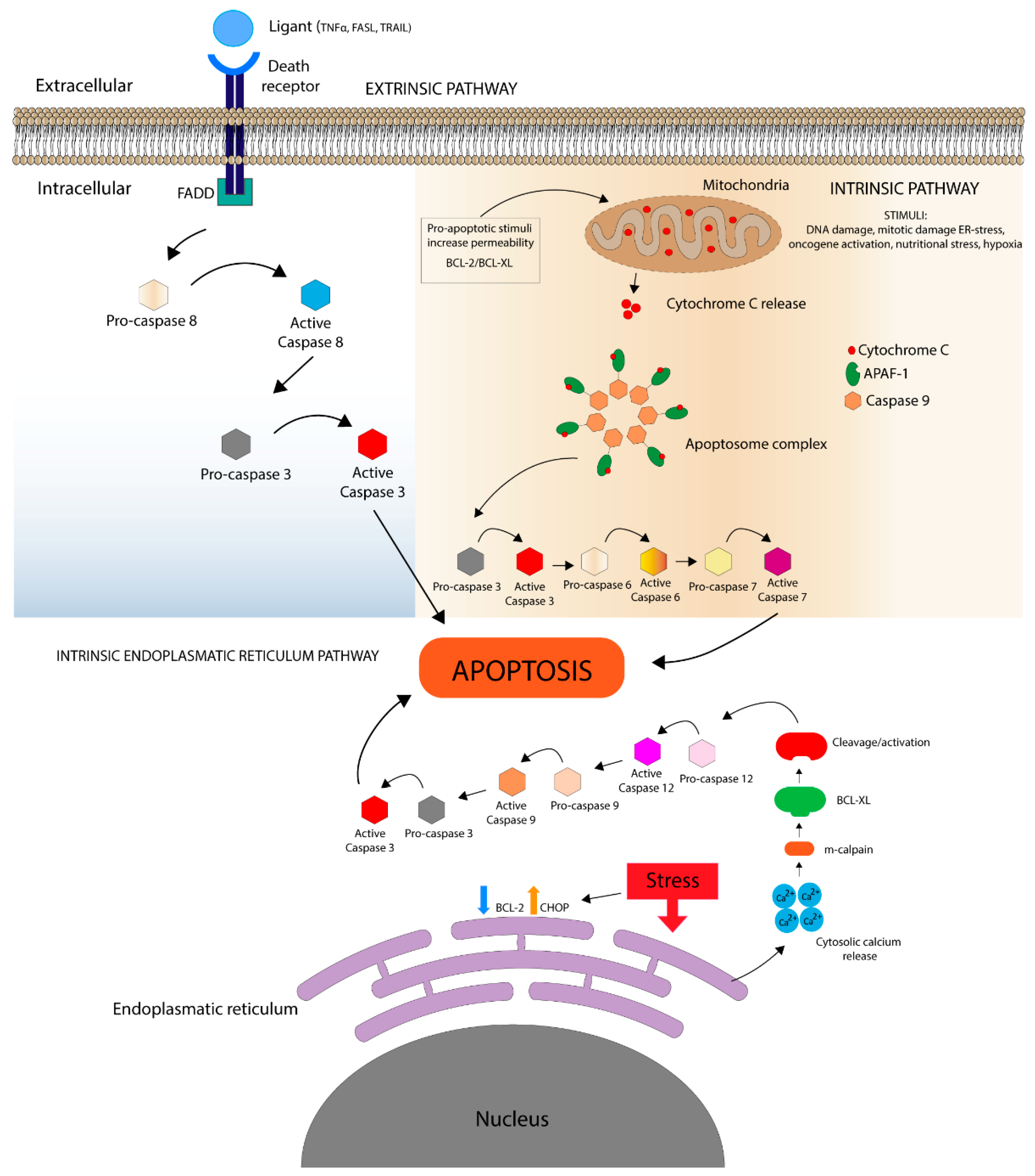
1.1. Death Receptor Pathways
1.2. Mitochondrial Pathway
Bcl Protein Family
1.3. Intrinsic Endoplasmic Reticulum Pathway
2. Apoptosis in Cancer
3. Roles of Ion Channels in Apoptosis: Targets to Induce Cancer Cell Death
3.1. Voltage-Dependent Calcium Channels
3.2. Voltage-Dependent Potassium Channels
3.3. Voltage-Dependent Chloride Channels
3.4. Voltage-Dependent Sodium Channels
4. Scorpion Toxins and Their Applications in Cancer Therapy
5. Anti-Proliferative and Cytotoxic Scorpion Toxins to Cancer Cells
5.1. Chlorotoxin (ClTx)
5.2. AaCtx (Androctonus Australis Chlorotoxin)
5.3. BmKCT
5.4. BmK AGAP
5.5. Charybdotoxin (ChTX)
5.6. Iberiotoxin (IbTx)
5.7. Margatoxin (MgTx)
5.8. Tamapin
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Häcker, G. The morphology of apoptosis. Cell Tissue Res. 2000, 301, 5–17. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Strasser, A.; McDunn, J.E.; Swanson, P.E. Cell death. N. Engl. J. Med. 2009, 361, 1570–1583. [Google Scholar] [CrossRef] [Green Version]
- Kondratskyi, A.; Kondratska, K.; Skryma, R.; Prevarskaya, N. Ion channels in the regulation of apoptosis. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2532–2546. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef]
- Nakagawa, T.; Yuan, J. Cross-talk between two cysteine protease families. activation of caspase-12 by calpain in apoptosis. J. Cell Biol. 2000, 150, 887–894. [Google Scholar] [CrossRef] [Green Version]
- Szegezdi, E.; Fitzgerald, U.; Samali, A. Caspase-12 and er-stress-mediated apoptosis: The story so far. Ann. N. Y. Acad. Sci. 2003, 1010, 186–194. [Google Scholar] [CrossRef]
- Leist, M.; Jäättelä, M. Four deaths and a funeral: From caspases to alternative mechanisms. Nat. Rev. Mol. Cell Biol. 2001, 2, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Bröker, L.E.; Kruyt, F.A.; Giaccone, G. Cell death independent of caspases: A review. Clin. Cancer Res. 2005, 11, 3155–3162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cruchten, S.; Van Den Broeck, W. Morphological and biochemical aspects of apoptosis, oncosis and necrosis. Anat. Histol. Embryol. 2002, 31, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Srinivasula, S.M.; Wang, L.; Talanian, R.V.; Litwack, G.; Fernandes-Alnemri, T.; Alnemri, E.S. CRADD, a novel human apoptotic adaptor molecule for caspase-2, and fasl/tumor necrosis factor receptor-interacting protein rip. Cancer Res. 1997, 57, 615–619. [Google Scholar] [PubMed]
- Hoppins, S.; Nunnari, J. Cell biology. Mitochondrial dynamics and apoptosis--the ER connection. Science 2012, 337, 1052–1054. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Riedl, S.J.; Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004, 5, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The BCL-2 protein family: Arbiters of cell survival. Science 1998, 281, 1322–1326. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Can. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Jemal, A.; Grey, N.; Ferlay, J.; Forman, D. Global cancer transitions according to the human development index (2008-2030): A population-based study. Lancet Oncol. 2012, 13, 790–801. [Google Scholar] [CrossRef]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer. Res. 2011, 30, 87. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect Biol. 2010, 2, a001107. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Vucic, D. Targeting iap proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Fan, T.; Yu, M. Inhibitor of apoptosis proteins and apoptosis. Acta Biochim. Biophys. Sin. 2008, 40, 278–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S.; Küfer, M.U.; Meyer, E.; van Valen, F.; Dockhorn-Dworniczak, B.; Debatin, K.M. Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene 2001, 20, 5865–5877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opferman, J.T. Attacking cancer’s achilles heel: Antagonism of anti-apoptotic BCL-2 family members. FEBS J. 2016, 283, 2661–2675. [Google Scholar] [CrossRef]
- Min, K.A.; Maharjan, P.; Ham, S.; Shin, M.C. Pro-apoptotic peptides-based cancer therapies: Challenges and strategies to enhance therapeutic efficacy. Arch. Pharm. Res. 2018, 41, 594–616. [Google Scholar] [CrossRef]
- Chabner, B.A.; Roberts, T.G. Timeline: Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the eortc-ncic trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Chávez-Galán, L.; Arenas-Del Angel, M.C.; Zenteno, E.; Chávez, R.; Lascurain, R. Cell death mechanisms induced by cytotoxic lymphocytes. Cell Mol. Immunol. 2009, 6, 15–25. [Google Scholar] [CrossRef]
- Lewis-Wambi, J.S.; Jordan, V.C. Estrogen regulation of apoptosis: How can one hormone stimulate and inhibit? Breast Cancer Res. 2009, 11, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.; Gong, W.H.; Li, X.C.; Zou, C.P.; Jiang, G.J.; Li, X.H.; Feng, D.P. Induction of apoptosis by a combination of paclitaxel and carboplatin in the presence of hyperthermia. Asian Pac. J. Cancer Prev. 2012, 13, 81–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robilotto, A.T.; Baust, J.M.; Van Buskirk, R.G.; Gage, A.A.; Baust, J.G. Temperature-dependent activation of differential apoptotic pathways during cryoablation in a human prostate cancer model. Prostate Cancer Prostatic Dis. 2013, 16, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Kischkel, F.C.; Hellbardt, S.; Behrmann, I.; Germer, M.; Pawlita, M.; Krammer, P.H.; Peter, M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (disc) with the receptor. EMBO J. 1995, 14, 5579–5588. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.; Makhson, A.; Gligorov, J.; Lichinitser, M.; Lluch, A.; Semiglazov, V.; Scotto, N.; Mitchell, L.; Tjulandin, S. Phase II study of bevacizumab in combination with trastuzumab and capecitabine as first-line treatment for HER-2-positive locally recurrent or metastatic breast cancer. Oncologist 2012, 17, 469–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-García, D.; Manero-Rupérez, N.; Quesada, R.; Korrodi-Gregório, L.; Soto-Cerrato, V. Therapeutic strategies involving survivin inhibition in cancer. Med. Res. Rev. 2019, 39, 887–909. [Google Scholar] [CrossRef]
- Zottel, A.; VidetičPaska, A.; Jovčevska, I. Nanotechnology meets oncology: Nanomaterials in brain cancer research, diagnosis and therapy. Materials 2019, 12, 1588. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.; Föller, M.; Lang, K.S.; Lang, P.A.; Ritter, M.; Gulbins, E.; Vereninov, A.; Huber, S.M. Ion channels in cell proliferation and apoptotic cell death. J. Membr. Biol. 2005, 205, 147–157. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels and the hallmarks of cancer. Trends. Mol. Med. 2010, 16, 107–121. [Google Scholar] [CrossRef]
- Lehen’kyi, V.; Shapovalov, G.; Skryma, R.; Prevarskaya, N. Ion channnels and transporters in cancer. 5. Ion channels in control of cancer and cell apoptosis. Am. J. Physiol. Cell Physiol. 2011, 301, C1281-9. [Google Scholar] [CrossRef]
- Varghese, E.; Samuel, S.M.; Sadiq, Z.; Kubatka, P.; Liskova, A.; Benacka, J.; Pazinka, P.; Kruzliak, P.; Büsselberg, D. Anti-cancer agents in proliferation and cell death: The calcium connection. Int. J. Mol. Sci. 2019, 20, 3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, F.; Chen, H.; Zhou, C.; Liu, S.; Guo, M.; Chen, P.; Zhuang, H.; Xie, D.; Wu, S. T-type Ca2+ channel expression in human esophageal carcinomas: A functional role in proliferation. Cell Calcium 2008, 43, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antal, L.; Martin-Caraballo, M. T-type calcium channels in cancer. Cancers 2019, 11, 134. [Google Scholar] [CrossRef] [Green Version]
- Lehen’kyi, V.; Raphaël, M.; Prevarskaya, N. The role of the TRPV6 channel in cancer. J. Physiol. 2012, 590, 1369–1376. [Google Scholar] [CrossRef] [Green Version]
- Haustrate, A.; Hantute-Ghesquier, A.; Prevarskaya, N.; Lehen’kyi, V. TRPV6 calcium channel regulation, downstream pathways, and therapeutic targeting in cancer. Cell Calcium 2019, 80, 117–124. [Google Scholar] [CrossRef]
- Dhennin-Duthille, I.; Gautier, M.; Faouzi, M.; Guilbert, A.; Brevet, M.; Vaudry, D.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. High expression of transient receptor potential channels in human breast cancer epithelial cells and tissues: Correlation with pathological parameters. Cell Physiol. Biochem. 2011, 28, 813–822. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef]
- Roth, K.G.; Mambetsariev, I.; Kulkarni, P.; Salgia, R. The mitochondrion as an emerging therapeutic target in cancer. Trends Mol. Med. 2019. [Google Scholar] [CrossRef]
- Rasola, A.; Sciacovelli, M.; Pantic, B.; Bernardi, P. Signal transduction to the permeability transition pore. FEBS Lett. 2010, 584, 1989–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, P.; Rasola, A.; Forte, M.; Lippe, G. The mitochondrial permeability transition pore: Channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol. Rev. 2015, 95, 1111–1155. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Zhao, J.; Wiedmer, T.; Sims, P.J. Normal hemostasis but defective hematopoietic response to growth factors in mice deficient in phospholipid scramblase 1. Blood 2002, 99, 4030–4038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woon, L.A.; Holland, J.W.; Kable, E.P.; Roufogalis, B.D. Ca2+ sensitivity of phospholipid scrambling in human red cell ghosts. Cell Calcium 1999, 25, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, D.W.; Comfurius, P.; Bevers, E.M.; Zwaal, R.F. Comparison between Ca2+-induced scrambling of various fluorescently labelled lipid analogues in red blood cells. Biochem. J. 2002, 362, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. Roles of K+ channels in regulating tumour cell proliferation and apoptosis. Pflug. Arch. 2004, 448, 274–286. [Google Scholar] [CrossRef]
- Binggeli, R.; Cameron, I.L. Cellular potentials of normal and cancerous fibroblasts and hepatocytes. Cancer Res. 1980, 40, 1830–1835. [Google Scholar]
- Wonderlin, W.F.; Woodfork, K.A.; Strobl, J.S. Changes in membrane potential during the progression of MCF-7 human mammary tumor cells through the cell cycle. J. Cell Physiol. 1995, 165, 177–185. [Google Scholar] [CrossRef]
- Maeno, E.; Ishizaki, Y.; Kanaseki, T.; Hazama, A.; Okada, Y. Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 9487–9492. [Google Scholar] [CrossRef] [Green Version]
- Abdul, M.; Hoosein, N. Voltage-gated potassium ion channels in colon cancer. Oncol. Rep. 2002, 9, 961–964. [Google Scholar] [CrossRef]
- Yu, S.P.; Yeh, C.H.; Sensi, S.L.; Gwag, B.J.; Canzoniero, L.M.; Farhangrazi, Z.S.; Ying, H.S.; Tian, M.; Dugan, L.L.; Choi, D.W. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science 1997, 278, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Remillard, C.V.; Yuan, J.X. Activation of K+ channels: An essential pathway in programmed cell death. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 286, L49–L67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, F.M.; Cidlowski, J.A. Potassium is a critical regulator of apoptotic enzymes in vitro and in vivo. Adv. Enzym. Regul. 1999, 39, 157–171. [Google Scholar] [CrossRef]
- Abdul, M.; Santo, A.; Hoosein, N. Activity of potassium channel-blockers in breast cancer. Anticancer Res. 2003, 23, 3347–3351. [Google Scholar]
- Woodfork, K.A.; Wonderlin, W.F.; Peterson, V.A.; Strobl, J.S. Inhibition of ATP-sensitive potassium channels causes reversible cell-cycle arrest of human breast cancer cells in tissue culture. J. Cell Physiol. 1995, 162, 163–171. [Google Scholar] [CrossRef]
- Suzuki, T.; Takimoto, K. Selective expression of HERG and Kv2 channels influences proliferation of uterine cancer cells. Int. J. Oncol. 2004, 25, 153–159. [Google Scholar] [CrossRef]
- Lai, Q.; Wang, T.; Guo, Q.; Zhang, Y.; Wang, Y.; Yuan, L.; Ling, R.; He, Y.; Wang, W. Positive correlation between the expression of hEag1 and HIF-1α in breast cancers: An observational study. BMJ Open 2014, 4, e005049. [Google Scholar] [CrossRef] [Green Version]
- Restrepo-Angulo, I.; Sánchez-Torres, C.; Camacho, J. Human EAG1 potassium channels in the epithelial-to-mesenchymal transition in lung cancer cells. Anticancer Res. 2011, 31, 1265–1270. [Google Scholar]
- Skryma, R.N.; Prevarskaya, N.B.; Dufy-Barbe, L.; Odessa, M.F.; Audin, J.; Dufy, B. Potassium conductance in the androgen-sensitive prostate cancer cell line, lncap: Involvement in cell proliferation. Prostate 1997, 33, 112–122. [Google Scholar] [CrossRef]
- Meyer, R.; Heinemann, S.H. Characterization of an eag-like potassium channel in human neuroblastoma cells. J. Physiol. 1998, 508, 49–56. [Google Scholar] [CrossRef]
- Gómez-Varela, D.; Zwick-Wallasch, E.; Knötgen, H.; Sánchez, A.; Hettmann, T.; Ossipov, D.; Weseloh, R.; Contreras-Jurado, C.; Rothe, M.; Stühmer, W.; et al. Monoclonal antibody blockade of the human Eag1 potassium channel function exerts antitumor activity. Cancer Res. 2007, 67, 7343–7349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, K.S.; Yuspa, S.H. Intracellular chloride channels: Critical mediators of cell viability and potential targets for cancer therapy. Curr. Pharm. Des. 2005, 11, 2753–2764. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, L.; Zhu, L.; Nie, S.; Zhang, J.; Zhong, P.; Cai, B.; Luo, H.; Jacob, T.J. Cell cycle-dependent expression of volume-activated chloride currents in nasopharyngeal carcinoma cells. Am. J. Physiol. Cell Physiol. 2002, 283, C1313-23. [Google Scholar] [CrossRef] [PubMed]
- Wondergem, R.; Gong, W.; Monen, S.H.; Dooley, S.N.; Gonce, J.L.; Conner, T.D.; Houser, M.; Ecay, T.W.; Ferslew, K.E. Blocking swelling-activated chloride current inhibits mouse liver cell proliferation. J. Physiol. 2001, 532, 661–672. [Google Scholar] [CrossRef]
- Nilius, B. Chloride channels go cell cycling. J. Physiol. 2001, 532, 581. [Google Scholar] [CrossRef]
- Kang, M.K.; Kang, S.K. Pharmacologic blockade of chloride channel synergistically enhances apoptosis of chemotherapeutic drug-resistant cancer stem cells. Biochem. Biophys. Res. Commun. 2008, 373, 539–544. [Google Scholar] [CrossRef]
- Kang, M.K.; Kang, S.K. Tumorigenesis of chemotherapeutic drug-resistant cancer stem-like cells in brain glioma. Stem Cells Dev. 2007, 16, 837–847. [Google Scholar] [CrossRef]
- Shen, M.R.; Droogmans, G.; Eggermont, J.; Voets, T.; Ellory, J.C.; Nilius, B. Differential expression of volume-regulated anion channels during cell cycle progression of human cervical cancer cells. J. Physiol. 2000, 529, 385–394. [Google Scholar] [CrossRef]
- Olsen, M.L.; Schade, S.; Lyons, S.A.; Amaral, M.D.; Sontheimer, H. Expression of voltage-gated chloride channels in human glioma cells. J. Neurosci. 2003, 23, 5572–5582. [Google Scholar] [CrossRef] [Green Version]
- Rønnov-Jessen, L.; Villadsen, R.; Edwards, J.C.; Petersen, O.W. Differential expression of a chloride intracellular channel gene, CLIC4, in transforming growth factor-beta1-mediated conversion of fibroblasts to myofibroblasts. Am. J. Pathol. 2002, 161, 471–480. [Google Scholar] [CrossRef]
- Smallbone, K.; Gavaghan, D.J.; Gatenby, R.A.; Maini, P.K. The role of acidity in solid tumour growth and invasion. J. Biol. 2005, 235, 476–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, Y.; Shimizu, T.; Maeno, E.; Tanabe, S.; Wang, X.; Takahashi, N. Volume-sensitive chloride channels involved in apoptotic volume decrease and cell death. J. Membr. Biol. 2006, 209, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Lyons, J.C.; Ohtsubo, T.; Song, C.W. Acidic environment causes apoptosis by increasing caspase activity. Br. J. Cancer 1999, 80, 1892–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, M.; Gasparoli, L.; Arcangeli, A. Potassium channels: Novel emerging biomarkers and targets for therapy in cancer. Recent. Pat. Anticancer Drug Discov. 2013, 8, 53–65. [Google Scholar] [CrossRef]
- Kischel, P.; Girault, A.; Rodat-Despoix, L.; Chamlali, M.; Radoslavova, S.; Abou Daya, H.; Lefebvre, T.; Foulon, A.; Rybarczyk, P.; Hague, F.; et al. Ion channels: New actors playing in chemotherapeutic resistance. Cancers 2019, 11, 376. [Google Scholar] [CrossRef] [Green Version]
- Ullrich, N.; Sontheimer, H. Cell cycle-dependent expression of a glioma-specific chloride current: Proposed link to cytoskeletal changes. Am. J. Physiol. 1997, 273, C1290-7. [Google Scholar] [CrossRef]
- Li, M.; Wu, D.B.; Wang, J. Effects of volume-activated chloride channels on the invasion and migration of human endometrial cancer cells. Eur. J. Gynaecol. Oncol. 2013, 34, 60–64. [Google Scholar] [PubMed]
- Iitaka, D.; Shiozaki, A.; Ichikawa, D.; Kosuga, T.; Komatsu, S.; Okamoto, K.; Fujiwara, H.; Ishii, H.; Nakahari, T.; Marunaka, Y.; et al. Blockade of chloride ion transport enhances the cytocidal effect of hypotonic solution in gastric cancer cells. J. Surg. Res. 2012, 176, 524–534. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, L.; Zhang, J.; Zhang, L.; Yan, X.; Su, J. Suppression of chloride voltage-gated channel 3 expression increases sensitivity of human glioma U251 cells to cisplatin through lysosomal dysfunction. Oncol. Lett. 2018, 16, 835–842. [Google Scholar] [CrossRef]
- Nelson, M.; Yang, M.; Millican-Slater, R.; Brackenbury, W.J. Nav1.5 regulates breast tumor growth and metastatic dissemination in vivo. Oncotarget 2015, 6, 32914–32929. [Google Scholar] [CrossRef] [Green Version]
- Zhivotovsky, B.; Orrenius, S. Calcium and cell death mechanisms: A perspective from the cell death community. Cell Calcium 2011, 50, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Zhang, J.; Körner, H.; Jiang, Y.; Ying, S. The emerging role of voltage-gated sodium channels in tumor biology. Front. Oncol. 2019, 9, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diss, J.K.; Stewart, D.; Pani, F.; Foster, C.S.; Walker, M.M.; Patel, A.; Djamgoz, M.B. A potential novel marker for human prostate cancer: Voltage-gated sodium channel expression in vivo. Prostate Cancer Prostatic Dis. 2005, 8, 266–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roger, S.; Rollin, J.; Barascu, A.; Besson, P.; Raynal, P.I.; Iochmann, S.; Lei, M.; Bougnoux, P.; Gruel, Y.; Le Guennec, J.Y. Voltage-gated sodium channels potentiate the invasive capacities of human non-small-cell lung cancer cell lines. Int. J. Biochem Cell Biol. 2007, 39, 774–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, S.P.; Diss, J.K.; Chioni, A.M.; Mycielska, M.E.; Pan, H.; Yamaci, R.F.; Pani, F.; Siwy, Z.; Krasowska, M.; Grzywna, Z.; et al. Voltage-gated sodium channel expression and potentiation of human breast cancer metastasis. Clin. Cancer Res. 2005, 11, 5381–5389. [Google Scholar] [CrossRef] [Green Version]
- Brackenbury, W.J. Voltage-gated sodium channels and metastatic disease. Channels 2012, 6, 352–361. [Google Scholar] [CrossRef] [Green Version]
- Nelson, M.; Yang, M.; Dowle, A.A.; Thomas, J.R.; Brackenbury, W.J. The sodium channel-blocking antiepileptic drug phenytoin inhibits breast tumour growth and metastasis. Mol. Cancer 2015, 14, 13. [Google Scholar] [CrossRef] [Green Version]
- Rhana, P.; Trivelato, R.R.; Beirão, P.S.L.; Cruz, J.S.; Rodrigues, A.L.P. Is there a role for voltage-gated Na+ channels in the aggressiveness of breast cancer? Braz. J. Med. Biol. Res. 2017, 50, e6011. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, E.; Gurrola, G.B.; Schwartz, E.F.; Possani, L.D. Scorpion venom components as potential candidates for drug development. Toxicon 2015, 93, 125–135. [Google Scholar] [CrossRef]
- Lewis, R.J.; Garcia, M.L. Therapeutic potential of venom peptides. Nat. Rev. Drug Discov. 2003, 2, 790–802. [Google Scholar] [CrossRef]
- King, G.F. Venoms as a platform for human drugs: Translating toxins into therapeutics. Expert Opin. Biol. 2011, 11, 1469–1484. [Google Scholar] [CrossRef] [PubMed]
- Uzair, B.; Bint-E-Irshad, S.; Khan, B.A.; Azad, B.; Mahmood, T.; Rehman, M.U.; Braga, V.A. Scorpion venom peptides as a potential source for human drug candidates. Protein. Pept. Lett. 2018, 25, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Chua, P.J.; Bay, B.H.; Gopalakrishnakone, P. Scorpion venoms as a potential source of novel cancer therapeutic compounds. Exp. Biol. Med. 2014, 239, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Zhang, P.Y. Scorpion venoms in gastric cancer. Oncol. Lett. 2016, 12, 3683–3686. [Google Scholar] [CrossRef] [Green Version]
- Mahadevappa, R.; Ma, R.; Kwok, H.F. Venom peptides: Improving specificity in cancer therapy. Trends Cancer 2017, 3, 611–614. [Google Scholar] [CrossRef]
- Srairi-Abid, N.; Othman, H.; Aissaoui, D.; BenAissa, R. Anti-tumoral effect of scorpion peptides: Emerging new cellular targets and signaling pathways. Cell Calcium 2019, 80, 160–174. [Google Scholar] [CrossRef]
- DeBin, J.A.; Maggio, J.E.; Strichartz, G.R. Purification and characterization of chlorotoxin, a chloride channel ligand from the venom of the scorpion. Am. J. Physiol. 1993, 264, C361-9. [Google Scholar] [CrossRef]
- Lippens, G.; Najib, J.; Wodak, S.J.; Tartar, A. NMR sequential assignments and solution structure of chlorotoxin, a small scorpion toxin that blocks chloride channels. Biochemistry 1995, 34, 13–21. [Google Scholar] [CrossRef]
- Lyons, S.A.; O’Neal, J.; Sontheimer, H. Chlorotoxin, a scorpion-derived peptide, specifically binds to gliomas and tumors of neuroectodermal origin. Glia 2002, 39, 162–173. [Google Scholar] [CrossRef]
- Deshane, J.; Garner, C.C.; Sontheimer, H. Chlorotoxin inhibits glioma cell invasion via matrix metalloproteinase-2. J. Biol. Chem. 2003, 278, 4135–4144. [Google Scholar] [CrossRef] [Green Version]
- Mamelak, A.N.; Jacoby, D.B. Targeted delivery of antitumoral therapy to glioma and other malignancies with synthetic chlorotoxin (TM-601). Expert Opin. Drug Deliv. 2007, 4, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, D.B.; Dyskin, E.; Yalcin, M.; Kesavan, K.; Dahlberg, W.; Ratliff, J.; Johnson, E.W.; Mousa, S.A. Potent pleiotropic anti-angiogenic effects of TM601, a synthetic chlorotoxin peptide. Anticancer Res. 2010, 30, 39–46. [Google Scholar]
- Qin, C.; He, B.; Dai, W.; Zhang, H.; Wang, X.; Wang, J.; Zhang, X.; Wang, G.; Yin, L.; Zhang, Q. Inhibition of metastatic tumor growth and metastasis via targeting metastatic breast cancer by chlorotoxin-modified liposomes. Mol. Pharm. 2014, 11, 3233–3241. [Google Scholar] [CrossRef] [PubMed]
- Kesavan, K.; Ratliff, J.; Johnson, E.W.; Dahlberg, W.; Asara, J.M.; Misra, P.; Frangioni, J.V.; Jacoby, D.B. Annexin A2 is a molecular target for TM601, a peptide with tumor-targeting and anti-angiogenic effects. J. Biol. Chem. 2010, 285, 4366–4374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGonigle, S.; Majumder, U.; Kolber-Simonds, D.; Wu, J.; Hart, A.; Noland, T.; TenDyke, K.; Custar, D.; Li, D.; Du, H.; et al. Neuropilin-1 drives tumor-specific uptake of chlorotoxin. Cell Commun. Signal 2019, 17, 67. [Google Scholar] [CrossRef] [Green Version]
- Graf, N.; Mokhtari, T.E.; Papayannopoulos, I.A.; Lippard, S.J. Platinum(IV)-chlorotoxin (CTX) conjugates for targeting cancer cells. J. Inorg. Biochem. 2012, 110, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Patil, R.; Galstyan, A.; Sun, T.; Shatalova, E.S.; Butte, P.; Mamelak, A.N.; Carico, C.; Kittle, D.S.; Grodzinski, Z.B.; Chiechi, A.; et al. Polymalic acid chlorotoxin nanoconjugate for near-infrared fluorescence guided resection of glioblastoma multiforme. Biomaterials 2019, 206, 146–159. [Google Scholar] [CrossRef]
- Rjeibi, I.; Mabrouk, K.; Mosrati, H.; Berenguer, C.; Mejdoub, H.; Villard, C.; Laffitte, D.; Bertin, D.; Ouafik, L.; Luis, J.; et al. Purification, synthesis and characterization of AaCtx, the first chlorotoxin-like peptide from Androctonus australis scorpion venom. Peptides 2011, 32, 656–663. [Google Scholar] [CrossRef]
- Othman, H.; Wieninger, S.A.; ElAyeb, M.; Nilges, M.; Srairi-Abid, N. In silico prediction of the molecular basis of ClTx and AaCTx interaction with matrix metalloproteinase-2 (MMP-2) to inhibit glioma cell invasion. J. Biomol. Struct. Dyn. 2017, 35, 2815–2829. [Google Scholar] [CrossRef]
- Zeng, X.C.; Li, W.X.; Zhu, S.Y.; Peng, F.; Zhu, Z.H.; Wu, K.L.; Yiang, F.H. Cloning and characterization of a cDNA sequence encoding the precursor of a chlorotoxin-like peptide from the chinese scorpion Buthus martensii Karsch. Toxicon 2000, 38, 1009–1014. [Google Scholar] [CrossRef]
- Fan, S.; Sun, Z.; Jiang, D.; Dai, C.; Ma, Y.; Zhao, Z.; Liu, H.; Wu, Y.; Cao, Z.; Li, W. BmKCT toxin inhibits glioma proliferation and tumor metastasis. Cancer Lett. 2010, 291, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, N.; Bordey, A.; Gillespie, G.Y.; Sontheimer, H. Expression of voltage-activated chloride currents in acute slices of human gliomas. Neuroscience 1998, 83, 1161–1173. [Google Scholar] [CrossRef]
- Li, Z.; Hu, P.; Wu, W.; Wang, Y. Peptides with therapeutic potential in the venom of the scorpion Buthus martensii Karsch. Peptides 2019, 115, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.F.; Ma, R.L.; Wang, S.L.; Duan, Z.Y.; Zhang, J.H.; Wu, L.J.; Wu, C.F. Expression of an antitumor-analgesic peptide from the venom of Chinese scorpion Buthus martensii Karsch in Escherichia coli. Protein Expr. Purif. 2003, 27, 253–258. [Google Scholar] [CrossRef]
- Zhao, Y.; Cai, X.; Ye, T.; Huo, J.; Liu, C.; Zhang, S.; Cao, P. Analgesic-antitumor peptide inhibits proliferation and migration of SHG-44 human malignant glioma cells. J. Cell Biochem. 2011, 112, 2424–2434. [Google Scholar] [CrossRef]
- Gu, Y.; Liu, S.-L.; Ju, W.-Z.; Li, C.-Y.; Cao, P. Analgesic–antitumor peptide induces apoptosis and inhibits the proliferation of SW480 human colon cancer cells. Oncol. Lett. 2013, 5, 483–488. [Google Scholar] [CrossRef] [Green Version]
- Guo, G.; Cui, Y.; Chen, H.; Zhang, L.; Zhao, M.; Chen, B.; Zhang, J.; Liu, Y. Analgesic–antitumor peptide inhibits the migration and invasion of HepG2 cells by an upregulated VGSC β1 subunit. Tumor. Biol. 2016, 37, 3033–3041. [Google Scholar] [CrossRef] [PubMed]
- Kampo, S.; Ahmmed, B.; Zhou, T.; Owusu, L.; Anabah, T.W.; Doudou, N.R.; Kuugbee, E.D.; Cui, Y.; Lu, Z.; Yan, Q.; et al. Scorpion venom analgesic peptide, BmK AGAP inhibits stemness, and epithelial-mesenchymal transition by down-regulating PTX3 in breast cancer. Front. Oncol. 2019, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Gallego, G.; Navia, M.A.; Reuben, J.P.; Katz, G.M.; Kaczorowski, G.J.; Garcia, M.L. Purification, sequence, and model structure of charybdotoxin, a potent selective inhibitor of calcium-activated potassium channels. Proc. Natl. Acad. Sci. USA 1988, 85, 3329–3333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, A.; Hanley, P.; Fabian, A.; Stock, C. Potassium channels keep mobile cells on the go. Physiology 2008, 23, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Schwab, A.; Wojnowski, L.; Gabriel, K.; Oberleithner, H. Oscillating activity of a Ca2+-sensitive K+ channel. A prerequisite for migration of transformed Madin-Darby canine kidney focus cells. J. Clin. Investig. 1994, 93, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
- Schwab, A.; Reinhardt, J.; Schneider, S.W.; Gassner, B.; Schuricht, B. K+ channel-dependent migration of fibroblasts and human melanoma cells. Cell Physiol. Biochem. 1999, 9, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Galvez, A.; Gimenez-Gallego, G.; Reuben, J.P.; Roy-Contancin, L.; Feigenbaum, P.; Kaczorowski, G.J.; Garcia, M.L. Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium-activated potassium channel from venom of the scorpion Buthus tamulus. J. Biol. Chem. 1990, 265, 11083–11090. [Google Scholar] [PubMed]
- Weaver, A.K.; Liu, X.; Sontheimer, H. Role for calcium-activated potassium channels (BK) in growth control of human malignant glioma cells. J. Neurosci. Res. 2004, 78, 224–234. [Google Scholar] [CrossRef] [Green Version]
- Schickling, B.M.; England, S.K.; Aykin-Burns, N.; Norian, L.A.; Leslie, K.K.; Frieden-Korovkina, V.P. BKCa channel inhibitor modulates the tumorigenic ability of hormone-independent breast cancer cells via the Wnt pathway. Oncol. Rep. 2015, 33, 533–538. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Calvo, M.; Leonard, R.J.; Novick, J.; Stevens, S.P.; Schmalhofer, W.; Kaczorowski, G.J.; Garcia, M.L. Purification, characterization, and biosynthesis of margatoxin, a component of Centruroides margaritatus venom that selectively inhibits voltage-dependent potassium channels. J. Biol. Chem. 1993, 268, 18866–18874. [Google Scholar]
- Bartok, A.; Toth, A.; Somodi, S.; Szanto, T.G.; Hajdu, P.; Panyi, G.; Varga, Z. Margatoxin is a non-selective inhibitor of human Kv1.3 K+ channels. Toxicon 2014, 87, 6–16. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.H.; Choi, S.Y.; Ryu, P.D.; Lee, S.Y. Anti-proliferative effect of Kv1.3 blockers in A549 human lung adenocarcinoma in vitro and in vivo. Eur. J. Pharm. 2011, 651, 26–32. [Google Scholar] [CrossRef]
- Pedarzani, P.; D’hoedt, D.; Doorty, K.B.; Wadsworth, J.D.; Joseph, J.S.; Jeyaseelan, K.; Kini, R.M.; Gadre, S.V.; Sapatnekar, S.M.; Stocker, M.; et al. Tamapin, a venom peptide from the indian red scorpion (Mesobuthus tamulus) that targets small conductance Ca2+-activated K+ channels and afterhyperpolarization currents in central neurons. J. Biol. Chem. 2002, 277, 46101–46109. [Google Scholar] [CrossRef] [Green Version]
- Ramírez-Cordero, B.; Toledano, Y.; Cano-Sánchez, P.; Hernández-López, R.; Flores-Solis, D.; Saucedo-Yáñez, A.L.; Chávez-Uribe, I.; Brieba, L.G.; del Río-Portilla, F. Cytotoxicity of recombinant tamapin and related toxin-like peptides on model cell lines. Chem. Res. Toxicol. 2014, 27, 960–967. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Ion Channel | Toxin | Species | Accession Number | Cell Lineage | Biological Effects | References |
|---|---|---|---|---|---|---|
| Chlorotoxin | Leiurus quinquestriatus quinquestriatus | P45639 | Human glioblastoma (D54-MG) | Affects glioma cell invasion | [110] | |
| Cl− | AaCtx | Androctonus australis | P86436 | Human glioma (U87) | Prevents the invasion and migration of human glioma cells | [118] |
| BmKCT | Mesobuthus martensii Karsch | Q9UAD0 | Human glioma (SHG-44) | Inhibits glioma cell migration and invasion in vitro and in vivo | [121] | |
| Na+ | BmK AGAP | Mesobuthus martensii Karsch | G4V3T9 | Ehrlich ascites and S-180 fibrosarcoma in vivo Human breast adenocarcinoma (MCF7) Human malignant glioma cells (SHG-44) Human colon adenocarcinoma cells (SW480) Human hepatoma cells (HepG2) Human breast adenocarcinoma (MCF-7 and MDA-MB-231) | Might inhibit SHG-44 cells proliferation and migration through BCL-2, NF-κB/p65, AKT, and MAPK signaling pathways Increases apoptosis and inhibits the proliferation of human colon adenocarcinoma SW480 cells Suppresses the migration and invasion of HepG2 cells via a voltage-gated sodium channel (VGSC) β1 subunit Inhibits cancer cell stemness, epithelial-mesenchymal transition (EMT), migration, and invasion in breast cancer cells | [124,125,126,127,128] |
| K+ | Charybdotoxin | Leiurus quinquestriatus hebreus | P13487 | Human A7 melanoma cells | Induces reduction in melanoma cell migration in a dose-dependent manner | [132] |
| Iberiotoxin | Mesobuthus tamulus | P24663 | Human glioblastoma (D54-MG) Human breast adenocarcinoma (MDA-MB-231) Human ductal mammary gland carcinoma (UACC893) | Induces decrease in the growth of glioblastoma multiforme in a dose-dependent manner Might specifically attenuate tumorigenicity in breast cancer models by transmembrane depolarization and downregulation of β-catenin and (phospho)Akt and HER-2/neu protein levels | [134,135] | |
| Margatoxin | Centruroides margaritatus | P40755 | Human lung adenocarcinoma (A549) | Exhibits anti-proliferative effects on human lung adenocarcinoma A549 cells and also causes a reduction of tumor volume in a xenograft model using nude mice | [138] | |
| Tamapin | Mesobuthus tamulus | P59869 | Human Leukaemic T cell lymphoblast (Jurkat E6-1) Human breast adenocarcinoma (MDA-MB-231) | Might induce cell death by apoptosis | [140] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dueñas-Cuellar, R.A.; Santana, C.J.C.; Magalhães, A.C.M.; Pires, O.R., Jr.; Fontes, W.; Castro, M.S. Scorpion Toxins and Ion Channels: Potential Applications in Cancer Therapy. Toxins 2020, 12, 326. https://doi.org/10.3390/toxins12050326
Dueñas-Cuellar RA, Santana CJC, Magalhães ACM, Pires OR Jr., Fontes W, Castro MS. Scorpion Toxins and Ion Channels: Potential Applications in Cancer Therapy. Toxins. 2020; 12(5):326. https://doi.org/10.3390/toxins12050326
Chicago/Turabian StyleDueñas-Cuellar, Rosa Amalia, Carlos José Correia Santana, Ana Carolina Martins Magalhães, Osmindo Rodrigues Pires, Jr., Wagner Fontes, and Mariana S. Castro. 2020. "Scorpion Toxins and Ion Channels: Potential Applications in Cancer Therapy" Toxins 12, no. 5: 326. https://doi.org/10.3390/toxins12050326
APA StyleDueñas-Cuellar, R. A., Santana, C. J. C., Magalhães, A. C. M., Pires, O. R., Jr., Fontes, W., & Castro, M. S. (2020). Scorpion Toxins and Ion Channels: Potential Applications in Cancer Therapy. Toxins, 12(5), 326. https://doi.org/10.3390/toxins12050326