Role of Nrf2 Nucleus Translocation in Beauvericin-Induced Cell Damage in Rat Hepatocytes

 ,
,  and
and
Abstract
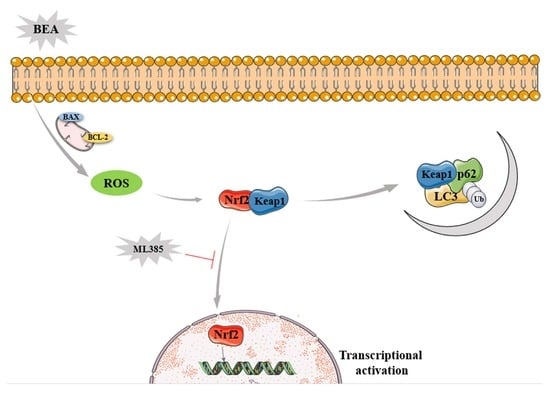
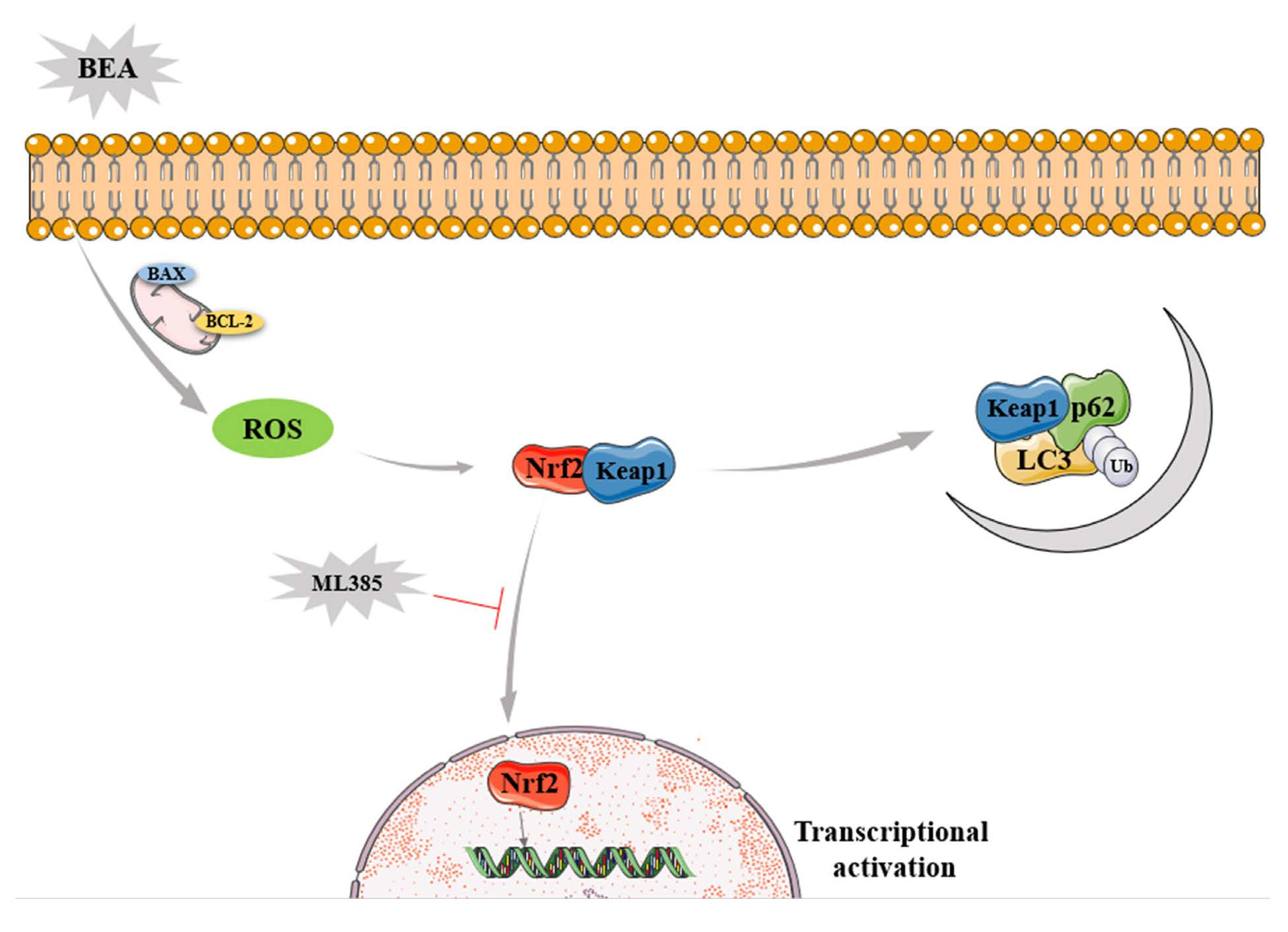
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
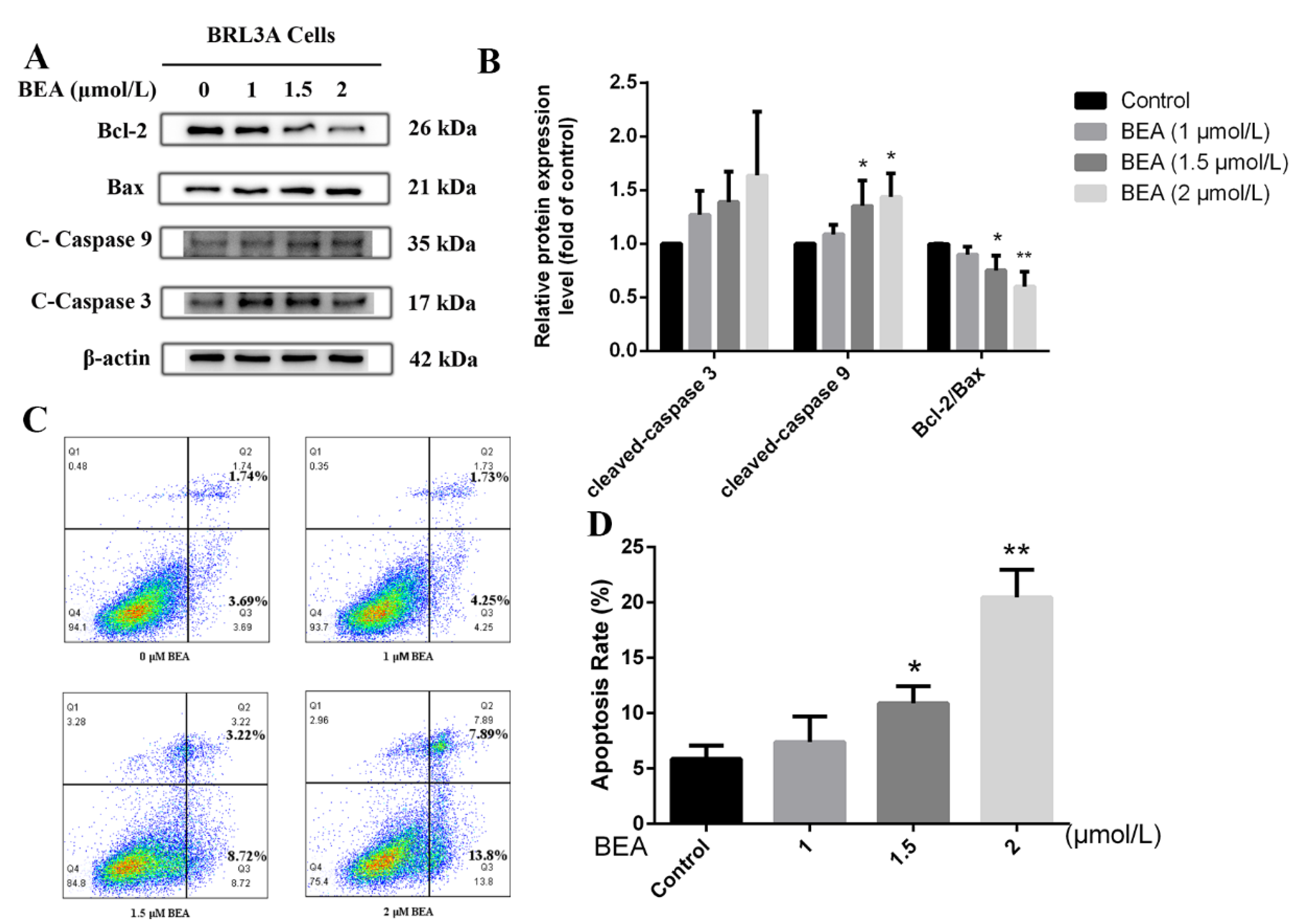
2.1. Liver Cell Injury and Apoptosis Induced by BEA
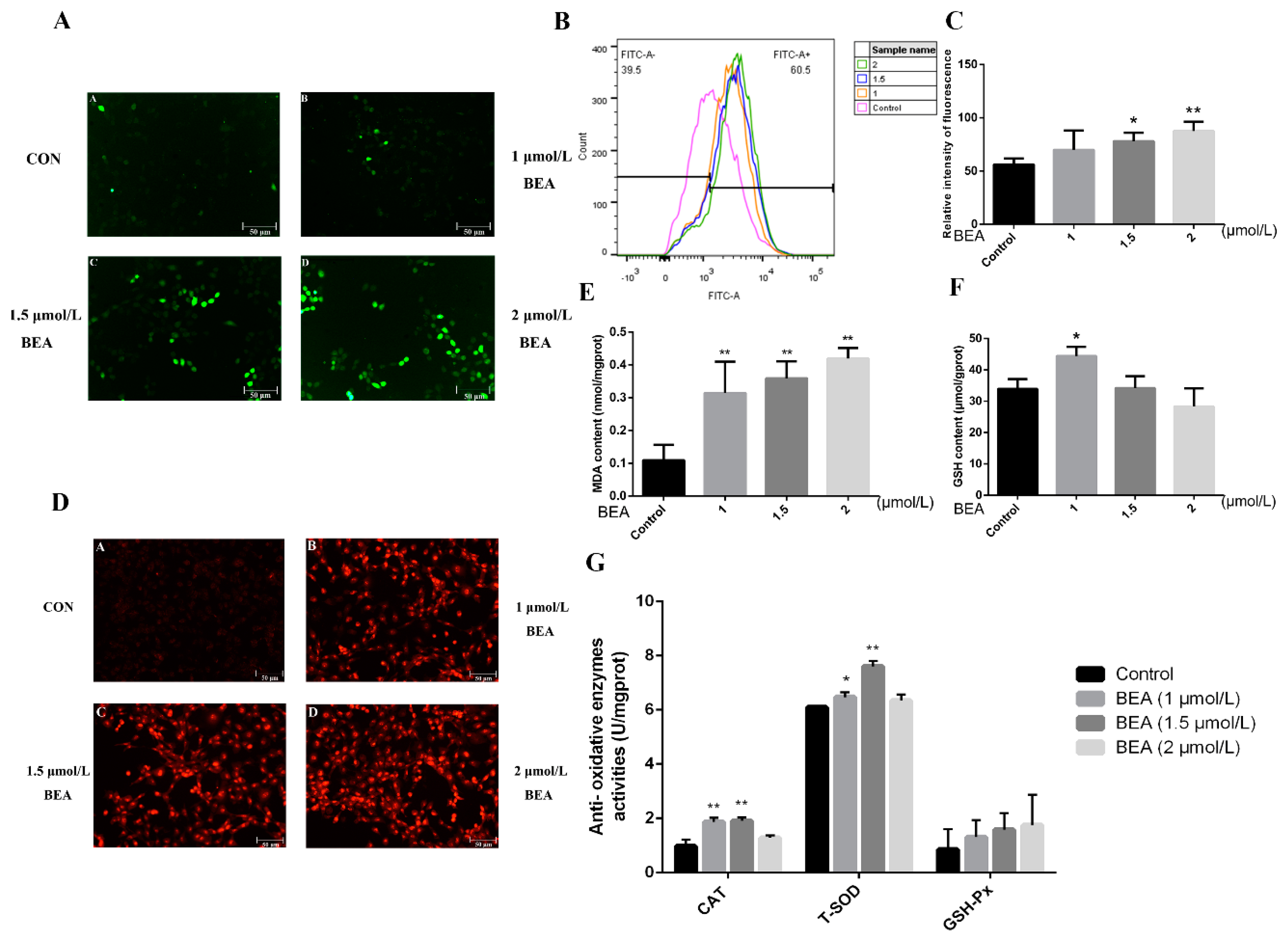
2.2. Effect of BEA on the Oxidative Damage in BRL3A Cells
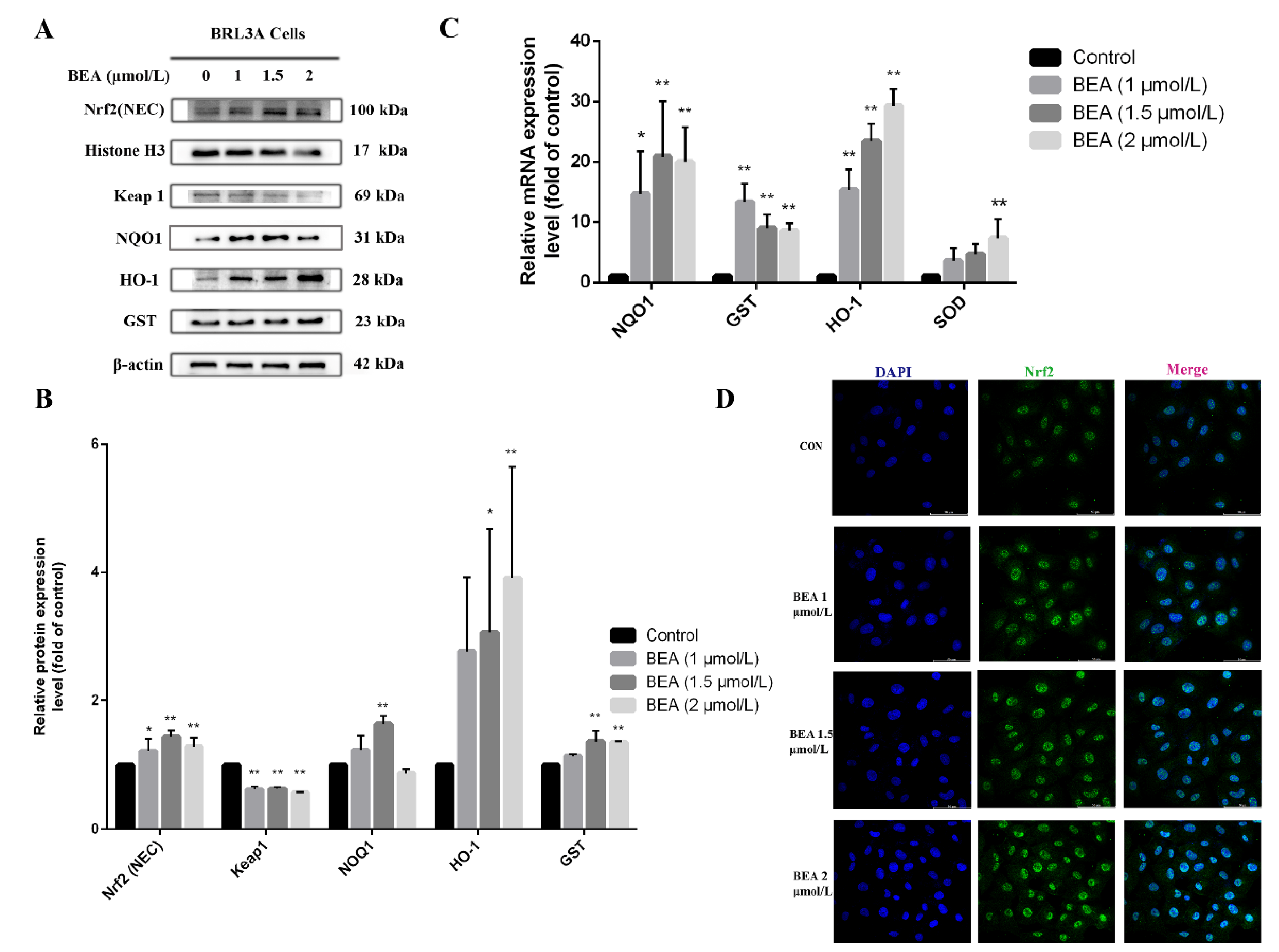
2.3. Effect of BEA on the Nrf2 Signaling Pathway of BRL3A Cells
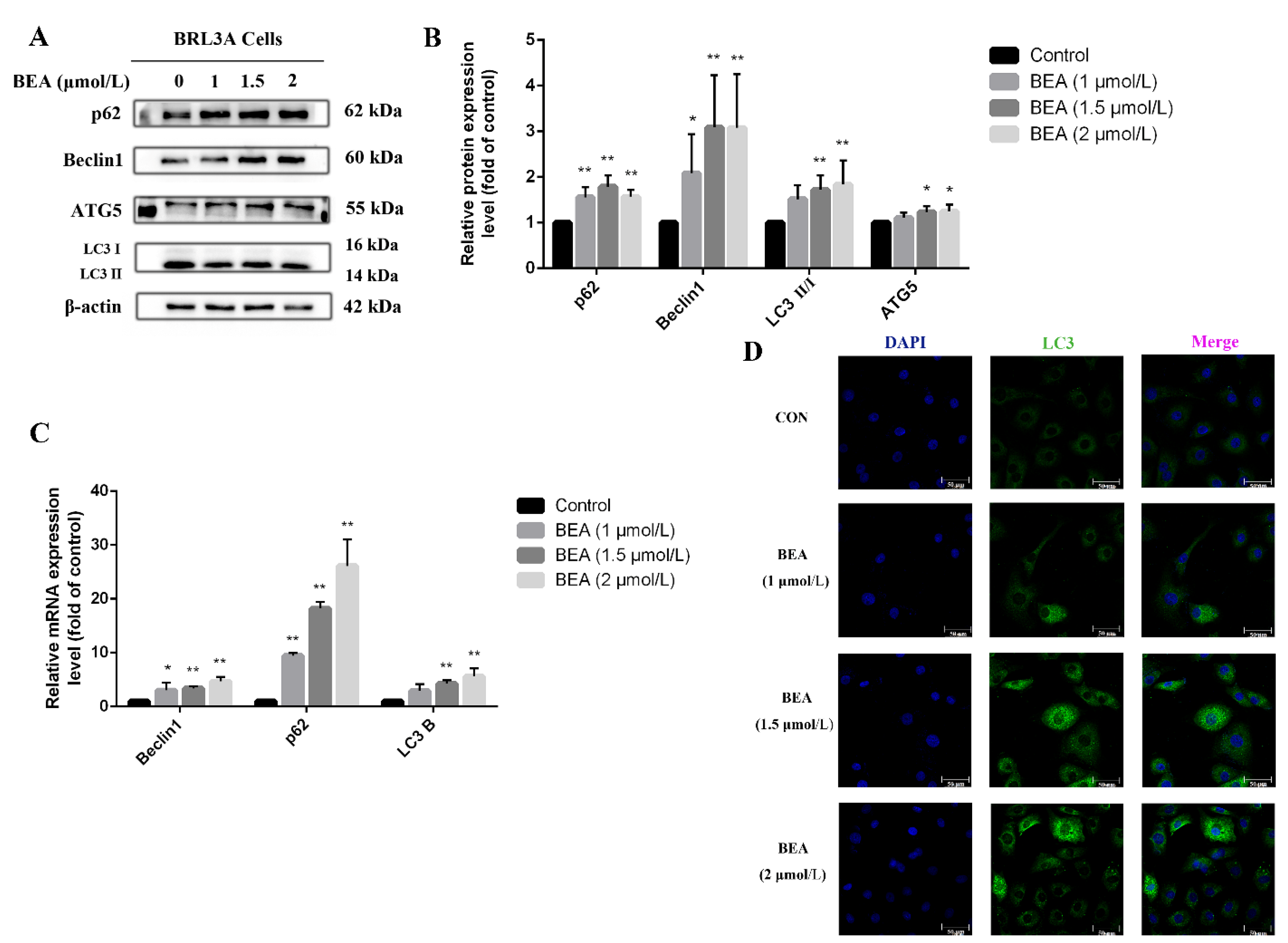
2.4. Effect of BEA on the Autophagy in BRL3A Cells
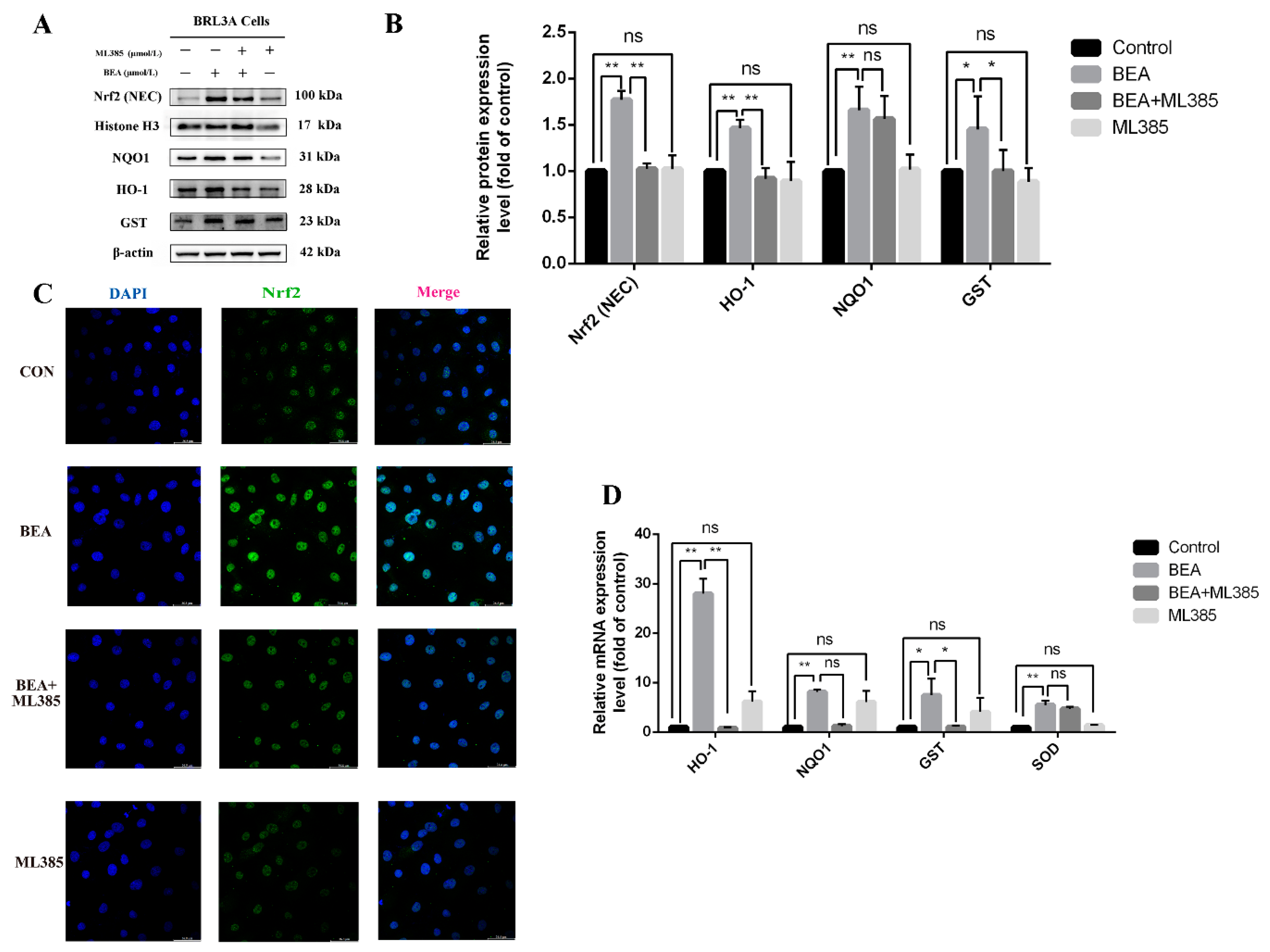
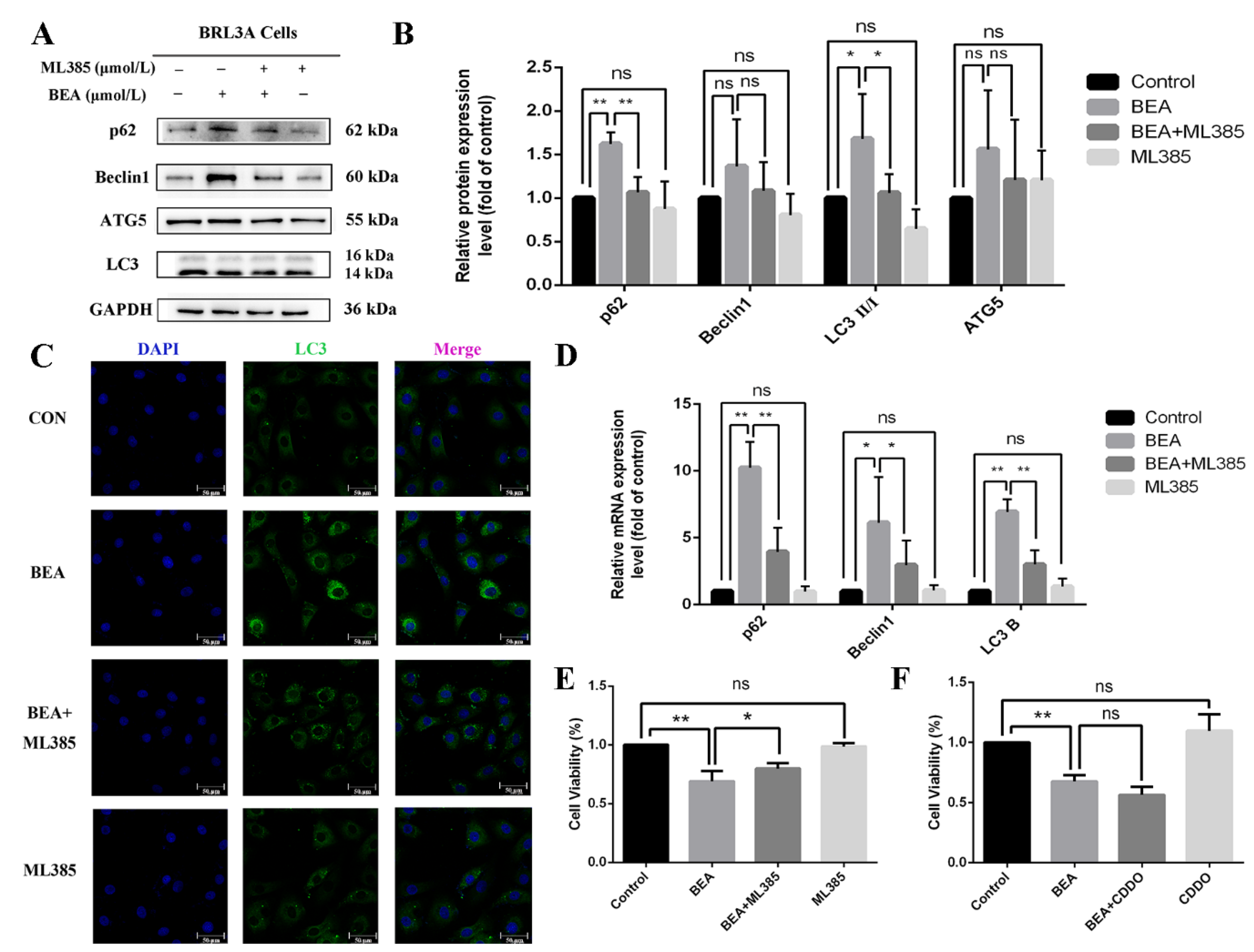
2.5. Effect of ML385 on the Alleviation of Cell Injury in BEA-Exposed BRL3A Cells
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Chemicals and Reagents
5.2. Cell Culture
5.3. Cell Proliferation Assay
5.4. LDH Release Assay
5.5. Measurement of Apoptosis Rate by Flow Cytometry
5.6. Detection of Superoxide Anions
5.7. Measurement of ROS
5.8. Measurement of Oxidants and Antioxidants
5.9. Western Blot Analysis
5.10. RNA Isolation, cDNA Synthesis and Quantitative Real-Time PCR (qRT-PCR)
5.11. Immunofluorescence Analysis
5.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hamill, R.L.; Higgens, C.E.; Boaz, H.E.; Gorman, M. The structure op beauvericin, a new depsipeptide antibiotic toxic to artemia salina. Tetrahedron Lett. 1969, 10, 4255–4258. [Google Scholar] [CrossRef]
- Jow, G.; Chou, C.; Chen, B.; Tsai, J. Beauvericin induces cytotoxic effects in human acute lymphoblastic leukemia cells through cytochrome c release, caspase 3 activation: The causative role of calcium. Cancer Lett. 2004, 216, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Søderstrøm, S.; Lie, K.; Lundebye, A.; Søfteland, L. Beauvericin (BEA) and enniatin B (ENNB)-induced impairment of mitochondria and lysosomes—Potential sources of intracellular reactive iron triggering ferroptosis in Atlantic salmon primary hepatocytes. Food Chem. Toxicol. 2022, 161, 112819. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xu, L. Beauvericin, a bioactive compound produced by fungi: A short review. Molecules 2012, 17, 2367–2377. [Google Scholar] [CrossRef] [PubMed]
- Mallebrera, B.; Prosperini, A.; Font, G.; Ruiz, M. In vitro mechanisms of Beauvericin toxicity: A review. Food Chem. Toxicol. 2018, 111, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Jestoi, M. Emerging fusarium-mycotoxins fusaproliferin, beauvericin, enniatins, and moniliformin: A review. Crit. Rev. Food Sci. 2008, 48, 21–49. [Google Scholar] [CrossRef]
- Klarić, M.; Pepeljnjak, S.; Domijan, A.; Petrik, J. Lipid peroxidation and glutathione levels in porcine kidney PK15 cells after individual and combined treatment with fumonisin B(1), beauvericin and ochratoxin A. Basic Clin. Pharmacol. 2007, 100, 157–164. [Google Scholar] [CrossRef]
- Svingen, T.; Lund Hansen, N.; Taxvig, C.; Vinggaard, A.; Jensen, U.; Have Rasmussen, P. Enniatin B and beauvericin are common in Danish cereals and show high hepatotoxicity on a high-content imaging platform. Environ. Toxicol. 2017, 32, 1658–1664. [Google Scholar] [CrossRef]
- Rodríguez-Carrasco, Y.; Heilos, D.; Richter, L.; Süssmuth, R.; Heffeter, P.; Sulyok, M.; Kenner, L.; Berger, W.; Dornetshuber-Fleiss, R. Mouse tissue distribution and persistence of the food-born fusariotoxins Enniatin B and Beauvericin. Toxicol. Lett. 2016, 247, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.; Dinkova-Kostova, A.; Tew, K. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Hass, D.; Barnstable, C. Uncoupling proteins in the mitochondrial defense against oxidative stress. Prog. Retin. Eye Res. 2021, 83, 100941. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Wang, X.; Zhao, F.; Villeneuve, N.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Shaikh, Z. Activation of Nrf2 by cadmium and its role in protection against cadmium-induced apoptosis in rat kidney cells. Toxicol. Appl. Pharm. 2009, 241, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.; Awuh, J.; Øvervatn, A.; McMahon, M.; Hayes, J.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Silva-Islas, C.; Maldonado, P. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol. Res. 2018, 134, 92–99. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.; Ueno, I.; Sakamoto, A.; Tong, K.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Elazar, Z. Oxidation as a post-translational modification that regulates autophagy. Autophagy 2007, 3, 371–373. [Google Scholar] [CrossRef] [Green Version]
- Portal-Núñez, S.; Esbrit, P.; Alcaraz, M.; Largo, R. Oxidative stress, autophagy, epigenetic changes and regulation by miRNAs as potential therapeutic targets in osteoarthritis. Biochem. Pharmacol. 2016, 108, 1–10. [Google Scholar] [CrossRef]
- Ichimura, Y.; Waguri, S.; Sou, Y.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Xu, X.; Zhang, F.; Wang, M.; Xu, Y.; Tang, D.; Wang, J.; Qin, Y.; Liu, Y.; Tang, C.; et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017, 11, 297–311. [Google Scholar] [CrossRef]
- Dong, W.; Liu, G.; Zhang, K.; Tan, Y.; Zou, H.; Yuan, Y.; Gu, J.; Song, R.; Zhu, J.; Liu, Z. Cadmium exposure induces rat proximal tubular cells injury via p62-dependent Nrf2 nucleus translocation mediated activation of AMPK/AKT/mTOR pathway. Ecotox. Environ. Saf. 2021, 214, 112058. [Google Scholar] [CrossRef] [PubMed]
- Emmanuel, K.T.; Els, V.; Bart, H.; Evelyne, D.; Els, V.; Els, D. Carry-over of some Fusarium mycotoxins in tissues and eggs of chickens fed experimentally mycotoxin-contaminated diets. Food Chem. Toxicol. 2020, 145, 111715. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Patocka, J.; Nepovimova, E.; Kuca, K. FusariumA Review on the Synthesis and Bioactivity Aspects of Beauvericin, a Mycotoxin. Front. Pharmacol. 2018, 9, 1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimian, G.; Buist-Homan, M.; Faber, K.; Moshage, H. Pertussis toxin, an inhibitor of G(αi) PCR, inhibits bile acid- and cytokine-induced apoptosis in primary rat hepatocytes. PLoS ONE 2012, 7, e43156. [Google Scholar] [CrossRef] [Green Version]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, Q.; Ci, X.; Chen, S.; Xie, Z.; Li, H.; Zhang, H.; Chen, F.; Xie, Q. Evaluation of the efficacy of chlorogenic acid in reducing small intestine injury, oxidative stress, and inflammation in chickens challenged with Clostridium perfringens type A. Poult Sci. 2020, 99, 6606–6618. [Google Scholar] [CrossRef]
- Mallebrera, B.; Font, G.; Ruiz, M. Disturbance of antioxidant capacity produced by beauvericin in CHO-K1 cells. Toxicol. Lett. 2014, 226, 337–342. [Google Scholar] [CrossRef]
- Dornetshuber, R.; Heffeter, P.; Lemmens-Gruber, R.; Elbling, L.; Marko, D.; Micksche, M.; Berger, W. Oxidative stress and DNA interactions are not involved in Enniatin- and Beauvericin-mediated apoptosis induction. Mol. Nutr. Food Res. 2009, 53, 1112–1122. [Google Scholar] [CrossRef]
- Thiruvengadam, M.; Venkidasamy, B.; Subramanian, U.; Samynathan, R.; Ali Shariati, M.; Rebezov, M.; Girish, S.; Thangavel, S.; Dhanapal, A.R.; Fedoseeva, N.; et al. Bioactive Compounds in Oxidative Stress-Mediated Diseases: Targeting the NRF2/ARE Signaling Pathway and Epigenetic Regulation. Antioxidants 2021, 10, 1859. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.; Wells, G.; Hayes, J.; Cousin, S.; Rumsey, W.; Attucks, O.; Franklin, S.; Levonen, A.; Kensler, T.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [Green Version]
- Mallebrera, B.; Juan-Garcia, A.; Font, G.; Ruiz, M. Mechanisms of beauvericin toxicity and antioxidant cellular defense. Toxicol. Lett. 2016, 246, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ye, T.; Xue, B.; Yang, M.; Li, R.; Xu, X.; Zeng, X.; Tian, N.; Bao, L.; Huang, Y. Mitochondrial GRIM-19 deficiency facilitates gastric cancer metastasis through oncogenic ROS-NRF2-HO-1 axis via a NRF2-HO-1 loop. Gastric Cancer 2021, 24, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Feng, C.; Jiang, H. Novel target for treating Alzheimer’s Diseases: Crosstalk between the Nrf2 pathway and autophagy. Ageing Res. Rev. 2021, 65, 101207. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Tang, K.; Wang, Z.; Wang, L. Persistent activation of Nrf2 promotes a vicious cycle of oxidative stress and autophagy inhibition in cadmium-induced kidney injury. Toxicology 2021, 464, 152999. [Google Scholar] [CrossRef]
- Liu, B.; Yan, L.; Jiao, X.; Sun, X.; Zhao, Z.; Yan, J.; Guo, M.; Zang, Y. Lycopene Alleviates Hepatic Hypoxia/Reoxygenation Injury Through Nrf2/HO-1 Pathway in AML12 Cell. J. Interf. Cytok Res. 2020, 40, 406–417. [Google Scholar] [CrossRef]
- Atilano-Roque, A.; Aleksunes, L.; Joy, M. Bardoxolone methyl modulates efflux transporter and detoxifying enzyme expression in cisplatin-induced kidney cell injury. Toxicol. Lett. 2016, 259, 52–59. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, J.; Wang, Y.; Xu, W.; Cai, G.; Zou, H.; Yuan, Y.; Gu, J.; Liu, Z.; Bian, J. Role of Nrf2 Nucleus Translocation in Beauvericin-Induced Cell Damage in Rat Hepatocytes. Toxins 2022, 14, 367. https://doi.org/10.3390/toxins14060367
Shi J, Wang Y, Xu W, Cai G, Zou H, Yuan Y, Gu J, Liu Z, Bian J. Role of Nrf2 Nucleus Translocation in Beauvericin-Induced Cell Damage in Rat Hepatocytes. Toxins. 2022; 14(6):367. https://doi.org/10.3390/toxins14060367
Chicago/Turabian StyleShi, Jiabin, Yaling Wang, Wenlin Xu, Guodong Cai, Hui Zou, Yan Yuan, Jianhong Gu, Zongping Liu, and Jianchun Bian. 2022. "Role of Nrf2 Nucleus Translocation in Beauvericin-Induced Cell Damage in Rat Hepatocytes" Toxins 14, no. 6: 367. https://doi.org/10.3390/toxins14060367
APA StyleShi, J., Wang, Y., Xu, W., Cai, G., Zou, H., Yuan, Y., Gu, J., Liu, Z., & Bian, J. (2022). Role of Nrf2 Nucleus Translocation in Beauvericin-Induced Cell Damage in Rat Hepatocytes. Toxins, 14(6), 367. https://doi.org/10.3390/toxins14060367