The Protein Kinase C Agonist PEP005 (Ingenol 3-Angelate) in the Treatment of Human Cancer: A Balance between Efficacy and Toxicity


Abstract
:1. Introduction
2. The Protein Kinase C Family
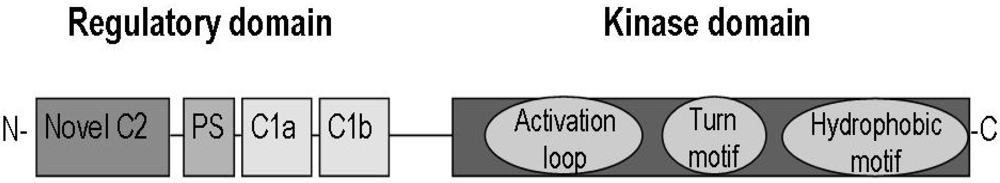
2.1. Classification and Characterization of Protein Kinase C Isoenzymes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classical isoforms cPKC | Novel isoforms nPKC | Atypical isoforms aPKC | |
|---|---|---|---|
| Members | α, βI, βII, γ | δ, ε, η, μ, θ | ζ, ι/λ |
| Phorbol ester activation | Yes | Yes | No |
| Regulatory cofactors | Diacylglycerol | Diacylglycerol | Independent of Ca and diacylglycerol |
| Phosphatidyl-serine | Ca-independent | ||
| Ca | |||
| Effect on apoptosis | Antiapoptotic: α, βI, βII | Antiapoptotic: ε | Antiapoptotic: ζ |
| Proapoptotic: δ |
| PKC isoform | Tumor Type | Expression | References |
|---|---|---|---|
| Classical | |||
| PKC-α | Bladder | Increased | [12] |
| Brain | Decreased | [13] | |
| Brain | Increased | [14] | |
| Breast | Decreased | [15,16] | |
| Ovarian | Decreased | [17] | |
| Renal | Decreased | [18] | |
| Colon | Decreased | [19] | |
| T-cell leukemia | Decreased | [20] | |
| PKC-β | Bladder | Decreased | [12] |
| Colon | Decreased | [21,22,23] | |
| Prostate | Decreased | [24] | |
| T-cell leukemia | Decreased | [20] | |
| Melanoma | Decreased | [25] | |
| PKC-βI | Bladder | Decreased | [26] |
| PKC-βII | Bladder | Decreased | [27] |
| Colon | Decreased | [28] | |
| DLBCL | Increased | [29] | |
| Novel | |||
| PKC-δ | Bladder | Decreased | [12,26,27] |
| Brain | Decreased | [14] | |
| Colon | Increased | [23] | |
| Squamous cell carcinoma | Decreased | [30] | |
| PKC-ε | Bladder | Increased | [12] |
| Brain | Increased | [31] | |
| Breast | Increased | [32] | |
| Colon | Decreased | [23] | |
| Prostate | Increased | [24] | |
| Thyroid | Decreased | [33] | |
| PKC-η | Breast | Decreased | [34,35] |
| Colon | Decreased | [21] | |
| Renal | Increased | [18] | |
| PKC-θ | Gastrointestinal stromal tumor | Increased | [36] |
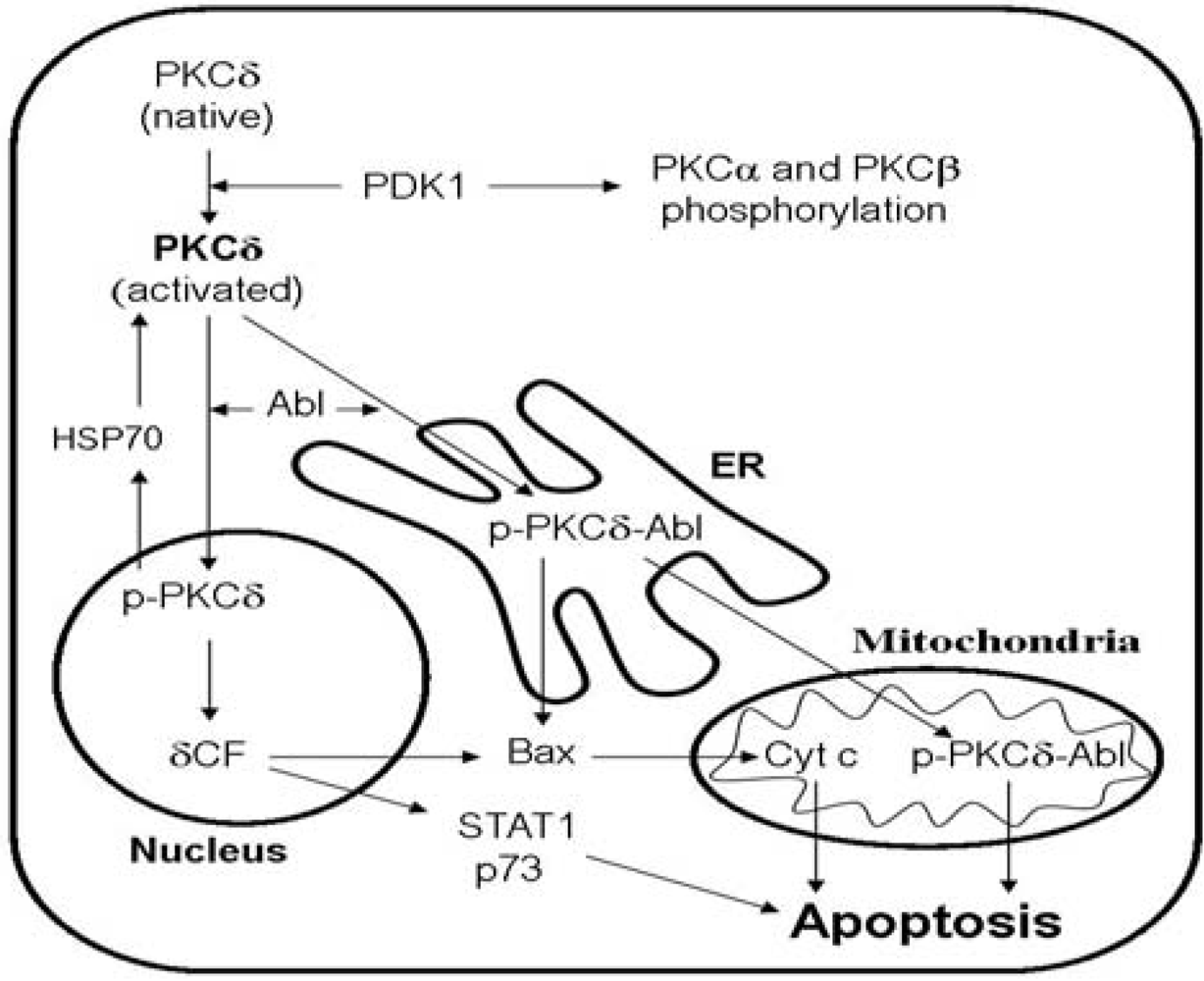
2.2. PKCδ and the Effects of PEP005


2.3. The Phenotype of PKCδ Null Mice
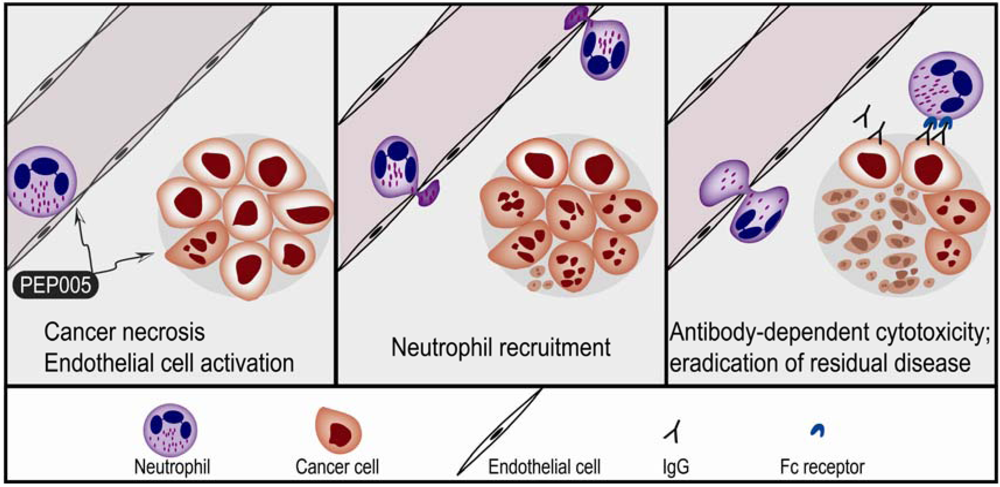
3. The Importance of Neutrophil Recruitment and Humoral Immunity after Topical Application of Pep005 for Skin Cancer
3.1. PEP005 EFFECTS on Endothelial Cells
3.2. The Anticancer Effect of Neutrophils after Topical Application of PEP005
- - Topical application of PEP005 can cure implanted skin cancers without later relapse in the T cell deficient Foxn1nu mice. This effect is associated with local macroscopic inflammation due to leukocyte infiltration dominated by neutrophils. After antibody-depletion of neutrophils topical PEP005 treatment caused a similar initial ablation, but tumors later re-emerged.
- - The neutrophil extravasation into the inflamed sites is severely impaired in CD18-deficient mice; topical treatment of implanted tumors in these animals was associated with initial cure followed by a weak local inflammation and later tumor relapse.
- - NK cells and macrophages are present in Foxn1nu mice, and macrophages are seen in PEP005 induced infiltrates. The local inflammation and relapse rate were not altered by depletion of NK cells. Neither inflammation nor relapse risk was altered for tumors implanted in Csfmop/Csfmop mice that lack functional M-CSF and therefore are severely monocytopenic.
- - The effect of topical PEP005 was investigated for LK2 tumors implanted in SCID mice that lack a humoral immune system [55]. Tumors grew at similar rates and the initial tumor-ablative effect and local inflammatory reactions were similar to Foxn1nu mice, but a high relapse rate was observed for the B cell-depleted mice.
3.3. Clinical Studies of PEP005 in the Treatment of Skin Cancer

4. Antileukemic Effects of Pep005
4.1. Effects of PEP005 on Acute Myelogenous Leukemia Cells
- - Chemokine release. Primary human AML cells show constitutive release of a wide range of chemokines [61,62]. PEP005 causes increased release of both CCL and CXCL chemokines, including CXCL8 that also was released at increased levels by skin cells after topical application (see above). The chemokines released at increased levels are pro-angiogenic and chemotactic not only for neutrophils but also for T cells and monocytes.
- - Chemokine receptor expression. PEP005 has only minor effects on the expression of most CCR and CXCR receptors (CCR1-3, CCR5, CXCR2, 3), the only exception being CXCR4 that shows decreased expression. CXCR4 is one of the two receptors for the CXCL12 chemokine that is usually not released or only released at low levels by primary human AML cells [61]. However, it is released by bone marrow stromal cells [62]. The CXCL12/CXCR4 system is important for AML cell migration and CXCR4 expression seems to have an adverse prognostic impact in AML [63]. For this reason the PEP005 induced reduction in CXCR4 expression should possibly be regarded as an anti-leukemic effect.
- - Differentiation. PEP005 decreases the expression of stem cell markers (including CXCR4) and increases the expression of lineage-associated markers, an observation consistent with differentiation induction.
- - Apoptosis regulation. PEP005 increases the expression of Bax and the activation of caspase 3. These pro-apoptotic effects are seen over a wide concentration range, whereas no induction of apoptosis was evident for normal CD34+ hematopoietic cells when testing concentrations up to 200 nM.
- - Intracellular signaling. The effect in AML cells is mediated through a PKCδ agonistic effect. The ERK1/2 pathway then seems to be important for the increased chemokine release together with increased expression of the NFκB subunits p50, p52 and p65.
4.2. The Role of PKC in Other Leukemias
5. Effects of PEP005 in Solid Tumors
5.1. Pharmacological in Vitro Studies
5.2. Studies in Animal Models
5.3. The Possibility of Topical Application for Other Cancers
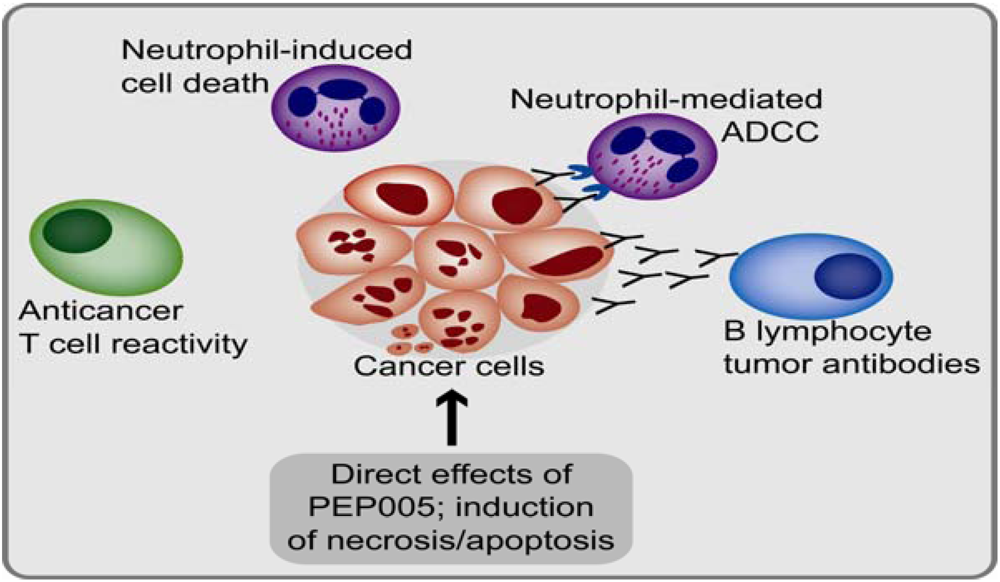
6. Immunomodulatory Effects of PEP005

7. Concluding Remarks: Efficiency versus Toxicity in the future Use of PEP005 in Cancer Treatment
7.1. Combination of PEP005 with Conventional Chemotherapy
7.2. The proinflammatory Effects of PEP005, A Possible Risk during Systemic Therapy
7.3. Cancer-Directed Delivery of PEP005 in Systemic Therapy
7.4. Sequential Treatment with Intensive Chemotherapy and PEP005; Decreased Risk for Proinflammatory-Induced Adverse Events?
7.5. PEP005 effects on the Chemokine System-Advantage or Disadvantage?
7.6. Final Comment
Acknowledgements
References and Notes
- Reikvam, H.; Ersvaer, E.; Bruserud, O. Heat shock protein 90 - a potential target in the treatment of human acute myelogenous leukemia. Curr. Cancer Drug Targets 2009, 9, 761–776. [Google Scholar] [PubMed]
- Reikvam, H.; Olsnes, A.M.; Gjertsen, B.T.; Ersvær, E.; Bruserud, Ø. Nuclear Factor-κB signaling - a contributor in leukemogenesis and a target for pharmacological intervention in human acute myelogenous leukemia. Curr. Rev. Oncogen. 2009, in press. [Google Scholar]
- Ogbourne, S.M.; Hampson, P.; Lord, J.M.; Parsons, P.; De Witte, P.A.; Suhrbier, A. Proceedings of the First International Conference on PEP005. Anticancer Drugs 2007, 18, 357–362. [Google Scholar] [PubMed]
- Kedei, N.; Lundberg, D.J.; Toth, A.; Welburn, P.; Garfield, S.H.; Blumberg, P.M. Characterization of the interaction of ingenol 3-angelate with protein kinase C. Cancer Res. 2004, 64, 3243–3255. [Google Scholar] [PubMed]
- Hug, H.; Sarre, T.F. Protein kinase C isoenzymes: Divergence in signal transduction? Biochem. J. 1993, 291, 329–343. [Google Scholar] [PubMed]
- Ohno, S.; Akita, Y.; Konno, Y.; Imajoh, S.; Suzuki, K. A novel phorbol ester receptor/protein kinase, nPKC, distantly related to the protein kinase C family. Cell 1988, 53, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Mellor, H.; Parker, P.J. The extended protein kinase C superfamily. Biochem. J. 1998, 332, 281–292. [Google Scholar] [PubMed]
- Redig, A.J.; Platanias, L.C. Protein kinase C signaling in leukemia. Leuk. Lymphoma 2008, 49, 1255–1262. [Google Scholar] [PubMed]
- Selvatici, R.; Falzarano, S.; Mollica, A.; Spisani, S. Signal transduction pathways triggered by selective formylpeptide analogues in human neutrophils. Eur. J. Pharmacol. 2006, 534, 1–11. [Google Scholar] [PubMed]
- Le Good, J.A.; Ziegler, W.H.; Parekh, D.B.; Alessi, D.R.; Cohen, P.; Parker, P.J. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science 1998, 281, 2042–2045. [Google Scholar] [PubMed]
- Reyland, M.E. Protein kinase Cδ and apoptosis. Biochem. Soc. Transact. 2007, 35, 1001–1004. [Google Scholar]
- Varga, A.; Czifra, G.; Tállai, B.; Németh, T.; Kovács, I.; Kovács, L.; Bíró, T. Tumor grade-dependent alterations in the protein kinase C isoform pattern in urinary bladder carcinomas. Eur. Urol. 2004, 46, 462–465. [Google Scholar] [PubMed]
- Benzil, D.L.; Finkelstein, S.D.; Epstein, M.H.; Finch, P.W. Expression pattern of alpha-protein kinase C in human astrocytomas indicates a role in malignant progression. Cancer Res. 1992, 52, 2951–2956. [Google Scholar] [PubMed]
- Mandil, R.; Ashkenazi, E.; Blass, M.; Kronfeld, I.; Kazimirsky, G.; Rosenthal, G.; Umansky, F.; Lorenzo, P.S.; Blumberg, P.M.; Brodie, C. Protein kinase Calpha and protein kinase Cdelta play opposite roles in the proliferation and apoptosis of glioma cells. Cancer Res. 2001, 61, 4612–4619. [Google Scholar] [PubMed]
- Ainsworth, P.D.; Winstanley, J.H.; Pearson, J.M.; Bishop, H.M.; Garrod, D.R. Protein kinase C alpha expression in normal breast, ductal carcinoma in situ and invasive ductal carcinom. Eur. J. Cancer 2004, 40, 2269–2273. [Google Scholar] [PubMed]
- Kerfoot, C.; Huang, W.; Rotenberg, S.A. Immunohistochemical analysis of advanced human breast carcinomas reveals downregulation of protein kinase C alpha. J. Histochem. Cytochem. 2004, 52, 419–422. [Google Scholar] [PubMed]
- Weichert, W.; Gekeler, V.; Denkert, C.; Dietel, M, Hauptmann. Protein kinase C isoform expression in ovarian carcinoma correlates with indicators of poor prognosis. Int. J. Oncol. 2003, 23, 633–639. [Google Scholar] [PubMed]
- Brenner, W.; Färber, G.; Herget, T.; Wiesner, C.; Hengstler, J.G.; Thüroff, J.W. Protein kinase C eta is associated with progression of renal cell carcinoma (RCC). Anticancer Res. 2003, 23, 4001–4006. [Google Scholar] [PubMed]
- Verstovsek, G.; Byrd, A.; Frey, M.R.; Petrelli, N.J.; Black, J.D. Colonocyte differentiation is associated with increased expression and altered distribution of protein kinase C isozymes. Gastroenterology 1998, 115, 75–85. [Google Scholar] [PubMed]
- Hidaka, M.; Nakakuma, H.; Kawaguchi, T.; Nagakura, S.; Horikawa, K.; Okuno, Y.; Kagimoto, T.; Takatsuki, K. Altered expression of protein kinase C in adult T-cell leukemia cells. Int. J. Hematol. 1992, 56, 135–141. [Google Scholar] [PubMed]
- Doi, S.; Goldstein, D.; Hug, H.; Weinstein, I.B. Expression of multiple isoforms of protein kinase C in normal human colon mucosa and colon tumors and decreased levels of protein kinase C beta and eta mRNAs in the tumors. Mol. Carcinog. 1994, 11, 197–203. [Google Scholar] [PubMed]
- Levy, M.F.; Pocsidio, J.; Guillem, J.G.; Forde, K.; LoGerfo, P.; Weinstein, I.B. Decreased levels of protein kinase C enzyme activity and protein kinase C mRNA in primary colon tumors. Dis. Colon. Rectum. 1993, 36, 913–921. [Google Scholar] [PubMed]
- Pongracz, J.; Clark, P.; Neoptolemos, J.P.; Lord, J.M. Expression of protein kinase C isoenzymes in colorectal cancer tissue and their differential activation by different bile acids. Int. J. Cancer 1995, 61, 35–39. [Google Scholar] [PubMed]
- Cornford, P.; Evans, J.; Dodson, A.; Parsons, K.; Woolfenden, A.; Neoptolemos, J.; Foster, C.S. Protein kinase C isoenzyme patterns characteristically modulated in early prostate cancer. Am. J. Pathol. 1999, 154, 137–144. [Google Scholar] [PubMed]
- Gilhooly, E.M.; Morse-Gaudio, M.; Bianchi, L.; Reinhart, L.; Rose, D.P.; Connolly, J.M.; Reed, J.A.; Albino, A.P. Loss of expression of protein kinase C beta is a common phenomenon in human malignant melanoma: A result of transformation or differentiation? Melanoma Res. 2001, 11, 355–369. [Google Scholar] [PubMed]
- Koren, R.; Langzam, L.; Paz, A.; Livne, P.M.; Gal, R.; Sampson, S.R. Protein kinase C (PKC) isoenzymes immunohistochemistry in lymph node revealing solution-fixed, paraffin-embedded bladder tumors. Appl. Immunohistochem. Mol. Morphol. 2000, 8, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Langzam, L.; Koren, R.; Gal, R.; Kugel, V.; Paz, A.; Farkas, A.; Sampson, S.R. Patterns of protein kinase C isoenzyme expression in transitional cell carcinoma of bladder. Relation to degree of malignancy. Am. J. Clin. Pathol. 2001, 116, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Kuranami, M.; Powell, C.T.; Hug, H.; Zeng, Z.; Cohen, A.M.; Guillem, J.G. Differential expression of protein kinase C isoforms in human colorectal cancers. J. Surg. Res. 1995, 58, 233–239. [Google Scholar] [PubMed]
- Hans, C.P.; Weisenburger, D.D.; Greiner, T.C.; Chan, W.C.; Aoun, P.; Cochran, G.T.; Pan, Z.; Smith, L.M.; Lynch, J.C.; Bociek, R.G.; Bierman, P.J.; Vose, J.M.; Armitage, J.O. Expression of PKC-beta or cyclin D2 predicts for inferior survival in diffuse large B-cell lymphoma. Mod. Pathol. 2005, 18, 1377–1384. [Google Scholar] [PubMed]
- D'Costa, A.M.; Robinson, J.K.; Maududi, T.; Chaturvedi, V.; Nickoloff, B.J.; Denning, M.F. The proapoptotic tumor suppressor protein kinase C-delta is lost in human squamous cell carcinomas. Oncogene 2006, 25, 378–386. [Google Scholar] [PubMed]
- Sharif, T.R.; Sharif, M. Overexpression of protein kinase C epsilon in astroglial brain tumor derived cell lines and primary tumor samples. Int. J. Oncol. 1999, 15, 237–243. [Google Scholar] [PubMed]
- Pan, Q.; Bao, L.W.; Kleer, C.G.; Sabel, M.S.; Griffith, K.A.; Teknos, T.N.; Merajver, S.D. Protein kinase C epsilon is a predictive biomarker of aggressive breast cancer and a validated target for RNA interference anticancer therapy. Cancer Res. 2005, 65, 8366–8371. [Google Scholar] [PubMed]
- Knauf, J.A.; Ward, L.S.; Nikiforov, Y.E.; Nikiforova, M.; Puxeddu, E.; Medvedovic, M.; Liron, T.; Mochly-Rosen, D.; Fagin, J.A. Isozyme-specific abnormalities of PKC in thyroid cancer: Evidence for post-transcriptional changes in PKC epsilon. J. Clin. Endocrinol. Metab. 2002, 87, 2150–2159. [Google Scholar] [PubMed]
- Beck, J.; Bohnet, B.; Brügger, D.; Bader, P.; Dietl, J.; Scheper, R.J.; Kandolf, R.; Liu, C.; Niethammer, D.; Gekeler, V. Multiple gene expression analysis reveals distinct differences between G2 and G3 stage breast cancers, and correlations of PKC eta with MDR1, MRP and LRP gene expression. Br. J. Cancer 1998, 77, 87–91. [Google Scholar] [PubMed]
- Masso-Welch, P.A.; Winston, J.S.; Edge, S.; Darcy, K.M.; Asch, H.; Vaughan, M.M.; Ip, M.M. Altered expression and localization of PKC eta in human breast tumors. Breast Cancer Res. Treat 2001, 68, 211–223. [Google Scholar] [PubMed]
- Blay, P.; Astudillo, A.; Buesa, J.M.; Campo, E.; Abad, M.; García-García, J.; Miquel, R.; Marco, V.; Sierra, M.; Losa, R.; Lacave, A.; Braña, A.; Balbín, M.; Freije, J.M. Protein kinase C theta is highly expressed in gastrointestinal stromal tumors but not in other mesenchymal neoplasias. Clin. Cancer Res. 2004, 10, 4089–4095. [Google Scholar] [PubMed]
- Koivunen, J.; Aaltonen, V.; Peltonen, J. Protein kinase C (PKC) family in cancer progression. Cancer letters 2006, 235, 1–10. [Google Scholar] [PubMed]
- Bassini, A.; Zauli, G.; Migliaccio, G.; Migliaccio, A.R.; Pascuccio, M.; Pierpaoli, S.; Guidotti, L.; Capitani, S.; Vitale, M. Lineage-restricted expression of protein kinase C isoforms in hematopoiesis. Blood 1999, 93, 1178–1188. [Google Scholar] [PubMed]
- Steinberg, S.F. Distinctive activation mechanisms and functions for protein kinase Cdelta. Biochem. J. 2004, 384, 49–459. [Google Scholar]
- Yamaguchi, T.; Miki, Y.; Yoshida, K. Protein kinase C delta activates IkappaB-kinase alpha to induce the p53 tumor suppressor in response to oxidative stress. Cell Signal 2007, 19, 2088–2097. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; White, D.; Hui, L.; Yoshida, K.; Foster, D.A.; Bargonetti, J. Inhibition of human p53 basal transcription by down-regulation of protein kinase Cdelta. J. Biol. Chem. 2004, 279, 9970–9977. [Google Scholar] [PubMed]
- Pearn, L.; Fisher, J.; Burnett, A.K.; Darley, R.L. The role of PKC and PDK1 in monocyte lineage specification by Ras. Blood 2007, 109, 4461–4469. [Google Scholar] [PubMed]
- Jackson, D.N.; Foster, D.A. The enigmatic protein kinase Cdelta: Complex roles in cell proliferation and survival. FASEB J. 2004, 18, 627–636. [Google Scholar] [PubMed]
- Leitges, M.; Mayr, M.; Braun, U.; Mayr, U.; Li, C.; Pfister, G.; Ghaffari-Tabrizi, N.; Baier, G.; Hu, Y.; Xu, Q. Exacerbated vein graft arteriosclerosis in protein kinase Cdelta-null mice. J. Clin. Invest 2001, 108, 1505–1512. [Google Scholar] [PubMed]
- Newton, A.C. Regulation of the ABC kinases by phosphorylation: Protein kinase C as a paradigm. Biochem. J. 2003, 370, 361–371. [Google Scholar] [PubMed]
- Zhu, T.; Tsuji, T.; Chen, C. Roles of PKC isoforms in the induction of apoptosis elicited by aberant Ras. Oncogene 2009, 29, 1050–1061. [Google Scholar] [PubMed]
- Qi, X.; Mochly-Rosen, D. The PKCδ-Abl complex communicates ER stress to the mitochondria - an essential step in subsequent apoptosis. J. Cell Sci. 2007, 121, 804–813. [Google Scholar]
- Humphries, M.J.; Limesand, K.H.; Schneider, J.C.; Nakayama, K.I.; Anderson, S.M.; Reyland, M.E. Suppression of apoptosis in the protein kinase Cdelta null mouse in vivo. J. Biol. Chem. 2006, 281, 9728–9737. [Google Scholar] [PubMed]
- Miyamoto, A.; Nakayama, K.; Imaki, H.; Hirose, S.; Jiang, Y.; Abe, M.; Tsukiyama, T.; Nagahama, H.; Ohno, S.; Hatakeyama, S.; Nakayama, K.I. Increased proliferation of B cells and auto-immunity in mice lacking protein kinase Cdelta. Nature 2002, 416, 865–869. [Google Scholar] [PubMed]
- Mecklenbräuker, I.; Saijo, K.; Zheng, N.Y.; Leitges, M.; Tarakhovsky, A. Protein kinase Cdelta controls self-antigen-induced B-cell tolerance. Nature 2002, 416, 860–865. [Google Scholar] [PubMed]
- Zabarovsky, E.R.; Lerman, M.I.; Minna, J.D. Tumor suppressor genes on chromosome 3p involved in the pathogenesis of lung and other cancers. Oncogene 2002, 21, 6915–6935. [Google Scholar] [PubMed]
- Zimmerman, G.A.; Prescott, S.M.; McIntyre, T.M. Endothelial cell interactions with granulocytes: Tethering and signaling molecules. Immunol. Today 1992, 13, 93–100. [Google Scholar] [PubMed]
- Imhof, B.A.; Dunon, D. Leukocyte migration and adhesion. Adv. Immunol. 1995, 58, 345–416. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.A. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu. Rev. Physiol. 1995, 57, 827–872. [Google Scholar] [PubMed]
- Challacombe, J.M.; Suhrbier, A.; Parsons, P.G.; Jones, B.; Hampson, P.; Kavanagh, D.; Rainger, G.E.; Morris, M.; Lord, J.M.; Le, T.T.; Hoang-Le, D.; Ogbourne, S.M. Neutrophils are a key component of the antitumor efficacy of topical chemotherapy with ingenol-3-angelate. J. Immunol. 2006, 177, 8123–8132. [Google Scholar] [PubMed]
- Hampson, P.; Kavanagh, D.; Smith, E.; Wang, K.; Lord, J.M.; Ed Rainger, G. The anti-tumor agent, ingenol-3-angelate (PEP005), promotes the recruitment of cytotoxic neutrophils by activation of vascular endothelial cells in a PKC-delta dependent manner. Cancer Immunol. Immunother. 2008, 57, 1241–1251. [Google Scholar] [PubMed]
- Siller, G.; Gebauer, K.; Welburn, P.; Katsamas, J.; Ogbourne, S.M. PEP005 (ingenol mebutate) gel, a novel agent for the treatment of actinic keratosis: Results of a randomized, double-blind, vehicle-controlled, multicentre, phase IIa study. Austral. J. Dermatol. 2009, 50, 16–22. [Google Scholar]
- Anderson, L.; Schmieder, G.J.; Werschler, W.P.; Tschen, E.H.; Ling, M.R.; Stough, D.B.; Katsamas, J. Randomized, double-blind, double-dummy, vehicle-controlled study of ingenol mebutate gel 0.025% and 0.05% for actinic keratosis. J. Am. Acad. Dermatol. 2009, 60, 934–943. [Google Scholar] [PubMed]
- Olsnes, A.M.; Ersvaer, E.; Ryningen, A.; Paulsen, K.; Hampson, P.; Lord, J.M.; Gjertsen, B.T.; Kristoffersen, E.K.; Bruserud, Ø. The protein kinase C agonist PEP005 increases NF-kappaB expression, induces differentiation and increases constitutive chemokine release by primary acute myeloid leukemia cells. Br. J. Haematol. 2009, 145, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Hampson, P.; Chahal, H.; Khanim, F.; Hayden, R.; Mulder, A.; Assi, L.K.; Bunce, C.M.; Lord, J.M. PEP005, a selective small-molecule activator of protein kinase C, has potent antileukemic activity mediated via the delta isoform of PKC. Blood 2005, 106, 1362–1368. [Google Scholar] [PubMed]
- Bruserud, Ø.; Ryningen, A.; Olsnes, A.M.; Stordrange, L.; Øyan, A.M.; Kalland, K.H.; Gjertsen, B.T. Subclassification of patients with acute myelogenous leukemia based on chemokine responsiveness and constitutive chemokine release by their leukemic cells. Haematologica 2007, 92, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Olsnes, A.M.; Hatfield, K.J.; Bruserud, Ø. The chemokine system and its contribution to leukemogenesis and treatment responsiveness in patients with acute myelogenous leukemia. J. BUON. 2009, 14, 131–140. [Google Scholar] [PubMed]
- Kittang, A.M.O.; Hatfield, K.J.; Sand, K.E.; Reikvam, H.; Bruserud, Ø. The chemokine network in acute myelogenous leukemia: Molecular mechanisms involved in leukemogenesis and their therapeutic implications. Cur. Microbiol. Immunol. Rev. 2010, in press. [Google Scholar]
- Nakagawa, R.; Soh, J.W.; Michie, A.M. Subversion of protein kinase C alpha signaling in hematopoietic progenitor cells results in the generation of a B -cell chronic lymphocytic leukemia-like population in vivo. Cancer Res. 2006, 66, 527–534. [Google Scholar] [PubMed]
- Ghoul, A.; Serova, M.; Astorgues-Xerri, L.; Bieche, I.; Bousquet, G.; Varna, M.; Vidaud, M.; Phillips, E.; Weill, S.; Benhadji, K.A.; Lokiec, F.; Cvitkovic, E.; Faivre, S.; Raymond, E. Epithelial-to-mesenchymal transition and resistance to ingenol 3-angelate, a novel protein kinase C modulator, in colon cancer cells. Cancer Res. 2009, 69, 4260–4269. [Google Scholar] [PubMed]
- Gillespie, S.K.; Zhang, X.D.; Hersey, P. Ingenol 3-angelate induces dual modes of cell death and differentially regulates tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in melanoma cells. Mol. Cancer Ther. 2004, 3, 1651–1658. [Google Scholar] [PubMed]
- Cozzi, S.J.; Parsons, P.G.; Ogbourne, S.M.; Pedley, J.; Boyle, G.M. Induction of senescence in diterpene ester-treated melanoma cells via protein kinase C-dependent hyperactivation of the mitogen-activated protein kinase pathway. Cancer Res. 2006, 66, 10083–10091. [Google Scholar] [PubMed]
- Benhadji, K.A.; Serova, M.; Ghoul, A.; Cvitkovic, E.; Le Tourneau, C.; Ogbourne, S.M.; Lokiec, F.; Calvo, F.; Hammel, P.; Faivre, S.; Raymond, E. Antiproliferative activity of PEP005, a novel ingenol angelate that modulates PKC functions, alone and in combination with cytotoxic agents in human colon cancer cells. Br. J. Cancer 2008, 99, 1808–1815. [Google Scholar] [PubMed]
- Serova, M.; Ghoul, A.; Benhadji, K.A.; Faivre, S.; Le Tourneau, C.; Cvitkovic, E.; Lokiec, F.; Lord, J.; Ogbourne, S.M.; Calvo, F.; Raymond, E. Effects of protein kinase C modulation by PEP005, a novel ingenol angelate, on mitogen-activated protein kinase and phosphatidylinositol 3-kinase signaling in cancer cells. Mol. Cancer Ther. 2008, 7, 915–922. [Google Scholar] [PubMed]
- Ogbourne, S.M.; Suhrbier, A.; Jones, B.; Cozzi, S.J.; Boyle, G.M.; Morris, M.; McAlpine, D.; Johns, J.; Scott, T.M.; Sutherland, K.P.; Gardner, J.M.; Le, T.T.; Lenarczyk, A.; Aylward, J.H.; Parsons, P.G. Antitumor activity of 3-ingenyl angelate: Plasma membrane and mitochondrial disruption and necrotic cell death. Cancer Res. 2004, 64, 2833–2839. [Google Scholar] [PubMed]
- Ersvaer, E.; Hampson, P.; Wendelbo, Ø.; Lord, J.M.; Gjertsen, B.T.; Bruserud, Ø. Circulating T cells in patients with untreated acute myelogenous leukemia are heterogeneous and can be activated through the CD3/TCR complex. Hematology 2007, 12, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Ersvaer, E.; Hampson, P.; Hatfield, K.; Ulvestad, E.; Wendelbo, Ø.; Lord, J.M.; Gjertsen, B.T.; Bruserud, Ø. T cells remaining after intensive chemotherapy for acute myelogenous leukemia show a broad cytokine release profile including high levels of interferon-gamma that can be further increased by a novel protein kinase C agonist PEP005. Cancer Immunol. Immunother. 2007, 56, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.; Gardner, J.; Hoang-Le, D.; Schmidt, C.W.; MacDonald, K.P.; Lambley, E.; Schroder, W.A.; Ogbourne, S.M.; Suhrbier, A. Immunostimulatory cancer chemotherapy using local ingenol-3-angelate and synergy with immunotherapies. Vaccine 2009, 27, 3053–3062. [Google Scholar] [PubMed]
- Shin, S.Y.; Kim, C.G.; Ko, J.; Min, D.S.; Chang, J.S.; Ohba, M.; Kuroki, T.; Choi, Y.B.; Kim, Y.H.; Na, D.S.; Kim, J.W.; Lee, Y.H. Transcriptional and post-transcriptional regulation of the PKC delta gene by etoposide in L1210 murine leukemia cells: Implication of PKC delta autoregulation. J. Mol. Biol. 2004, 340, 681–693. [Google Scholar] [PubMed]
- Kaur, S.; Parmar, S.; Smith, J.; Katsoulidis, E.; Li, Y.; Sassano, A.; Majchrzak, B.; Uddin, S.; Tallman, M.S.; Fish, E.N.; Platanias, L.C. Role of protein kinase C-delta (PKC-delta) in the generation of the effects of IFN-alpha in chronic myelogenous leukemia cells. Exp. Hematol. 2005, 33, 550–557. [Google Scholar] [PubMed]
- Wood, A.J.; Darbyshire, J. Injury to research volunteers--the clinical-research nightmare. N. Engl. J. Med. 2006, 354, 1869–1871. [Google Scholar] [PubMed]
- Farzaneh, L.; Kasahara, N.; Farzaneh, F. The strange case of TGN1412. Cancer Immunol. Immunother. 2007, 56, 129–134. [Google Scholar] [PubMed]
- Dayan, C.M.; Wraith, D.C. Preparing for first-in-man studies: The challenges for translational immunology post-TGN1412. Clin. Exp. Immunol. 2008, 151, 231–234. [Google Scholar] [PubMed]
- Kenter, M.J.; Cohen, A.F. Establishing risk of human experimentation with drugs: Lessons from TGN1412. Lancet 2006, 368, 1387–1391. [Google Scholar] [PubMed]
- Winkler, U.; Jensen, M.; Manzke, O.; Schulz, H.; Diehl, V.; Engert, A. Cytokine-release syndrome in patients with B-cell chronic lymphocytic leukemia and high lymphocyte counts after treatment with an anti-CD20 monoclonal antibody (rituximab, IDEC-C2B8). Blood 1999, 94, 2217–2224. [Google Scholar] [PubMed]
- Maghazachi, A. Role of chemokines for natural killer cells. Curr. Topics Microbiol. Immunol. 2010, in press. [Google Scholar]
- Carter, P.J.; Senter, P.D. Antibody-drug conjugates for cancer therapy. Cancer J. 2008, 14, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Stasi, R. Gemtuzumab ozogamicin: An anti-CD33 immunoconjugate for the treatment of acute myeloid leukemia. Expert Opin. Biol. Ther. 2008, 8, 527–540. [Google Scholar] [PubMed]
- Wong, J.Y. Systemic targeted radionuclide therapy: potential new areas. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, 74–82. [Google Scholar]
- Postema, M.; Gilja, O.H. Ultrasound-directed drug delivery. Curr. Pharm. Biotechnol. 2007, 8, 355–361. [Google Scholar] [PubMed]
- Berg, K.; Folini, M.; Prasmickaite, L.; Selbo, P.K.; Bonsted, A.; Engesaeter, B.Ø.; Zaffaroni, N.; Weyergang, A.; Dietze, A.; Maelandsmo, G.M.; Wagner, E.; Norum, O.J.; Høgset, A. Photochemical internalization: A new tool for drug delivery. Curr. Pharm. Biotechnol. 2007, 8, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø.; Foss, B.; Petersen, H. Hematopoietic growth factors in patients receiving intensive chemotherapy for malignant disorders: Studies of granulocyte-colony stimulating factor (G-CSF),granulocyte-macrophage colony stimulating factor (GM-CSF),interleukin-3 (IL-3) and Flt-3 ligand (Flt3L). Eur. Cytokine Netw. 2001, 12, 231–238. [Google Scholar] [PubMed]
- Ersvaer, E.; Olsnes, A.M.; Bruserud, Ø. The immunological dilemma: Cellular innate and adaptive immune response versus human acute myeloid leukemia. Open Hematol Reviews 2007, 1, 1–14. [Google Scholar]
- Dimberg, A. Chemokines in angiogenesis. Curr. Topics Microbiol. Immunol. 2010, in press. [Google Scholar]
- Bruserud, Ø. The chemokine system in experimental and clinical hematology. Curr. Topics Microbiol. Immunol. 2010, in press. [Google Scholar]
- Bonecchi, R.; Savino, B.; Borroni, E.M.; Mantovani, A.; Locati, M. Chemokine decoy receptors: Structure-function and biological properties. Curr. Topics Microbiol. Immunol. 2010, in press. [Google Scholar]
- Løffler, J.; Mezger, M.; Ok, M.; Oliver Morton, C.; Einsele, H. Genetic polymorphisms in the cytokine and chemokine system - their possible importance in allogeneic stem cell transplantation. Curr. Topics Microbiol. Immunol. 2010, in press. [Google Scholar]
- Kittan, N.A.; Hildebrandt, G.C. The chemokine system, a possible therapeutic target in acute graft versus host disease. Curr. Topics Microbiol. Immunol. 2010, in press. [Google Scholar]
- Reikvam, H.; Hatfield, K.J.; Øyan, A.; Kalland, K.H.; Kittang, A.O.; Bruserud, Ø. Primary human acute myelogenous leukemia cells release matrix metalloproteases and their inhibitors: Release profile and pharmacological modulation. Eur. J. Haematol. 2010, in press. [Google Scholar]
- Engelhardt, B.G.; Crowe, J.E. Homing in acute graft-versus-host disease: Tissue-specific T regulatory and Th17 cells. Curr. Topics Microbiol. Immunol. 2010, in press. [Google Scholar]
- Sandset, P.M. The immunobiology of heparin-induced thrombocytopenia. Curr. Topics Microbiol. Immunol. 2010, in press. [Google Scholar]
- Calandra, G. CXCR4 in clinical hematology. Curr. Topics Microbiol. 2010, in press. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ersvaer, E.; Kittang, A.O.; Hampson, P.; Sand, K.; Gjertsen, B.T.; Lord, J.M.; Bruserud, Ø. The Protein Kinase C Agonist PEP005 (Ingenol 3-Angelate) in the Treatment of Human Cancer: A Balance between Efficacy and Toxicity. Toxins 2010, 2, 174-194. https://doi.org/10.3390/toxins2010174
Ersvaer E, Kittang AO, Hampson P, Sand K, Gjertsen BT, Lord JM, Bruserud Ø. The Protein Kinase C Agonist PEP005 (Ingenol 3-Angelate) in the Treatment of Human Cancer: A Balance between Efficacy and Toxicity. Toxins. 2010; 2(1):174-194. https://doi.org/10.3390/toxins2010174
Chicago/Turabian StyleErsvaer, Elisabeth, Astrid Olsnes Kittang, Peter Hampson, Kristoffer Sand, Bjørn Tore Gjertsen, Janet M. Lord, and Øystein Bruserud. 2010. "The Protein Kinase C Agonist PEP005 (Ingenol 3-Angelate) in the Treatment of Human Cancer: A Balance between Efficacy and Toxicity" Toxins 2, no. 1: 174-194. https://doi.org/10.3390/toxins2010174
APA StyleErsvaer, E., Kittang, A. O., Hampson, P., Sand, K., Gjertsen, B. T., Lord, J. M., & Bruserud, Ø. (2010). The Protein Kinase C Agonist PEP005 (Ingenol 3-Angelate) in the Treatment of Human Cancer: A Balance between Efficacy and Toxicity. Toxins, 2(1), 174-194. https://doi.org/10.3390/toxins2010174