PP2A Inhibition Assay Using Recombinant Enzyme for Rapid Detection of Okadaic Acid and Its Analogs in Shellfish

Abstract
:1. Introduction
2. Results
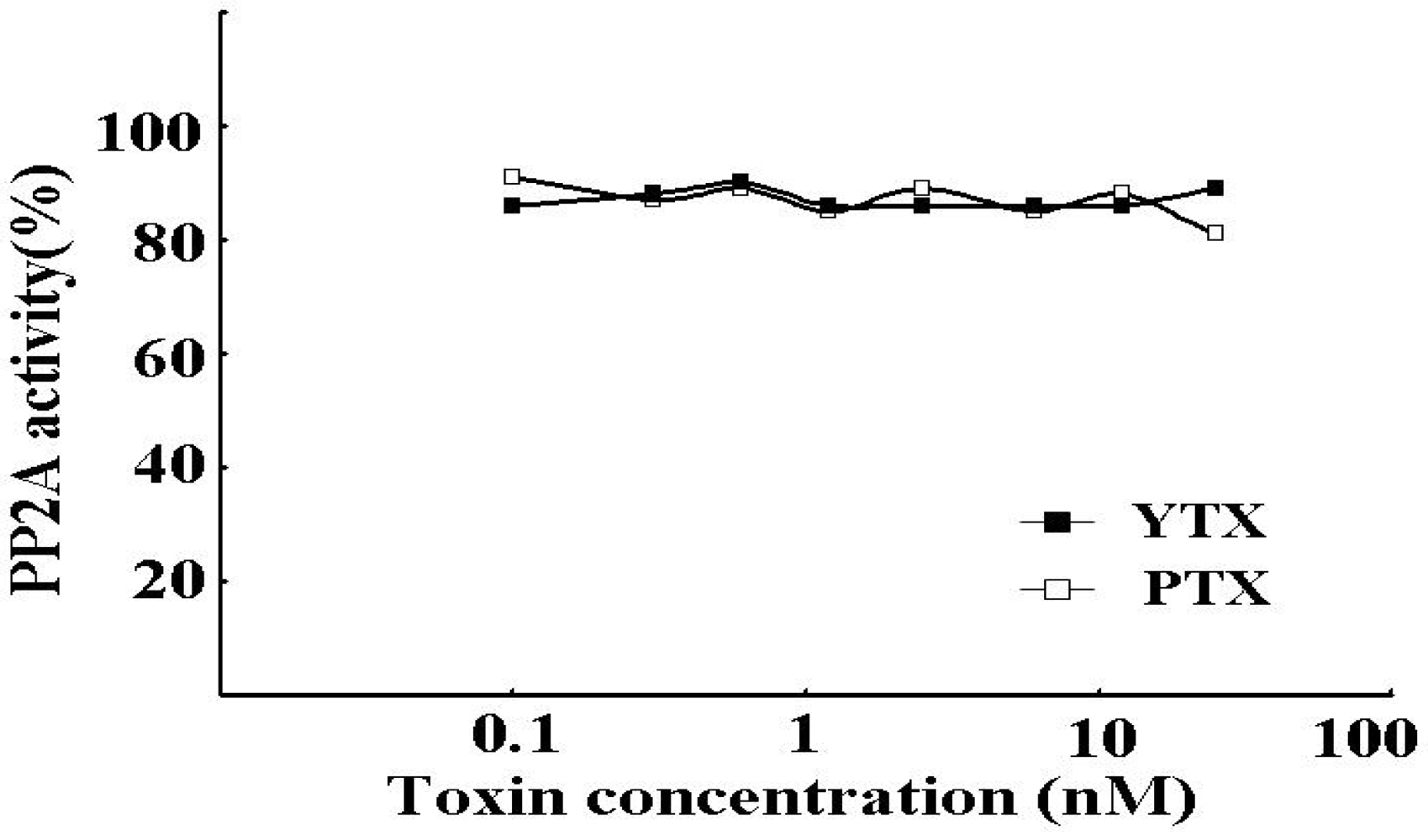
2.1. Comparison of the Inhibitory Effects of Lipophilic Toxins on rhPP2Ac Activity



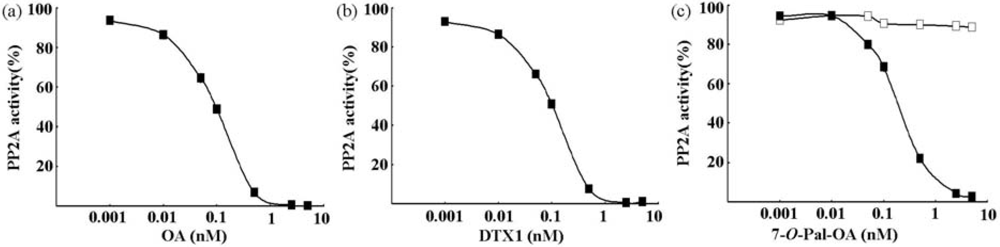{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxins | IC50(nM) |
|---|---|
| All assays were performed in triplicate. | |
| OA | 0.095 ± 0.007 |
| DTX | 0.104 ± 0.006 |
| 7-O-Pal-OA | >10 |
| Hydrolyzate of 7-O-Pal-OA | 0.135 ± 0.009 |
| TYX | > 20 |
| PTX-1 | > 20 |
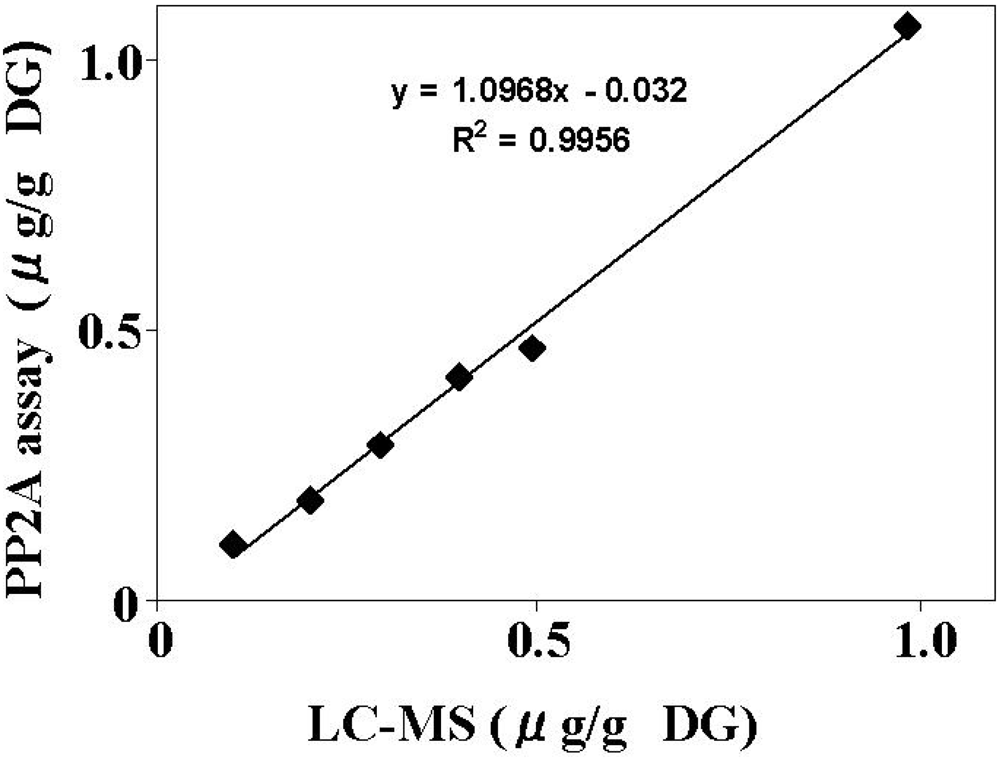
2.2. Method Validation of the PP2A Assay

| Matrix | Mussels | Scallops | Mean LOD (μg/g DG) | Mean LOQ (μg/g DG) | ||
|---|---|---|---|---|---|---|
| Unhydrolyzed | Hydrolyzed | Unhydrolyzed | Hydrolyzed | |||
| a Mean value of 6 Independent blank shellfish samples; b Mean blank value x 3 SD; c Mean blank value x 10 SD. | ||||||
| Meana(μg/g DG) | 0.0295 | 0.0282 | 0.0152 | 0.0215 | ||
| SD | ±0.0043 | ±0.0065 | ±0.0022 | ±0.002 | ||
| LODb(μg/g DG) | 0.0424 | 0.0476 | 0.0217 | 0.0274 | 0.0348 | |
| LOQc(μg/g DG) | 0.0725 | 0.0932 | 0.0372 | 0.0415 | 0.0611 | |
| Sample | OA concentration (μg/g DG) | SD | RSD | Recovery | |
|---|---|---|---|---|---|
| Spiked level | Mean | ||||
| All assays were performed in triplicate. | |||||
| (Detected by PP2A assay) | (%) | (%) | |||
| Mussels | 0.1 | 0.0996 | ±0.0045 | 4.5 | 99.6 |
| 0.2 | 0.1848 | ±0.0044 | 2.4 | 92.4 | |
| 0.3 | 0.2844 | ±0.0090 | 3.2 | 94.8 | |
| 0.4 | 0.4129 | ±0.0103 | 2.5 | 103.2 | |
| 0.5 | 0.4673 | ±0.0082 | 1.7 | 93.5 | |
| 1.0 | 1.0636 | ±0.0183 | 1.7 | 106.4 | |
| Scallops | 0.1 | 0.0708 | ±0.0050 | 7.1 | 70.8 |
| 0.2 | 0.1735 | ±0.0030 | 1.7 | 86.8 | |
| 0.3 | 0.2890 | ±0.0032 | 1.1 | 96.3 | |
| 0.4 | 0.3724 | ±0.0020 | 0.5 | 93.1 | |
| 0.5 | 0.4735 | ±0.0021 | 0.4 | 94.7 | |
| 1.0 | 1.0108 | ±0.0108 | 1.1 | 101.1 | |
| Sample | OA concentration (μg/g DG) | SD | RSD (%) | Replicates | |
|---|---|---|---|---|---|
| Spiked level | Mean (Detected by PP2A assay) | ||||
| All assays were performed in triplicate. | |||||
| Mussels | 0.1 | 0.1019 | ±0.0066 | 6.5 | 6 |
| 0.2 | 0.1898 | ±0.0121 | 6.4 | 6 | |
| 0.4 | 0.4079 | ±0.0194 | 4.8 | 6 | |
| Scallops | 0.1 | 0.0711 | ±0.0041 | 5.8 | 6 |
| 0.2 | 0.1713 | ±0.0122 | 7.1 | 6 | |
| 0.4 | 0.3596 | ±0.0184 | 5.1 | 6 | |

3. Discussion
4. Experimental Section
4.1. Lipophilic Toxins and Substrate Reagents and Solvents
4.2. Purification of rhPP2Ac and Phosphatase Activity Assay
4.3. PP2A Inhibition Assay
4.4. Preparation of Shellfish Samples for the PP2A Assay
4.5. Assay Procedure
4.6. LC-MS Determination
Acknowledgements
References and Notes
- Yasumoto, T.; Oshima, Y.; Yamaguchi, M. Occurrence of a new type of shellfish poisoning in the Tohoku District. Bull. Jpn. Soc. Sci. Fish 1978, 44, 1249–1255. [Google Scholar]
- Hamano, Y.; Kinoshita, Y.; Yasumoto, T. Toxic Dinoflagellates; Elsevier: New York,NY,USA, 1985; pp. 383–388. [Google Scholar]
- Fujiki, H.; Suganuma, M.; Suguri, H.; Yoshizawa, S.; Ojika, M.; Wakamatsu, K.; Yamada, K.; Sugimura, T. Induction of ornithine decarboxylase activity in mouse skin by a possible tumor promoter, okadaic acid. Proc. Jpn. Acad. Ser. B 1987, 63, 51–53. [Google Scholar]
- European Commission, Regulation (EC) No 853/2004. Off. J. Eur. Union L139 2004, 47, 55–105.
- Lee, J.S.; Igarashi, T.; Fraga, S.; Dahl, E.; Hovgaard, P.; Yasumoto, T. Determination of diarrhetic shellfish toxins in various dinoflagellates species. J. Appl. Phyco1. 1989, 1, 147–152. [Google Scholar] [CrossRef]
- Hu, T.; Doylc, J.; Jackson, D.; Marr, J.; Nixon, E.; Pleasance, S.; Quilliam, M.A.; Walter, J.A.; Wright, J.L.C. solation of a new diarrhetic shellfish poison from Irish mussels. J. Chem. Soc. Chem. Commun. 1992, 1, 39–41. [Google Scholar]
- Miles, C.O.; Wilkins, A.L.; Munday, R.; Dines, M.H.; Hawkes, A.D.; Briggs, L.R.; Sandvik, M.; Jensen, D.J.; Cooney, J.M.; Holland, P.T.; Quilliam, M.A.; Mackenzie, A.L.; Beuzenberg, V.; Towers, N.R. solation of pectenotoxin-2 from Dinophysis acuta and its conversion to pectenotoxins-2 seco acid, and preliminary assement of their acute toxicities. Toxicon 2004, 43, 1–9. [Google Scholar] [PubMed]
- Murakami, Y.; Oshima, Y.; Yasumoto, T. Identification of okadaic acid as a toxic component of a marine dinoflagellate Prorocentrum lima. Bull. Japan. Soc. Sci. Fish 1982, 48, 69–72. [Google Scholar]
- Bialojan, C.; Takai, A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatas. Biochem. J. 1988, 256, 283–290. [Google Scholar] [PubMed]
- Yasumoto, T.; Murata, M. Marine toxins. Chem. Rev. 1993, 93, 1897–1909. [Google Scholar]
- Cohen, P. The structure and regulation of protein phosphatases. Annu. Rev. Biochem. 1989, 58, 453–508. [Google Scholar] [PubMed]
- Cohen, P.; Holmes, C.F.B.; Tsukitani, Y. Okadaic acid: A new probe for the study of cellular regulation. Trends biochem. Sci. 1990, 15, 98–102. [Google Scholar]
- Takai, A.; Mieskes, G. Inhibitory effect of okadaic acid on the p-nitrophenyl phosphate phosphatase activity of protein phosphatases. Biochem. J. 1991, 275, 233–239. [Google Scholar] [PubMed]
- Simon, J.F.; Vernoux, J.P. Highly sensitive assay of okadaic acid using protein phosphatase and paranitrophenyl phosphate. Nat. Toxins 1994, 2, 293–301. [Google Scholar] [PubMed]
- Tubaro, A.; Florio, C.; Luxich, E.; Sosa, S.; Loggia, R.D.; Yasumoto, T. A protein phosphatase 2A inhibition assay for a fast and sensitive assessment of okadaic acid contamination in mussels. Toxicon 1996, 34, 743–752. [Google Scholar] [PubMed]
- Ikehara, T.; Shinjo, F.; Ikehara, S.; Imamura, S.; Yasumoto, T. Baculovirus expression, purification, and characterization of human protein phosphatase 2A catalytic subunits α and β . Protein Expr. Purif. 2006, 45, 150–156. [Google Scholar] [PubMed]
- Ikehara, T.; Imamura, S.; Oshiro, N.; Ikehara, S.; Shinjo, F.; Yasumoto, T. A protein phosphatase 2A (PP2A) inhibition assay using a recombinant enzyme for rapid detection of microcystins. Toxicon 2008, 51, 1368–1373. [Google Scholar] [PubMed]
- Wera, S.; Hemmings, B.A. Serine/threonine protein phosphatases. Biochem. J. 1995, 311, 17–29. [Google Scholar] [PubMed]
- Honkanen, R.E.; Zwiller, J.; Moore, R.E.; Daily, S.L.; Khatra, B.S.; Dukelow, M.; Boynton, A.L. Characterization of microcystin-LR, a potent inhibitor of type 1 and type 2A protein phosphatase. J. Biol. Chem. 1990, 265, 19401–19404. [Google Scholar] [PubMed]
- Albano, C.; Ronzitti, G.; Rossini, A.M.; Callegari, F.; Rossini, G.P. The total activity of a mixture of okadaic acid-group compounds can be calculated by those of individual analogues in a phosphoprotein phosphatase 2A assay. Toxicon 2009, 53, 631–637. [Google Scholar] [PubMed]
- Goto, H.; Igarashi, T.; Yamamoto, M.; Sekiguchi, R.; Watai, M.; Tanno, K.; Yasumoto, T. Quantitative determination of marine toxins associated with diarrhetic shellfish poisoning by liquid chromatography coupled with mass spectrometry. J. Chromatogr. A. 2001, 907, 181–189. [Google Scholar] [PubMed]
- Yanagi, T.; Murata, M.; Torigoe, K.; Yasumoto, T. Biological activities of semisynthetic analogs of Dinophysistoxin-3, the major diarrhetic shellfish toxin. Agric. Biol. Chem. 1989, 53, 525–529. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ikehara, T.; Imamura, S.; Yoshino, A.; Yasumoto, T. PP2A Inhibition Assay Using Recombinant Enzyme for Rapid Detection of Okadaic Acid and Its Analogs in Shellfish. Toxins 2010, 2, 195-204. https://doi.org/10.3390/toxins2010195
Ikehara T, Imamura S, Yoshino A, Yasumoto T. PP2A Inhibition Assay Using Recombinant Enzyme for Rapid Detection of Okadaic Acid and Its Analogs in Shellfish. Toxins. 2010; 2(1):195-204. https://doi.org/10.3390/toxins2010195
Chicago/Turabian StyleIkehara, Tsuyoshi, Shihoko Imamura, Atsushi Yoshino, and Takeshi Yasumoto. 2010. "PP2A Inhibition Assay Using Recombinant Enzyme for Rapid Detection of Okadaic Acid and Its Analogs in Shellfish" Toxins 2, no. 1: 195-204. https://doi.org/10.3390/toxins2010195
APA StyleIkehara, T., Imamura, S., Yoshino, A., & Yasumoto, T. (2010). PP2A Inhibition Assay Using Recombinant Enzyme for Rapid Detection of Okadaic Acid and Its Analogs in Shellfish. Toxins, 2(1), 195-204. https://doi.org/10.3390/toxins2010195