Molecular Mechanism of Ochratoxin A Transport in the Kidney

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. OTA Transport Systems in Classical Studies
2.1. Renal Slice Study
2.2. Membrane Vesicle Study
2.3. Micropuncture Study
2.4. Tubular Suspension Study
2.5. Isolated Tubular Study
2.6. Cell Monolayer Study

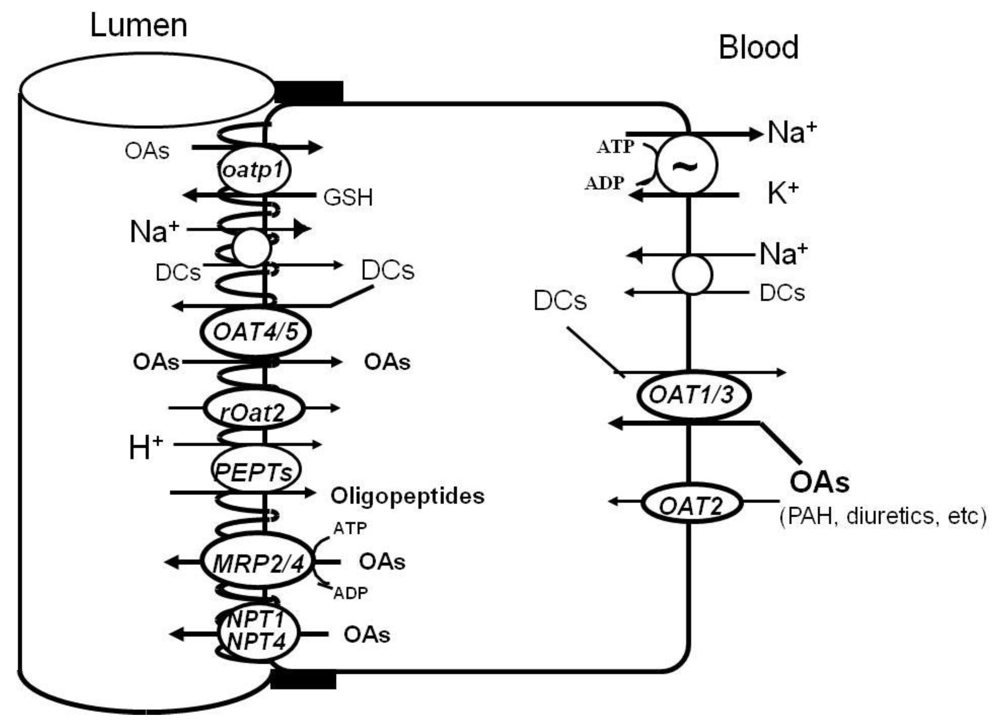
3. Molecular Identities of Renal Organic Anion Transporters

3.1. Organic Anion Transporter (OAT) Family SLC22
3.2. Organic Anion Transporting Polypeptide (OATP) Family SLC21/SLCO
3.3. Type I Sodium/Phosphate Transporter (NPT) Family SLC17
3.4. Oligopeptide Transporter (PEPT) Family SLC15
3.5. Multidrug Resistance-Associated Protein (MRP) Family ABCC
3.6. Breast Caner Resistance-Associated Protein (BCRP) ABCG2
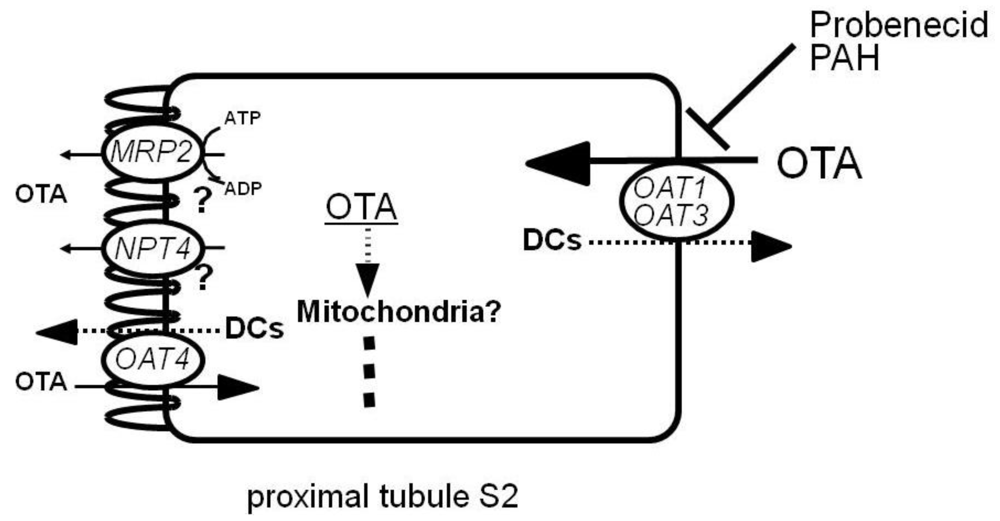
4. OTA Transport Properties in Cloned Organic Anion Transporters
4.1. Basolateral OATs
4.2. Apical OATs

4.3. OATPs
4.4. MRP2
5. Transport of OTA in Extrarenal Tissues
5.1. Intestine
5.2. Liver
5.3. Blood-brain Barrier
6. OTA as a Regulator of Transporter Expression
7. Conclusion
Acknowledgements
References
- van der Merwe, K.J.; Steyn, P.S.; Fourie, L.; Scott, D.B.; Theron, J.J. Ochratoxin A, a toxic metabolite produced by Aspergillus ochraceus Wilh. Nature 1965, 205, 1112–1113. [Google Scholar]
- Harwig, J.; Kuiper-Goodmann, T.; Scott, P.M.; Rechcigl, M. Handbook of Foodborne Diseases of Biological Origin; CRC Press: Boca Raton, FL, USA, 1983; pp. 193–238. [Google Scholar]
- Krogh, P.; Hald, B.; Pedersen, E.J. Occurrence of ochratoxin A and citrinin in cereals associated with mycotoxic porcine nephropathy. Acta Pathol. Microbio. Scand. Sect. B 1973, 81, 689–695. [Google Scholar]
- WHO, Selected mycotoxins: Ochratoxins, trichothecenes, and ergot. In Environmental Health Criteria; World Health Organization: Geneva, Switzerland, 1990; p. 105.
- Dietrich, D.R.; Heussner, A.H.; O'Brien, E. Ochratoxin A: Comparative pharmacokinetics and toxicological implications (experimental and domestic animals and humans). Food Addit. Contam. 2005, 22 (Suppl. 1), 45–52. [Google Scholar] [CrossRef] [PubMed]
- Clark, H.A.; Snedeker, S.M. Ochratoxin A: Its cancer risk and potential for exposure. J. Toxicol. Environ. Health B. Crit. Rev. 2006, 9, 265–296. [Google Scholar]
- Manderville, R.A.; Pfohl-Leszkowicz, A. Bioactivation and DNA adduction as a rationale for ochratoxin A carcinogenesis. World Mycotoxin J. 2008, 1, 357–367. [Google Scholar]
- Kuiper-Goodman, T.; Scott, P.M. Risk assessment of the mycotoxin ochratoxin A. Biomed. Environ. Sci. 1989, 2, 179–248. [Google Scholar]
- Pfohl-Leszkowicz, A.; Manderville, R.A. Ochratoxin A: An overview on toxicity and carcinogenicity in animals and humans. Mol. Nutr. Food Res. 2007, 51, 61–99. [Google Scholar]
- Simon, P. Ochratoxin and kidney disease in the human. J. Toxicol. 1996, 15, 239–249. [Google Scholar]
- Gekle, M.; Silbernagl, S. Mechanism of ochratoxin A-induced reduction of glomerular filtration rate in rats. J. Pharmacol. Exp. Ther. 1993, 267, 316–321. [Google Scholar]
- Gekle, M.; Silbernagl, S. The role of the proximal tubule in ochratoxin A nephrotoxicity in vivo: Toxicodynamic and toxokinetic aspects. Renal Physiol. Biochem. 1994, 17, 40–49. [Google Scholar]
- Dahlmann, A.; Dantzler, W.H.; Silbernagl, S.; Gekle, M. Detailed mapping of ochratoxin A reabsorption along the rat nephron in vivo: The nephrotoxin can be reabsorbed in all nephron segments by different mechanisms. J. Pharmacol. Exp. Ther. 1998, 286, 157–162. [Google Scholar]
- Stein, A.F.; Phillips, T.D.; Kubena, L.F.; Harvey, R.B. Renal tubular secretion and reabsorption as factors in ochratoxicosis: Effects of probenecid on nephrotoxicity. J. Toxicol. Environ. Health 1985, 16, 593–605. [Google Scholar]
- Stojković, R.; Hult, K.; Gamulin, S.; Plestina, R. High affinity binding of ochratoxin A to plasma constituents. Biochem. Int. 1984, 9, 33–38. [Google Scholar] [PubMed]
- Hagelberg, S.; Hult, K.; Fuchs, R. Toxicokinetics of ochratoxin A in several species and its plasma-binding properties. J. Appl. Toxicol. 1989, 9, 91–96. [Google Scholar]
- Gekle, M.; Sauvant, C.; Schwerdt, G. Ochratoxin A at nanomolar concentrations: A signal modulator in renal cells. Mol. Nutr. Food Res. 2005, 49, 118–130. [Google Scholar]
- Berndt, W.O.; Hayes, A.W. In vivo and in vitro changes in renal function caused by ochratoxin A in the rat. Toxicology 1979, 12, 5–17. [Google Scholar]
- Friis, C.; Brinn, R.; Hald, B. Uptake of ochratoxin A by slices of pig kidney cortex. Toxicology 1988, 52, 209–217. [Google Scholar]
- Sokol, P.P.; Ripich, G.; Holohan, P.D.; Ross, C.R. Mechanism of ochratoxin A transport in kidney. J. Pharmacol. Exp. Ther. 1988, 246, 460–465. [Google Scholar]
- Zingerle, M.; Silbernagl, S.; Gekle, M. Reabsorption of the nephrotoxin ochratoxin A along the rat nephron in vivo. J. Pharmacol. Exp. Ther. 1997, 280, 220–224. [Google Scholar] [PubMed]
- Groves, C.E.; Morales, M.; Wright, S.H. Peritubular transport of ochratoxin A in rabbit renal proximal tubules. J. Pharmacol. Exp. Ther. 1998, 284, 943–948. [Google Scholar]
- Groves, C.E.; Nowak, G.; Morales, M. Ochratoxin A secretion in primary cultures of rabbit renal proximal tubule cells. J. Am. Soc. Nephrol. 1999, 10, 13–20. [Google Scholar]
- Schwerdt, G.; Gekle, M.; Freudinger, R.; Mildenberger, S.; Silbernagl, S. Apical-to-basolateral transepithelial transport of Ochratoxin A by two subtypes of Madin-Darby canine kidney cells. Biochim. Biophys. Acta 1997, 1324, 191–199. [Google Scholar]
- Schwerdt, G.; Freudinger, R.; Silbernagl, S.; Gekle, M. Apical uptake of radiolabelled ochratoxin A into Madin-Darby canine kidney cells. Toxicology 1998, 131, 193–202. [Google Scholar]
- Bahnemann, E.; Kerling, H.P.; Ensminger, S.; Schwerdt, G.; Silbernagl, S.; Gekle, M. Renal transepithelial secretion of ochratoxin A in the non-filtering toad kidney. Toxicology 1997, 120, 11–17. [Google Scholar]
- Sauvant, C.; Silbernagl, S.; Gekle, M. Exposure to ochratoxin A impairs organic anion transport in proximal-tubule-derived opossum kidney cells. J. Pharmacol. Exp. Ther. 1998, 287, 13–20. [Google Scholar]
- Welborn, J.R.; Groves, C.E.; Wright, S.H. Peritubular transport of ochratoxin A by single rabbit renal proximal tubules. J. Am. Soc. Nephrol. 1998, 9, 1973–1982. [Google Scholar]
- Burckhardt, B.C.; Burckhardt, G. Transport of organic anions across the basolateral membrane of proximal tubule cells. Rev. Physiol. Biochem. Pharmacol. 2003, 146, 95–158. [Google Scholar]
- Wright, S.H.; Dantzler, W.H. Molecular and cellular physiology of renal organic cation and anion transport. Physiol. Rev. 2004, 84, 987–1049. [Google Scholar]
- Anzai, N.; Kanai, Y.; Endou, H. Organic anion transporter family: Current knowledge. J. Pharmacol. Sci. 2006, 100, 411–426. [Google Scholar]
- Anzai, N.; Endou, H. Drug Transport in the Kidney. In Drug Transporters Molecular Characterization and Role in Drug Disposition; You, G., Morris, M.E., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; pp. 463–493. [Google Scholar]
- Srimaroeng, C.; Perry, J.L.; Pritchard, J.B. Physiology, structure, and regulation of the cloned organic anion transporters. Xenobiotica 2008, 38, 889–935. [Google Scholar]
- Ahn, S.Y.; Bhatnagar, V. Update on the molecular physiology of organic anion transporters. Curr. Opin. Nephrol. Hypertens. 2008, 17, 499–505. [Google Scholar]
- Sekine, T.; Watanabe, N.; Hosoyamada, M.; Kanai, Y.; Endou, H. Expression cloning and characterization of a novel multispecific organic anion transporter. J. Biol. Chem. 1997, 272, 18526–18529. [Google Scholar]
- Sekine, T.; Cha, S.H.; Endou, H. The multispecific organic anion transporter (OAT) family. Pflugers Arch. 2000, 440, 337–350. [Google Scholar]
- Sekine, T.; Cha, S.H.; Tsuda, M.; Apiwattanakul, N.; Nakajima, N.; Kanai, Y.; Endou, H. Identification of multispecific organic anion transporter 2 expressed predominantly in the liver. FEBS Lett. 1998, 429, 179–182. [Google Scholar]
- Kusuhara, H.; Sekine, T.; Utsunomiya-Tate, N.; Tsuda, M.; Kojima, R.; Cha, S.H.; Sugiyama, Y.; Kanai, Y.; Endou, H. Molecular cloning and characterization of a new multispecific organic anion transporter from rat brain. J. Biol. Chem. 1999, 274, 13675–13680. [Google Scholar]
- Cha, S.H.; Sekine, T.; Kusuhara, H.; Yu, E.; Kim, J.Y.; Kim, D.K.; Sugiyama, Y.; Kanai, Y.; Endou, H. Molecular cloning and characterization of multispecific organic anion transporter 4 expressed in the placenta. J. Biol. Chem. 2000, 275, 4507–4512. [Google Scholar]
- Anzai, N.; Jutabha, P.; Enomoto, A.; Yokoyama, H.; Nonoguchi, H.; Hirata, T.; Shiraya, K.; He, X.; Cha, S.H.; Takeda, M.; Miyazaki, H.; Sakata, T.; Tomita, K.; Igarashi, T.; Kanai, Y.; Endou, H. Functional characterization of rat organic anion transporter 5(Slc22a19) at the apical membrane of renal proximal tubules. J. Pharmacol. Exp. Ther. 2005, 315, 534–544. [Google Scholar]
- Yokoyama, H.; Anzai, N.; Ljubojevic, M.; Ohtsu, N.; Sakata, T.; Miyazaki, H.; Nonoguchi, H.; Islam, R.; Onozato, M.; Tojo, A.; Tomita, K.; Kanai, Y.; Igarashi, T.; Sabolic, I.; Endou, H. Functional and immunochemical characterization of a novel organic anion transporter Oat8 (Slc22a9) in rat renal collecting duct. Cell. Physiol. Biochem. 2008, 21, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, H.; Anzai, N.; Shin, H.J.; Wempe, M.F.; Jutabha, P.; Enomoto, A.; Cha, S.H.; Satoh, T.; Ishida, M.; Sakurai, H.; Endou, H. Identification of a novel organic anion transporter mediating carnitine transport in mouse liver and kidney. Cell. Physiol. Biochem. 2010, 25, 511–522. [Google Scholar]
- Bahn, A.; Hagos, Y.; Reuter, S.; Balen, D.; Brzica, H.; Krick, W.; Burckhardt, B.C.; Sabolic, I.; Burckhardt, G. Identification of a new urate and high affinity nicotinate transporter, hOAT10 (SLC22A13). J. Biol. Chem. 2008, 283, 16332–16241. [Google Scholar]
- Jacquemin, E.; Hagenbuch, B.; Stieger, B.; Wolkoff, A.W.; Meier, P.J. Expression cloning of a rat liver Na+-independent organic anion transporter. Proc. Natl. Acad. Sci. USA 1994, 91, 133–137. [Google Scholar]
- Hagenbuch, B.; Meier, P.J. Organic anion transporting polypeptides of the OATP/ SLC21 family: Phylogenetic classification as OATP/ SLCO superfamily, new nomenclature and molecular/functional properties. Pfluegers Arch. 2004, 447, 653–665. [Google Scholar]
- Mikkaichi, T.; Suzuki, T.; Onogawa, T.; Tanemoto, M.; Mizutamari, H.; Okada, M.; Chaki, T.; Masuda, S.; Tokui, T.; Eto, N.; Abe, M.; Satoh, F.; Unno, M.; Hishinuma, T.; Inui, K.; Ito, S.; Goto, J.; Abe, T. Isolation and characterization of a digoxin transporter and its rat homologue expressed in the kidney. Proc. Natl. Acad. Sci. USA 2004, 101, 3569–3574. [Google Scholar]
- Reimer, R.J.; Edwards, R.H. Organic anion transport is the primary function of the SLC17/type I phosphate transporter family. Pfluegers Arch. 2004, 447, 629–635. [Google Scholar]
- Uchino, H.; Tamai, I.; Yamashita, K.; Minemoto, Y.; Sai, Y.; Yabuuchi, H.; Miyamoto, K.; Takeda, E.; Tsuji, A. p-aminohippuric acid transport at renal apical membrane mediated by human inorganic phosphate transporter NPT1. Biochem. Biophys. Res. Commun. 2000, 270, 254–259. [Google Scholar]
- Jutabha, P.; Anzai, N.; Kitamura, K.; Taniguchi, A.; Kaneko, S.; Yamada, H.; Shimada, H.; Uchimura, K.; Hayata, M.; Morinaga, J.; Urano, W.; Seki, G.; Uchida, S.; Endou, H.; Sakurai, H. Mutations in renal drug efflux transporter hOATv1 cause hyperuricemia. Endocrine J. 2010, 57 (Suppl. 2), S563. [Google Scholar]
- Terada, T.; Inui, K. Peptide transporters: structure, function, regulation and application for drug delivery. Curr. Drug Metab. 2004, 5, 85–94. [Google Scholar]
- Ford, J.M.; Hait, W.N. Pharmacology of drugs that alter multidrug resistance in cancer. Pharmacol. Rev. 1990, 42, 155–199. [Google Scholar]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer. 2002, 2, 48–58. [Google Scholar]
- Russel, F.G.; Masereeuw, R.; van Aubel, R.A. Molecular aspects of renal anionic drug transport. Annu. Rev. Physiol. 2002, 64, 563–594. [Google Scholar]
- Smeets, P.H.; van Aubel, R.A.; Wouterse, A.C.; van den Heuvel, J.J.; Russel, F.G. Contribution of multidrug resistance protein 2 (MRP2/ABCC2) to the renal excretion of p-aminohippurate (PAH) and identification of MRP4 (ABCC4) as a novel PAH transporter. J. Am. Soc. Nephrol. 2004, 15, 2828–2835. [Google Scholar]
- van Aubel, R.A.; Smeets, P.H.; van den Heuvel, J.J.; Russel, F.G. Human organic anion transporter MRP4 (ABCC4) is an efflux pump for the purine end metabolite urate with multiple allosteric substrate binding sites. Am. J. Physiol. Renal Physiol. 2005, 288, F327–F333. [Google Scholar]
- Ishikawa, T.; Nakagawa, H. Human ABC transporter ABCG2 in cancer chemotherapy and pharmacogenomics. J. Exp. Ther. Oncol. 2009, 8, 5–24. [Google Scholar]
- Tsuda, M.; Sekine, T.; Takeda, M.; Cha, S.H.; Kanai, Y.; Kimura, M.; Endou, H. Transport of ochratoxin A by renal multispecific organic anion transporter 1. J. Pharmacol. Exp. Ther. 1999, 289, 1301–1305. [Google Scholar]
- Jung, K.Y.; Takeda, M.; Kim, D.K.; Tojo, A.; Narikawa, S.; Yoo, B.S.; Hosoyamada, M.; Cha, S.H.; Sekine, T.; Endou, H. Characterization of ochratoxin A transport by human organic anion transporters. Life Sci. 2001, 69, 2123–2135. [Google Scholar]
- Zhang, X.; Groves, C.E.; Bahn, A.; Barendt, W.M.; Prado, M.D.; Rödiger, M.; Chatsudthipong, V.; Burckhardt, G.; Wright, S.H. Relative contribution of OAT and OCT transporters to organic electrolyte transport in rabbit proximal tubule. Am. J. Physiol. Renal Physiol. 2004, 287, F999–F1010. [Google Scholar]
- Babu, E.; Takeda, M.; Narikawa, S.; Kobayashi, Y.; Enomoto, A.; Tojo, A.; Cha, S.H.; Sekine, T.; Sakthisekaran, D.; Endou, H. Role of human organic anion transporter 4 in the transport of ochratoxin A. Biochim. Biophys. Acta 2002, 1590, 64–75. [Google Scholar]
- Ekaratanawong, S.; Anzai, N.; Jutabha, P.; Miyazaki, H.; Noshiro, R.; Takeda, M.; Kanai, Y.; Sophasan, S.; Endou, H. Human organic anion transporter 4 is a renal apical organic anion/dicarboxylate exchanger in the proximal tubules. J. Pharmacol. Sci. 2004, 94, 297–304. [Google Scholar]
- Jutabha, P.; Anzai, N.; Thammapratip, T.; Endou, H.; Sakurai, H. Ochratoxin A transport mediated by a novel human voltage-driven organic anion transporter hOATv1. J. Tox. Sci. 2010, 35 (Suppl. 1), S219. [Google Scholar]
- Takeuchi, A.; Masuda, S.; Saito, H.; Abe, T.; Inui, K. Multispecific substrate recognition of kidney-specific organic anion transporters OAT-K1 and OAT-K2. J. Pharmacol. Exp. Ther. 2001, 299, 261–267. [Google Scholar]
- Leier, I.; Hummel-Eisenbeiss, J.; Cui, Y.; Keppler, D. ATP-dependent para-aminohippurate transport by apical multidrug resistance protein MRP2. Kidney Int. 2000, 57, 1636–1642. [Google Scholar]
- Schrickx, J.; Lektarau, Y.; Fink-Gremmels, J. Ochratoxin A secretion by ATP-dependent membrane transporters in Caco-2 cells. Arch. Toxicol. 2006, 80, 243–249. [Google Scholar]
- Berger, V.; Gabriel, A.F.; Sergent, T.; Trouet, A.; Larondelle, Y.; Schneider, Y.J. Interaction of ochratoxin A with human intestinal Caco-2 cells: Possible implication of a multidrug resistance-associated protein (MRP2). Toxicol. Lett. 2003, 140–141, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Kontaxi, M.; Echkardt, U.; Hagenbuch, B.; Stieger, B.; Meier, P.J.; Petzinger, E. Uptake of the mycotoxin ochratoxin A in liver cells occurs via the cloned organic anion transporting polypeptide. J. Pharmacol. Exp. Ther. 1996, 279, 1507–1513. [Google Scholar]
- Ose, A.; Kusuhara, H.; Endo, C.; Tohyama, K.; Miyajima, M.; Kitamura, S.; Sugiyama, Y. Functional characterization of mouse organic anion transporting peptide 1a4 in the uptake and efflux of drugs across the blood-brain barrier. Drug Metab. Dispos. 2010, 38, 168–176. [Google Scholar]
- Zlender, V.; Breljak, D.; Ljubojević, M.; Flajs, D.; Balen, D.; Brzica, H.; Domijan, A.M.; Peraica, M.; Fuchs, R.; Anzai, N.; Sabolić, I. Low doses of ochratoxin A upregulate the protein expression of organic anion transporters Oat1, Oat2, Oat3 and Oat5 in rat kidney cortex. Toxicol. Appl. Pharmacol. 2009, 239, 284–296. [Google Scholar]
- Cihlar, T.; Lin, D.C.; Pritchard, J.B.; Fuller, M.D.; Mendel, D.B.; Sweet, D.H. The antiviral nucleotide analogs cidofovir and adefovir are novel substrates for human and rat renal organic anion transporter 1. Mol. Pharmacol. 1999, 56, 570–580. [Google Scholar]
- Lacy, S.A.; Hitchcock, M.J.; Lee, W.A.; Tellier, P.; Cundy, K.C. Effect of oral probenecid coadministration on the chronic toxicity and pharmacokinetics of intravenous cidofovir in cynomolgus monkeys. Toxicol. Sci. 1998, 44, 97–106. [Google Scholar]
- Dai, J.; Park, G.; Perry, J.L.; Il'ichev, Y.V.; Bow, D.A.; Pritchard, J.B.; Faucet, V.; Pfohl-Leszkowicz, A.; Manderville, R.A.; Simon, J.D. Molecular aspects of the transport and toxicity of ochratoxin A. Acc. Chem. Res. 2004, 37, 874–881. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Anzai, N.; Jutabha, P.; Endou, H. Molecular Mechanism of Ochratoxin A Transport in the Kidney. Toxins 2010, 2, 1381-1398. https://doi.org/10.3390/toxins2061381
Anzai N, Jutabha P, Endou H. Molecular Mechanism of Ochratoxin A Transport in the Kidney. Toxins. 2010; 2(6):1381-1398. https://doi.org/10.3390/toxins2061381
Chicago/Turabian StyleAnzai, Naohiko, Promsuk Jutabha, and Hitoshi Endou. 2010. "Molecular Mechanism of Ochratoxin A Transport in the Kidney" Toxins 2, no. 6: 1381-1398. https://doi.org/10.3390/toxins2061381
APA StyleAnzai, N., Jutabha, P., & Endou, H. (2010). Molecular Mechanism of Ochratoxin A Transport in the Kidney. Toxins, 2(6), 1381-1398. https://doi.org/10.3390/toxins2061381