Multiple Toxin-Antitoxin Systems in Mycobacterium tuberculosis

Abstract
:1. General Overview of TA Systems in M. tuberculosis


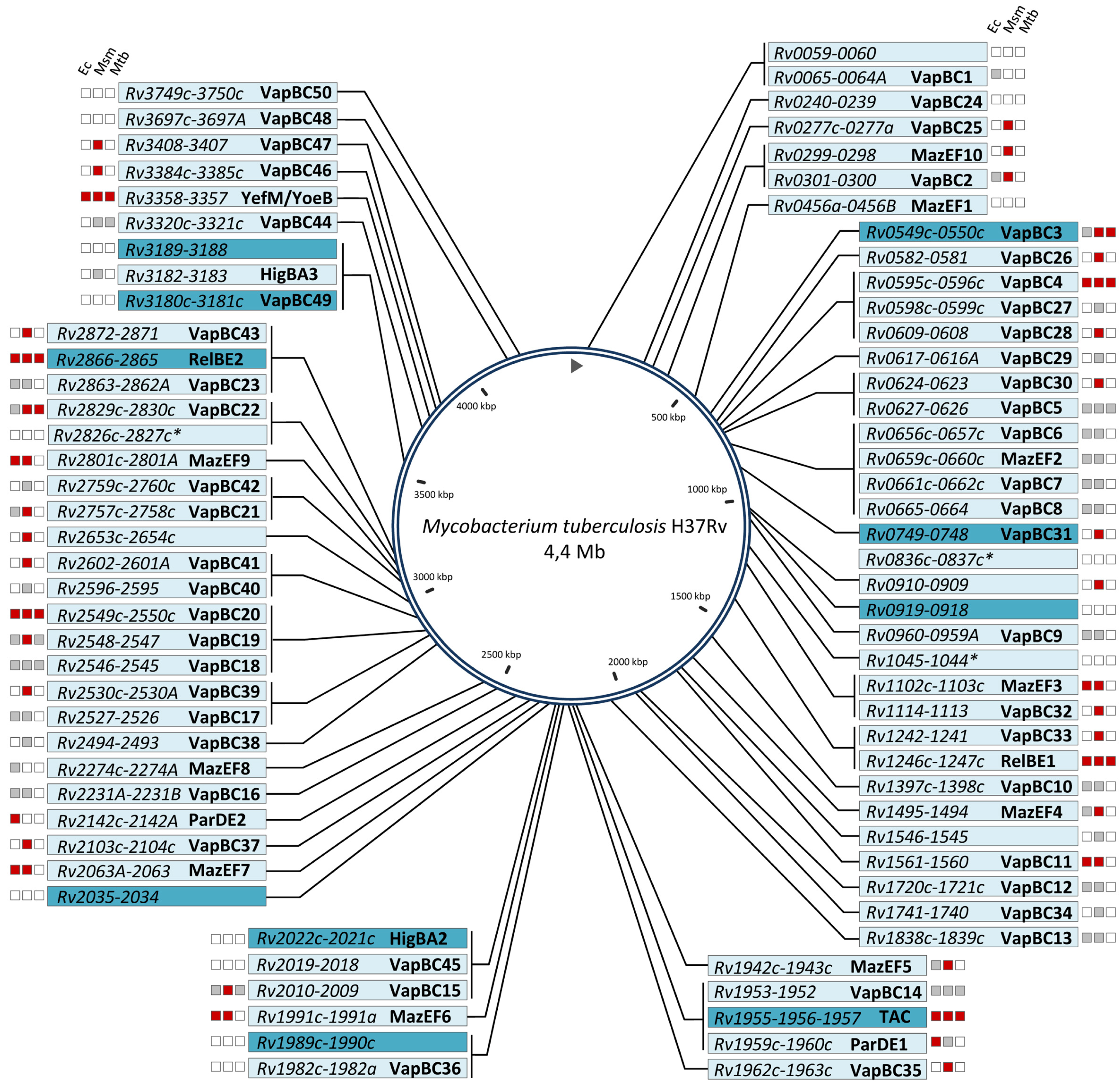{kind=link}
{kind=link}
| Genomes | VapBC | MazEF | RelBE | ParDE | YefM/YoeB | HigBA | TAC | Otherb | Total |
|---|---|---|---|---|---|---|---|---|---|
| M. tuberculosis H37Rv | 50 | 10 | 2 | 2 | 1 | 2 | 1 | 11 | 79 |
| M. smegmatis MC2155 | 1 | 1 | 1c | 2 | 4 | ||||
| M. marinum M | 1 | 1 | |||||||
| M. avium 104 | 1 | 2 | 3 | ||||||
| M. avium paratuberculosis K10 | 1 | 1 | 1 | 3 | |||||
| M. abscessus ATCC 19977 | 1 | 7 | 8 | ||||||
| M. ulcerans Agy99 | 1 | 1 | 2 | ||||||
| M. gilvum PYR-GCK | 3 | 1 | 1 | 9 | 14 |
2. The VapBC Family
3. The MazEF Family
4. The RelBE and YefM/YoeB Families
5. The HigBA Family
6. The Tripartite TAC System

7. The ParDE Family
8. Other TA Modules
9. Role of M. tuberculosis Proteases in TA Activation
10. Concluding Remarks
Acknowledgements
Conflicts of Interest
References
- Yamaguchi, Y.; Park, J.H.; Inouye, M. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 2011, 45, 61–79. [Google Scholar] [CrossRef]
- Pandey, D.P.; Gerdes, K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acid Res. 2005, 33, 966–976. [Google Scholar] [CrossRef]
- Gerdes, K.; Maisonneuve, E. Bacterial persistence and toxin-antitoxin loci. Annu. Rev. Microbiol. 2012, 66, 103–123. [Google Scholar] [CrossRef]
- Masuda, H.; Tan, Q.; Awano, N.; Wu, K.P.; Inouye, M. Yeeu enhances the bundling of cytoskeletal polymers of Mreb and Ftsz, antagonizing the Cbta (Yeev) toxicity in Escherichia coli. Mol. Microbiol. 2012, 84, 979–989. [Google Scholar] [CrossRef]
- Wang, X.; Lord, D.M.; Cheng, H.Y.; Osbourne, D.O.; Hong, S.H.; Sanchez-Torres, V.; Quiroga, C.; Zheng, K.; Herrmann, T.; Peti, W.; et al. A new type V toxin-antitoxin system where mRNA for toxin GhoT is cleaved by antitoxin GhoS. Nat. Chem. Biol. 2012, 8, 855–861. [Google Scholar] [CrossRef]
- Van Melderen, L. Toxin-antitoxin systems: Why so many, what for? Curr. Opin. Microbiol. 2010, 13, 781–785. [Google Scholar] [CrossRef]
- Lewis, K. Persister cells. Ann. Rev. Microbiol. 2010, 64, 357–372. [Google Scholar] [CrossRef]
- Maisonneuve, E.; Castro-Camargo, M.; Gerdes, K. (p)ppgpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 2013, 154, 1140–1150. [Google Scholar] [CrossRef]
- Barry, C.E., 3rd.; Boshoff, H.I.; Dartois, V.; Dick, T.; Ehrt, S.; Flynn, J.; Schnappinger, D.; Wilkinson, R.J.; Young, D. The spectrum of latent tuberculosis: Rethinking the biology and intervention strategies. Nat. Rev. Microbiol. 2009, 7, 845–855. [Google Scholar]
- Georgiades, K.; Raoult, D. Genomes of the most dangerous epidemic bacteria have a virulence repertoire characterized by fewer genes but more toxin-antitoxin modules. PLoS ONE 2011, 6, e17962. [Google Scholar] [CrossRef]
- Sala, A.; Bordes, P.; Fichant, G.; Genevaux, P. Toxin-antitoxin loci in Mycobacterium tuberculosis. In Prokaryotic Toxin-Antitoxin; Gerdes, K., Ed.; Springer: Berlin, Heidelberg, Germany, 2013; pp. 295–314. [Google Scholar]
- Keren, I.; Minami, S.; Rubin, E.; Lewis, K. Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. mBio 2011, 2, e00100–e00111. [Google Scholar]
- Fozo, E.M.; Makarova, K.S.; Shabalina, S.A.; Yutin, N.; Koonin, E.V.; Storz, G. Abundance of type I toxin-antitoxin systems in bacteria: Searches for new candidates and discovery of novel families. Nucleic Acid Res. 2010, 38, 3743–3759. [Google Scholar] [CrossRef]
- Blower, T.R.; Short, F.L.; Rao, F.; Mizuguchi, K.; Pei, X.Y.; Fineran, P.C.; Luisi, B.F.; Salmond, G.P. Identification and classification of bacterial type III toxin-antitoxin systems encoded in chromosomal and plasmid genomes. Nucleic Acids Res. 2012, 40, 6158–6173. [Google Scholar]
- Gupta, A. Killing activity and rescue function of genome-wide toxin-antitoxin loci of Mycobacterium tuberculosis. FEMS Microbiol. Lett. 2009, 290, 45–53. [Google Scholar] [CrossRef]
- Ramage, H.R.; Connolly, L.E.; Cox, J.S. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: Implications for pathogenesis, stress responses, and evolution. PLoS Genet. 2009, 5, e1000767. [Google Scholar] [CrossRef]
- Huang, F.; He, Z.G. Characterization of an interplay between a Mycobacterium tuberculosis MazF homolog, Rv1495 and its sole DNA topoisomerase I. Nucleic Acids Res. 2010, 38, 8219–8230. [Google Scholar] [CrossRef]
- Singh, R.; Barry, C.E., 3rd.; Boshoff, H.I. The three RelE homologs of Mycobacterium tuberculosis have individual, drug-specific effects on bacterial antibiotic tolerance. J. Bacteriol. 2010, 192, 1279–1291. [Google Scholar] [CrossRef]
- Zhu, L.; Sharp, J.D.; Kobayashi, H.; Woychik, N.A.; Inouye, M. Noncognate Mycobacterium tuberculosis toxin-antitoxins can physically and functionally interact. J. Biol. Chem. 2010, 285, 39732–39738. [Google Scholar] [CrossRef]
- Ahidjo, B.A.; Kuhnert, D.; McKenzie, J.L.; Machowski, E.E.; Gordhan, B.G.; Arcus, V.; Abrahams, G.L.; Mizrahi, V. VapC toxins from Mycobacterium tuberculosis are ribonucleases that differentially inhibit growth and are neutralized by cognate VapB antitoxins. PLoS ONE 2011, 6, e21738. [Google Scholar] [CrossRef]
- Pullinger, G.D.; Lax, A.J. A Salmonella dublin virulence plasmid locus that affects bacterial growth under nutrient-limited conditions. Mol. Microbiol. 1992, 6, 1631–1643. [Google Scholar] [CrossRef]
- Arcus, V.L.; McKenzie, J.L.; Robson, J.; Cook, G.M. The PIN-domain ribonucleases and the prokaryotic VapBC toxin-antitoxin array. Protein Eng. Des. Sel. 2011, 24, 33–40. [Google Scholar] [CrossRef]
- Miallau, L.; Faller, M.; Chiang, J.; Arbing, M.; Guo, F.; Cascio, D.; Eisenberg, D. Structure and proposed activity of a member of the VapBC family of toxin-antitoxin systems. VapBC-5 from Mycobacterium tuberculosis. J. Biol. Chem. 2009, 284, 276–283. [Google Scholar] [CrossRef]
- Winther, K.S.; Gerdes, K. Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc. Natl. Acad. Sci. USA 2011, 108, 7403–7407. [Google Scholar] [CrossRef]
- Sharp, J.D.; Cruz, J.W.; Raman, S.; Inouye, M.; Husson, R.N.; Woychik, N.A. Growth and translation inhibition through sequence specific RNA binding by a Mycobacterium tuberculosis VapC toxin. J. Biol. Chem. 2012. [Google Scholar] [CrossRef]
- McKenzie, J.L.; Duyvestyn, J.M.; Smith, T.; Bendak, K.; Mackay, J.; Cursons, R.; Cook, G.M.; Arcus, V.L. Determination of ribonuclease sequence-specificity using pentaprobes and mass spectrometry. RNA 2012, 18, 1267–1278. [Google Scholar] [CrossRef]
- Winther, K.S.; Brodersen, D.E.; Brown, A.K.; Gerdes, K. VapC20 of Mycobacterium tuberculosis cleaves the sarcin-ricin loop of 23S rRNA. Nat. Commun. 2013, 4, 2796. [Google Scholar]
- Min, A.B.; Miallau, L.; Sawaya, M.R.; Habel, J.; Cascio, D.; Eisenberg, D. The crystal structure of the Rv0301-Rv0300 VapBC-3 toxin-antitoxin complex from M. tuberculosis reveals a Mg2+ ion in the active site and a putative RNA-binding site. Protein Sci. 2012, 21, 1754–1767. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Comprehensive comparative-genomic analysis of type-2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct. 2009, 4, 19. [Google Scholar] [CrossRef]
- McKenzie, J.L.; Robson, J.; Berney, M.; Smith, T.C.; Ruthe, A.; Gardner, P.P.; Arcus, V.L.; Cook, G.M. A VapBC toxin-antitoxin module is a post-transcriptional regulator of metabolic flux in Mycobacteria. J. Bacteriol. 2012. [Google Scholar] [CrossRef]
- Albrethsen, J.; Agner, J.; Piersma, S.R.; Hojrup, P.; Pham, T.V.; Weldingh, K.; Jimenez, C.R.; Andersen, P.; Rosenkrands, I. Proteomic profiling of Mycobacterium tuberculosis identifies nutrient-starvation-responsive toxin-antitoxin systems. Mol. Cell. Proteomics 2013, 12, 1180–1191. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Inouye, M. Regulation of growth and death in Escherichia coli by toxin-antitoxin systems. Nat. Rev. Microbiol. 2011, 9, 779–790. [Google Scholar] [CrossRef]
- Kamada, K.; Hanaoka, F.; Burley, S.K. Crystal structure of the MazE/MazF complex: Molecular bases of antidote-toxin recognition. Mol. Cell. 2003, 11, 875–884. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Yamaguchi, Y.; Park, J.H.; Inouye, M.; Patel, D.J. Structural basis of mRNA recognition and cleavage by toxin MazF and its regulation by antitoxin MazE in Bacillus subtilis. Mol. Cell. 2013, 52, 447–458. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, L.; Zhang, J.; Inouye, M. Characterization of ChpbK, an mRNA interferase from Escherichia coli. J. Biol. Chem. 2005, 280, 26080–26088. [Google Scholar] [CrossRef]
- Park, J.H.; Yamaguchi, Y.; Inouye, M. Bacillus subtilis MazF-bs (EndoA) is a UACAU-specific mRNA interferase. FEBS Lett. 2011, 585, 2526–2532. [Google Scholar] [CrossRef]
- Zhu, L.; Zhang, Y.; Teh, J.S.; Zhang, J.; Connell, N.; Rubin, H.; Inouye, M. Characterization of mRNA interferases from Mycobacterium tuberculosis. J. Biol. Chem. 2006, 281, 18638–18643. [Google Scholar]
- Schifano, J.M.; Edifor, R.; Sharp, J.D.; Ouyang, M.; Konkimalla, A.; Husson, R.N.; Woychik, N.A. Mycobacterial toxin MazF-mt6 inhibits translation through cleavage of 23S rRNA at the ribosomal A site. Proc. Natl. Acad. Sci. USA 2013, 110, 8501–8506. [Google Scholar]
- Zhu, L.; Phadtare, S.; Nariya, H.; Ouyang, M.; Husson, R.N.; Inouye, M. The mRNA interferases, MazF-mt3 and MazF-mt7 from Mycobacterium tuberculosis target unique pentad sequences in single-stranded RNA. Mol. Microbiol. 2008, 69, 559–569. [Google Scholar] [CrossRef]
- Vesper, O.; Amitai, S.; Belitsky, M.; Byrgazov, K.; Kaberdina, A.C.; Engelberg-Kulka, H.; Moll, I. Selective translation of leaderless mRNAs by specialized ribosomes generated by MazF in Escherichia coli. Cell 2011, 147, 147–157. [Google Scholar] [CrossRef]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [Google Scholar] [CrossRef]
- Hazan, R.; Sat, B.; Engelberg-Kulka, H. Escherichia coli MazEF-mediated cell death is triggered by various stressful conditions. J. Bacteriol. 2004, 186, 3663–3669. [Google Scholar] [CrossRef]
- Betts, J.C.; Lukey, P.T.; Robb, L.C.; McAdam, R.A.; Duncan, K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 2002, 43, 717–731. [Google Scholar] [CrossRef]
- Dahl, J.L.; Kraus, C.N.; Boshoff, H.I.; Doan, B.; Foley, K.; Avarbock, D.; Kaplan, G.; Mizrahi, V.; Rubin, H.; Barry, C.E., 3rd. The role of RelMtb-mediated adaptation to stationary phase in long-term persistence of Mycobacterium tuberculosis in mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10026–10031. [Google Scholar] [CrossRef]
- Ramirez, M.V.; Dawson, C.C.; Crew, R.; England, K.; Slayden, R.A. MazF6 toxin of Mycobacterium tuberculosis demonstrates antitoxin specificity and is coupled to regulation of cell growth by a Soj-like protein. BMC Microbiol. 2013, 13, 240. [Google Scholar] [CrossRef]
- Diderichsen, B.; Fiil, N.P.; Lavalle, R. Genetics of the RelB locus in Escherichia coli. J. Bacteriol. 1977, 131, 30–33. [Google Scholar]
- Christensen, S.K.; Gerdes, K. Delayed-relaxed response explained by hyperactivation of RelE. Mol. Microbiol. 2004, 53, 587–597. [Google Scholar] [CrossRef]
- Grady, R.; Hayes, F. Axe-txe, a broad-spectrum proteic toxin-antitoxin system specified by a multidrug-resistant, clinical isolate of Enterococcus faecium. Mol. Microbiol. 2003, 47, 1419–1432. [Google Scholar] [CrossRef]
- Christensen, S.K.; Gerdes, K. RelE toxins from bacteria and archaea cleave mRNAs on translating ribosomes, which are rescued by tmRNA. Mol. Microbiol. 2003, 48, 1389–1400. [Google Scholar] [CrossRef]
- Kamada, K.; Hanaoka, F. Conformational change in the catalytic site of the ribonuclease YoeB toxin by YefM antitoxin. Mol. Cell. 2005, 19, 497–509. [Google Scholar] [CrossRef]
- Cherny, I.; Gazit, E. The YefM antitoxin defines a family of natively unfolded proteins: Implications as a novel antibacterial target. J. Biol. Chem. 2004, 279, 8252–8261. [Google Scholar] [CrossRef]
- Kumar, P.; Issac, B.; Dodson, E.J.; Turkenburg, J.P.; Mande, S.C. Crystal structure of Mycobacterium tuberculosis YefM antitoxin reveals that it is not an intrinsically unstructured protein. J. Mol. Biol. 2008, 383, 482–493. [Google Scholar] [CrossRef]
- Miallau, L.; Jain, P.; Arbing, M.A.; Cascio, D.; Phan, T.; Ahn, C.J.; Chan, S.; Chernishof, I.; Maxson, M.; Chiang, J.;et al. Comparative proteomics identifies the cell-associated lethality of M. tuberculosis RelBE-like toxin-antitoxin complexes. Structure 2013, 21, 627–637. [Google Scholar] [CrossRef]
- Blower, T.R.; Salmond, G.P.; Luisi, B.F. Balancing at survival's edge: The structure and adaptive benefits of prokaryotic toxin-antitoxin partners. Curr. Opin. Struct. Biol. 2011, 21, 109–118. [Google Scholar] [CrossRef]
- Korch, S.B.; Contreras, H.; Clark-Curtiss, J.E. Three Mycobacterium tuberculosis Rel toxin-antitoxin modules inhibit mycobacterial growth and are expressed in infected human macrophages. J. Bacteriol. 2009, 191, 1618–1630. [Google Scholar] [CrossRef]
- Yang, M.; Gao, C.; Wang, Y.; Zhang, H.; He, Z.G. Characterization of the interaction and cross-regulation of three Mycobacterium tuberculosis RelBE modules. PLoS ONE 2010, 5, e10672. [Google Scholar] [CrossRef]
- Tian, Q.B.; Ohnishi, M.; Tabuchi, A.; Terawaki, Y. A new plasmid-encoded proteic killer gene system: Cloning, sequencing, and analyzing hig locus of plasmid rts1. Biochem. Biophys. Res. Commun. 1996, 220, 280–284. [Google Scholar] [CrossRef]
- Gerdes, K.; Christensen, S.K.; Lobner-Olesen, A. Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 2005, 3, 371–382. [Google Scholar] [CrossRef]
- Hurley, J.M.; Woychik, N.A. Bacterial toxin HigB associates with ribosomes and mediates translation-dependent mRNA cleavage at A-rich sites. J. Biol. Chem. 2009, 284, 18605–18613. [Google Scholar] [CrossRef]
- Christensen-Dalsgaard, M.; Gerdes, K. Two HigBA loci in the Vibrio cholerae superintegron encode mRNA cleaving enzymes and can stabilize plasmids. Mol. Microbiol. 2006, 62, 397–411. [Google Scholar] [CrossRef]
- Schureck, M.A.; Maehigashi, T.; Miles, S.J.; Marquez, J.; Ei Cho, S.; Erdman, R.; Dunham, C.M. Structure of the P. vulgaris HigB-(HigA)2-HigB toxin-antitoxin complex. J. Biol. Chem. 2013. [Google Scholar] [CrossRef]
- Arbing, M.A.; Handelman, S.K.; Kuzin, A.P.; Verdon, G.; Wang, C.; Su, M.; Rothenbacher, F.P.; Abashidze, M.; Liu, M.; Hurley, J.M.; et al. Crystal structures of Phd-Doc, HigA, and YeeU establish multiple evolutionary links between microbial growth-regulating toxin-antitoxin systems. Structure 2010, 18, 996–1010. [Google Scholar] [CrossRef]
- Stinear, T.P.; Seemann, T.; Harrison, P.F.; Jenkin, G.A.; Davies, J.K.; Johnson, P.D.; Abdellah, Z.; Arrowsmith, C.; Chillingworth, T.; Churcher, C.; et al. Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. 2008, 18, 729–741. [Google Scholar] [CrossRef]
- Bordes, P.; Cirinesi, A.M.; Ummels, R.; Sala, A.; Sakr, S.; Bitter, W.; Genevaux, P. SecB-like chaperone controls a toxin-antitoxin stress-responsive system in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2011, 108, 8438–8443. [Google Scholar] [CrossRef]
- Fivian-Hughes, A.S.; Davis, E.O. Analyzing the regulatory role of the HigA antitoxin within Mycobacterium tuberculosis. J. Bacteriol. 2010, 192, 4348–4356. [Google Scholar] [CrossRef]
- Sala, A.; Calderon, V.; Bordes, P.; Genevaux, P. TAC from Mycobacterium tuberculosis: A paradigm for stress-responsive toxin-antitoxin systems controlled by SecB-like chaperones. Cell Stress Chap. 2013, 18, 129–135. [Google Scholar] [CrossRef]
- Schuessler, D.L.; Cortes, T.; Fivian-Hughes, A.S.; Lougheed, K.E.; Harvey, E.; Buxton, R.S.; Davis, E.O.; Young, D.B. Induced ectopic expression of HigB toxin in Mycobacterium tuberculosis results in growth inhibition, reduced abundance of a subset of mRNAs and cleavage of tmRNA. Mol. Microbiol. 2013, 90, 195–207. [Google Scholar]
- Botella, H.; Peyron, P.; Levillain, F.; Poincloux, R.; Poquet, Y.; Brandli, I.; Wang, C.; Tailleux, L.; Tilleul, S.; Charriere, G.M.; et al. Mycobacterial p(1)-type ATPases mediate resistance to zinc poisoning in human macrophages. Cell. Host Microbe 2011, 10, 248–259. [Google Scholar] [CrossRef]
- Smollett, K.L.; Fivian-Hughes, A.S.; Smith, J.E.; Chang, A.; Rao, T.; Davis, E.O. Experimental determination of translational start sites resolves uncertainties in genomic open reading frame predictions - application to Mycobacterium tuberculosis. Microbiology 2009, 155, 186–197. [Google Scholar] [CrossRef]
- Stewart, G.R.; Wernisch, L.; Stabler, R.; Mangan, J.A.; Hinds, J.; Laing, K.G.; Young, D.B.; Butcher, P.D. Dissection of the heat-shock response in Mycobacterium tuberculosis using mutants and microarrays. Microbiology 2002, 148, 3129–3138. [Google Scholar]
- Tailleux, L.; Waddell, S.J.; Pelizzola, M.; Mortellaro, A.; Withers, M.; Tanne, A.; Castagnoli, P.R.; Gicquel, B.; Stoker, N.G.; Butcher, P.D.; et al. Probing host pathogen cross-talk by transcriptional profiling of both Mycobacterium tuberculosis and infected human dendritic cells and macrophages. PLoS ONE 2008, 3, e1403. [Google Scholar] [CrossRef]
- Guo, M.; Feng, H.; Zhang, J.; Wang, W.; Wang, Y.; Li, Y.; Gao, C.; Chen, H.; Feng, Y.; He, Z.G. Dissecting transcription regulatory pathways through a new bacterial one-hybrid reporter system. Genome Res. 2009, 19, 1301–1308. [Google Scholar] [CrossRef]
- Driessen, A.J.; Nouwen, N. Protein translocation across the bacterial cytoplasmic membrane. Annu. Rev. Biochem. 2008, 77, 643–667. [Google Scholar] [CrossRef]
- Randall, L.L.; Hardy, S.J. SecB, one small chaperone in the complex milieu of the cell. Cell. Mol. Life Sci. 2002, 59, 1617–1623. [Google Scholar] [CrossRef]
- Castanie-Cornet, M.P.; Bruel, N.; Genevaux, P. Chaperone networking facilitates protein targeting to the bacterial cytoplasmic membrane. Biochim. Biophys. Acta 2013. [Google Scholar] [CrossRef]
- Zuber, B.; Chami, M.; Houssin, C.; Dubochet, J.; Griffiths, G.; Daffe, M. Direct visualization of the outer membrane of mycobacteria and corynebacteria in their native state. J. Bacteriol. 2008, 190, 5672–5680. [Google Scholar] [CrossRef]
- Niederweis, M.; Danilchanka, O.; Huff, J.; Hoffmann, C.; Engelhardt, H. Mycobacterial outer membranes: In search of proteins. Trends Microbiol. 2010, 18, 109–116. [Google Scholar] [CrossRef]
- Sani, M.; Houben, E.N.; Geurtsen, J.; Pierson, J.; de Punder, K.; van Zon, M.; Wever, B.; Piersma, S.R.; Jimenez, C.R.; Daffe, M.; et al. Direct visualization by cryo-EM of the mycobacterial capsular layer: A labile structure containing ESX-1-secreted proteins. PLoS Path. 2010, 6, e1000794. [Google Scholar] [CrossRef]
- Calloni, G.; Chen, T.; Schermann, S.M.; Chang, H.C.; Genevaux, P.; Agostini, F.; Tartaglia, G.G.; Hayer-Hartl, M.; Hartl, F.U. DnaK functions as a central hub in the E. coli chaperone network. Cell. Rep. 2012, 1, 251–264. [Google Scholar] [CrossRef]
- Bruel, N.; Castanie-Cornet, M.P.; Cirinesi, A.M.; Koningstein, G.; Georgopoulos, C.; Luirink, J.; Genevaux, P. Hsp33 controls elongation factor-Tu stability and allows Escherichia coli growth in the absence of the major DnaK and trigger factor chaperones. J. Biol Chem 2012, 287, 44435–44446. [Google Scholar] [CrossRef]
- Roberts, R.C.; Helinski, D.R. Definition of a minimal plasmid stabilization system from the broad-host-range plasmid rk2. J. Bacteriol. 1992, 174, 8119–8132. [Google Scholar]
- Jiang, Y.; Pogliano, J.; Helinski, D.R.; Konieczny, I. ParE toxin encoded by the broad-host-range plasmid rk2 is an inhibitor of Escherichia coli gyrase. Mol. Microbiol. 2002, 44, 971–979. [Google Scholar] [CrossRef]
- Sberro, H.; Leavitt, A.; Kiro, R.; Koh, E.; Peleg, Y.; Qimron, U.; Sorek, R. Discovery of functional toxin/antitoxin systems in bacteria by shotgun cloning. Mol. Cell. 2013, 50, 136–148. [Google Scholar] [CrossRef]
- Dy, R.L.; Przybilski, R.; Semeijn, K.; Salmond, G.P.; Fineran, P.C. A widespread bacteriophage abortive infection system functions through a type IV toxin-antitoxin mechanism. Nucleic Acid Res. 2014. [Google Scholar] [CrossRef]
- Wozniak, R.A.; Waldor, M.K. A toxin-antitoxin system promotes the maintenance of an integrative conjugative element. PLoS Genet. 2009, 5, e1000439. [Google Scholar] [CrossRef]
- Christensen, S.K.; Mikkelsen, M.; Pedersen, K.; Gerdes, K. RelE, a global inhibitor of translation, is activated during nutritional stress. Proc. Natl. Acad. Sci. USA 2001, 98, 14328–14333. [Google Scholar] [CrossRef]
- Aizenman, E.; Engelberg-Kulka, H.; Glaser, G. An Escherichia coli chromosomal “addiction module” regulated by guanosine [corrected] 3',5'-bispyrophosphate: A model for programmed bacterial cell death. Proc. Natl. Acad. Sci. USA 1996, 93, 6059–6063. [Google Scholar] [CrossRef]
- Christensen-Dalsgaard, M.; Jorgensen, M.G.; Gerdes, K. Three new RelE-homologous mRNA interferases of Escherichia coli differentially induced by environmental stresses. Mol. Microbiol. 2010, 75, 333–348. [Google Scholar] [CrossRef]
- Ning, D.; Liu, S.; Xu, W.; Zhuang, Q.; Wen, C.; Tang, X. Transcriptional and proteolytic regulation of the toxin-antitoxin locus VapBC10 (ssr2962/slr1767) on the chromosome of Synechocystis sp. Pcc 6803. PLoS ONE 2013, 8, e80716. [Google Scholar]
- Donegan, N.P.; Thompson, E.T.; Fu, Z.; Cheung, A.L. Proteolytic regulation of toxin-antitoxin systems by ClpPC in Staphylococcus aureus. J. Bacteriol. 2010, 192, 1416–1422. [Google Scholar] [CrossRef]
- Van Melderen, L.; Bernard, P.; Couturier, M. Lon-dependent proteolysis of CcdA is the key control for activation of CcdB in plasmid-free segregant bacteria. Mol. Microbiol. 1994, 11, 1151–1157. [Google Scholar] [CrossRef]
- Van Melderen, L.; Thi, M.H.; Lecchi, P.; Gottesman, S.; Couturier, M.; Maurizi, M.R. ATP-dependent degradation of CcdA by Lon protease. Effects of secondary structure and heterologous subunit interactions. J. Biol. Chem. 1996, 271, 27730–27738. [Google Scholar]
- Loris, R.; Marianovsky, I.; Lah, J.; Laeremans, T.; Engelberg-Kulka, H.; Glaser, G.; Muyldermans, S.; Wyns, L. Crystal structure of the intrinsically flexible addiction antidote MazE. J. Biol. Chem. 2003, 278, 28252–28257. [Google Scholar] [CrossRef]
- Hansen, S.; Vulic, M.; Min, J.; Yen, T.J.; Schumacher, M.A.; Brennan, R.G.; Lewis, K. Regulation of the Escherichia coli HipBA toxin-antitoxin system by proteolysis. PLoS ONE 2012, 7, e39185. [Google Scholar]
- Boutte, C.C.; Crosson, S. Bacterial lifestyle shapes stringent response activation. Trends Microbiol. 2013, 21, 174–180. [Google Scholar] [CrossRef]
- Ribeiro-Guimaraes, M.L.; Pessolani, M.C. Comparative genomics of mycobacterial proteases. Microb. Pathogen. 2007, 43, 173–178. [Google Scholar] [CrossRef]
- Pearce, M.J.; Mintseris, J.; Ferreyra, J.; Gygi, S.P.; Darwin, K.H. Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science 2008, 322, 1104–1107. [Google Scholar] [CrossRef]
- Festa, R.A.; McAllister, F.; Pearce, M.J.; Mintseris, J.; Burns, K.E.; Gygi, S.P.; Darwin, K.H. Prokaryotic ubiquitin-like protein (Pup) proteome of Mycobacterium tuberculosis [corrected]. PLoS ONE 2010, 5, e8589. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sala, A.; Bordes, P.; Genevaux, P. Multiple Toxin-Antitoxin Systems in Mycobacterium tuberculosis. Toxins 2014, 6, 1002-1020. https://doi.org/10.3390/toxins6031002
Sala A, Bordes P, Genevaux P. Multiple Toxin-Antitoxin Systems in Mycobacterium tuberculosis. Toxins. 2014; 6(3):1002-1020. https://doi.org/10.3390/toxins6031002
Chicago/Turabian StyleSala, Ambre, Patricia Bordes, and Pierre Genevaux. 2014. "Multiple Toxin-Antitoxin Systems in Mycobacterium tuberculosis" Toxins 6, no. 3: 1002-1020. https://doi.org/10.3390/toxins6031002
APA StyleSala, A., Bordes, P., & Genevaux, P. (2014). Multiple Toxin-Antitoxin Systems in Mycobacterium tuberculosis. Toxins, 6(3), 1002-1020. https://doi.org/10.3390/toxins6031002