
EDIN-B Promotes the Translocation of Staphylococcus aureus to the Bloodstream in the Course of Pneumonia


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
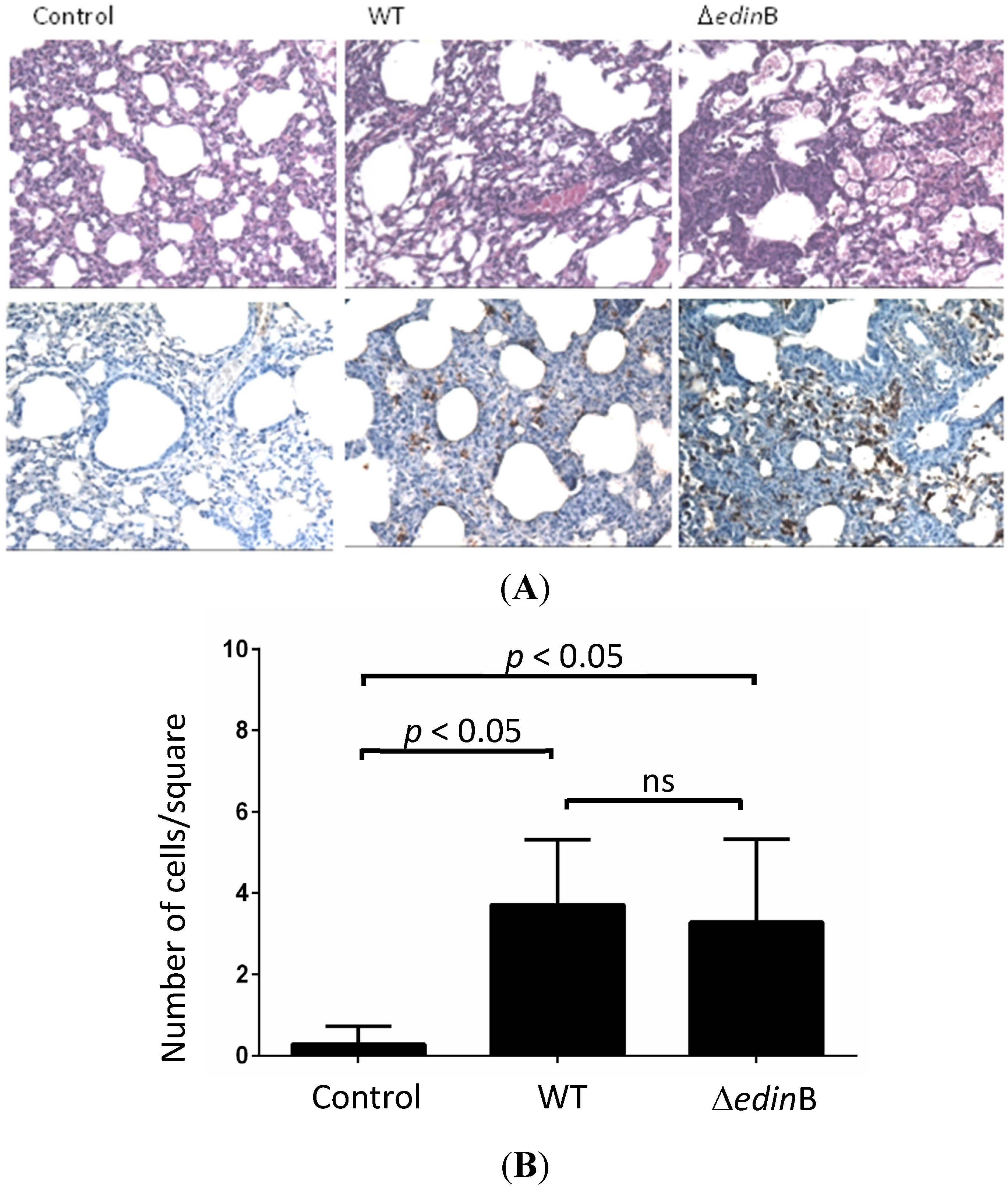
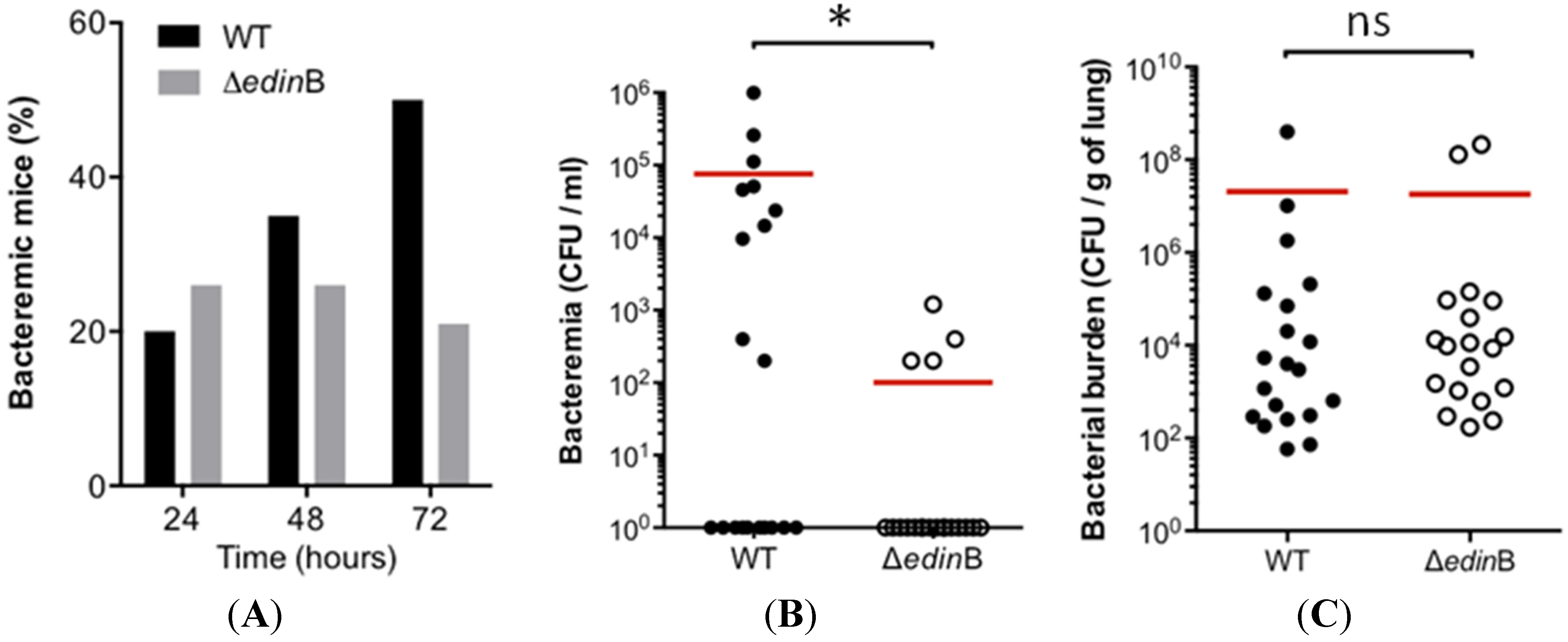
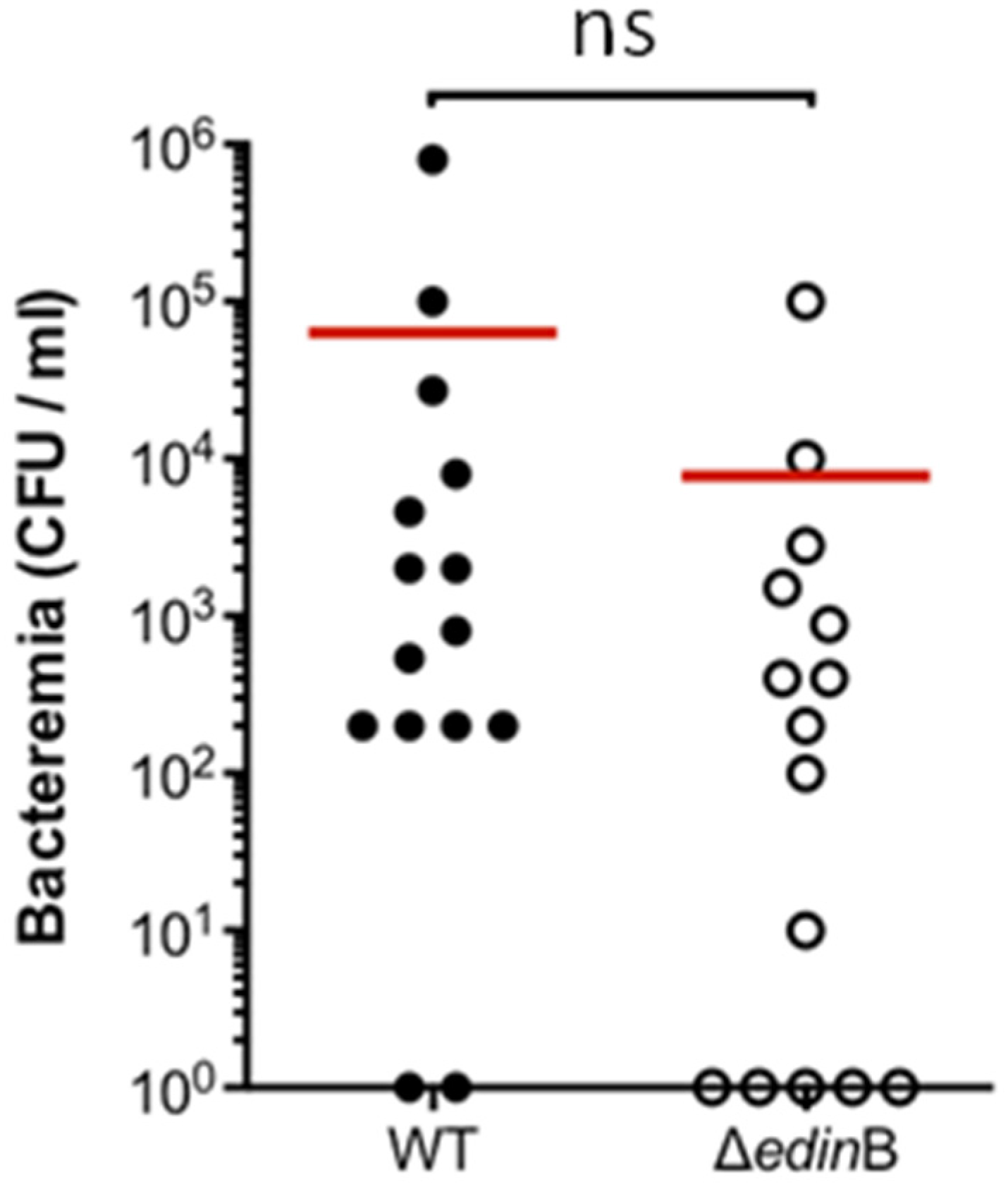
2.1. edinB Promotes Translocation of S. aureus into the Bloodstream During Pneumonia



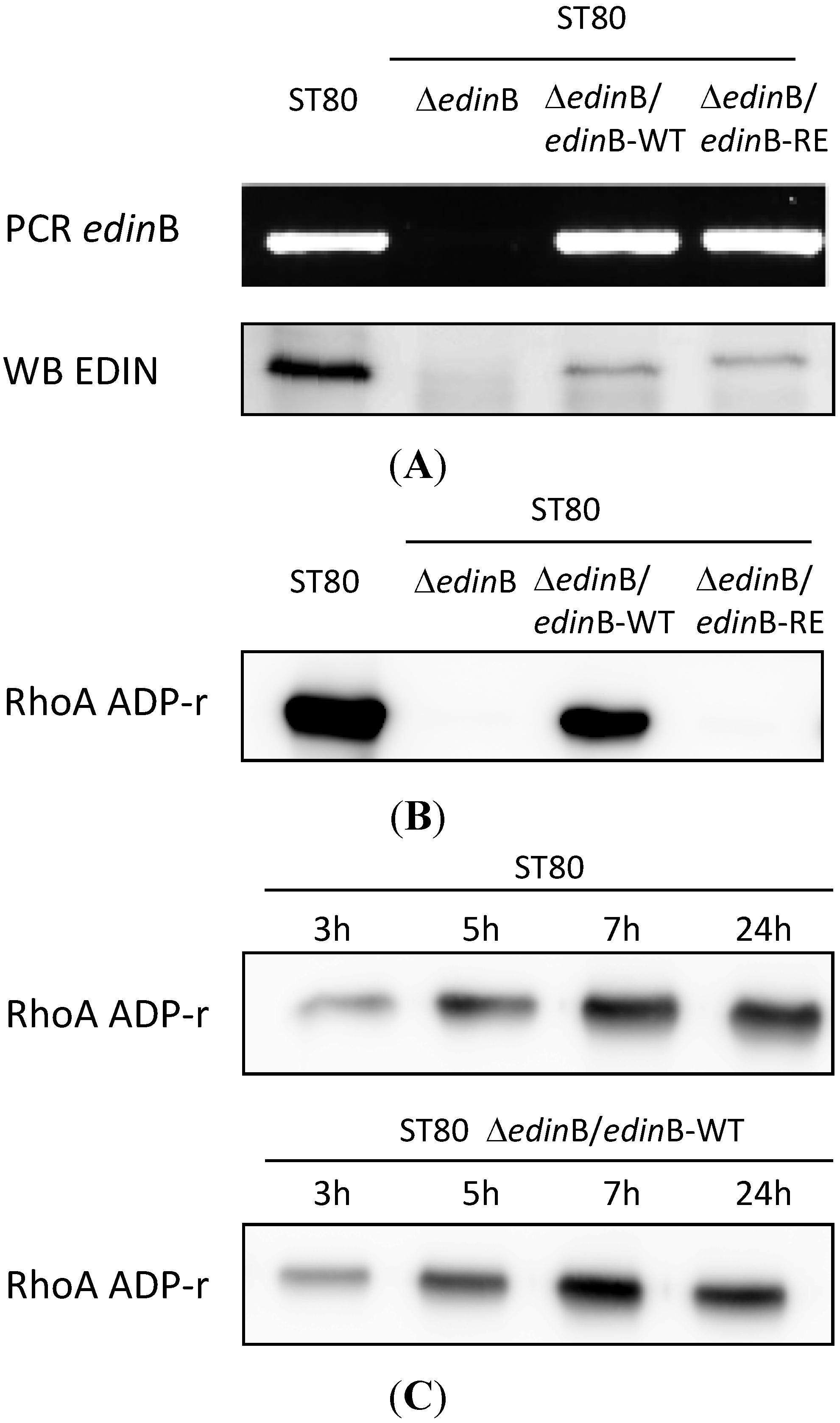
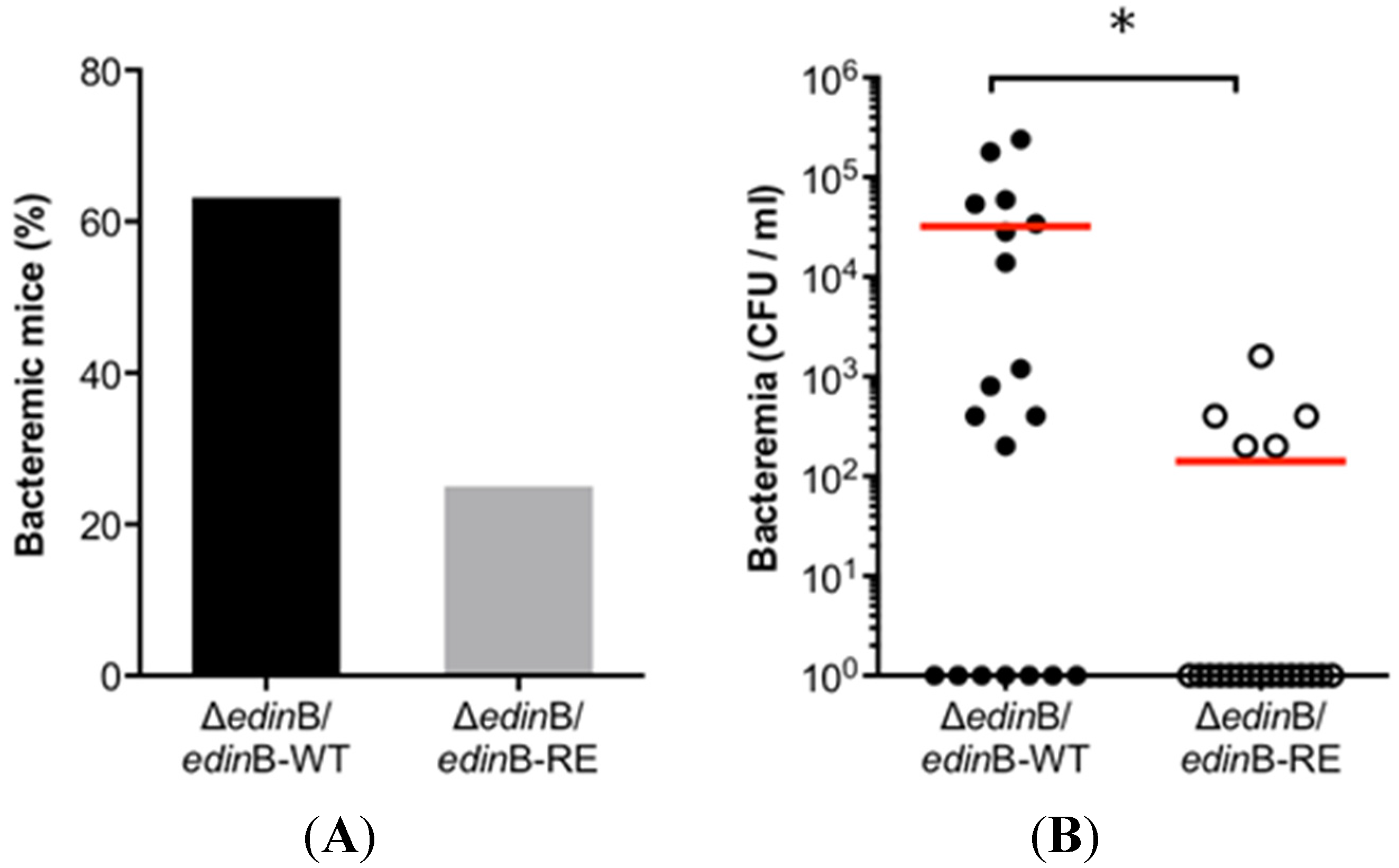
2.2. EDIN-B Activity Promotes the Translocation of S. aureus into the Bloodstream during Pneumonia

2.3. EDIN-B Activity and the Bacterial Persistence in the Blood

3. Discussion
4. Experimental Section
4.1. Ethics Statement
4.2. Bacterial Strains and DNA Constructs
4.3. Biochemical Assays and Products
4.4. Murine Model of Pneumonia and Bacteremia
4.5. Histopathology
4.6. Cell Culture and Immunofluorescence
4.7. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schreiber, M.P.; Chan, C.M.; Shorr, A.F. Bacteremia in Staphylococcus aureus pneumonia: Outcomes and epidemiology. J. Crit. Care 2011, 26, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Otter, J.A.; French, G.L. Molecular epidemiology of community-associated meticillin-resistant Staphylococcus aureus in Europe. Lancet Infect. Dis. 2010, 10, 227–239. [Google Scholar] [CrossRef]
- Lowy, F.D. How Staphylococcus aureus adapts to its host. N. Engl. J. Med. 2011, 364, 1987–1990. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.M.; Massey, R.C. How does Staphylococcus aureus escape the bloodstream? Trends Microbiol. 2011, 19, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Edwards, L.A.; O’Neill, C.; Furman, M.A.; Hicks, S.; Torrente, F.; Pérez-Machado, M.; Wellington, E.M.; Phillips, A.D.; Murch, S.H. Enterotoxin-producing staphylococci cause intestinal inflammation by a combination of direct epithelial cytopathy and superantigen-mediated T-cell activation. Inflamm. Bowel Dis. 2012, 18, 624–640. [Google Scholar] [CrossRef] [PubMed]
- Messad, N.; Landraud, L.; Canivet, B.; Lina, G.; Richard, J.L.; Sotto, A.; Lavigne, J.P.; Lemichez, E.; French Study Group on the Diabetic Foot. Distribution of edin in Staphylococcus aureus isolated from diabetic foot ulcers. Clin. Microbiol. Infect. 2013, 19, 875–880. [Google Scholar] [CrossRef] [PubMed]
- Sugai, M.; Chen, C.H.; Wu, H.C. Bacterial ADP-ribosyltransferase with a substrate specificity of the rho protein disassembles the Golgi apparatus in Vero cells and mimics the action of brefeldin A. Proc. Natl. Acad. Sci. USA 1992, 89, 8903–8907. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Hayashi, T.; Takami, H.; Ohnishi, M.; Murata, T.; Nakayama, K.; Asakawa, K.; Ohara, M.; Komatsuzawa, H.; Sugai, M. Complete nucleotide sequence of a Staphylococcus aureus exfoliative toxin B plasmid and identification of a novel ADP-ribosyltransferase, EDIN-C. Infect. Immun. 2001, 69, 7760–7771. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Nishifuji, K.; Sasaki, M.; Fudaba, Y.; Aepfelbacher, M.; Takata, T.; Ohara, M.; Komatsuzawa, H.; Amagai, M.; Sugai, M. Identification of the Staphylococcus aureus etd pathogenicity island which encodes a novel exfoliative toxin, ETD, and EDIN-B. Infect. Immun. 2002, 70, 5835–5845. [Google Scholar] [CrossRef] [PubMed]
- Aktories, K. Bacterial protein toxins that modify host regulatory GTPases. Nat. Rev. Microbiol. 2011, 9, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Chardin, P.; Boquet, P.; Madaule, P.; Popoff, M.R.; Rubin, E.J.; Gill, D.M. The mammalian G protein rhoC is ADP-ribosylated by Clostridium botulinum exoenzyme C3 and affects actin microfilaments in Vero cells. EMBO J. 1989, 8, 1087–1092. [Google Scholar] [PubMed]
- Paterson, H.F.; Self, A.J.; Garrett, M.D.; Just, I.; Aktories, K.; Hall, A. Microinjection of recombinant p21rho induces rapid changes in cell morphology. J. Cell Biol. 1990, 111, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Wilde, C.; Vogelsgesang, M.; Aktories, K. Rho-specific Bacillus cereus ADP-ribosyltransferase C3cer cloning and characterization. Biochemistry 2003, 42, 9694–9702. [Google Scholar] [CrossRef] [PubMed]
- Franke, G.C.; Bockenholt, A.; Sugai, M.; Rohde, H.; Aepfelbacher, M. Epidemiology, variable genetic organization and regulation of the EDIN-B toxin in Staphylococcus aureus from bacteraemic patients. Microbiology 2010, 156, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Lemichez, E.; Aktories, K. Hijacking of Rho GTPases during bacterial infection. Exp. Cell Res. 2013, 319, 2329–2336. [Google Scholar] [CrossRef] [PubMed]
- Aktories, K.; Lang, A.E.; Schwan, C.; Mannherz, H.G. Actin as target for modification by bacterial protein toxins. FEBS J. 2011, 278, 4526–4543. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.B.; Hall, A. RHO GTPases: Biochemistry and Biology. Annu. Rev. Cell Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Nusrat, A.; Giry, M.; Turner, J.R.; Colgan, S.P.; Parkos, C.A.; Carnes, D.; Lemichez, E.; Boquet, P.; Madara, J.L. Rho protein regulates tight junctions and perijunctional actin organization in polarized epithelia. Proc. Natl. Acad. Sci. USA 1995, 92, 10629–10633. [Google Scholar] [CrossRef] [PubMed]
- Lemichez, E.; Gonzalez-Rodriguez, D.; Bassereau, P.; Brochard-Wyart, F. Transcellular tunnel dynamics: Control of cellular dewetting by actomyosin contractility and I-BAR proteins. Biol. Cell 2012, 105, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Munro, P.; Benchetrit, M.; Nahori, M.A.; Stefani, C.; Clément, R.; Michiels, J.F.; Landraud, L.; Dussurget, O.; Lemichez, E. Staphylococcus aureus EDIN toxin promotes formation of infection foci in a mouse model of bacteremia. Infect. Immun. 2010, 78, 3404–3411. [Google Scholar] [CrossRef] [PubMed]
- Munro, P.; Clement, R.; Lavigne, J.P.; Pulcini, C.; Lemichez, E.; Landraud, L. High prevalence of edin-C encoding RhoA-targeting toxin in clinical isolates of Staphylococcus aureus. Eur. J. Clin. Microbiol. Infect. Dis. 2011, 30, 965–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perret, M.; Badiou, C.; Lina, G.; Burbaud, S.; Benito, Y.; Bes, M.; Cottin, V.; Couzon, F.; Juruj, C.; Dauwalder, O.; et al. Cross-talk between Staphylococcus aureus leukocidins-intoxicated macrophages and lung epithelial cells triggers chemokine secretion in an inflammasome-dependent manner. Cell. Microbiol. 2012, 14, 1019–1036. [Google Scholar] [CrossRef] [PubMed]
- Stegger, M.; Wirth, T.; Andersen, P.S.; Skov, R.L.; de Grassi, A.; Simões, P.M.; Tristan, A.; Petersen, A.; Aziz, M.; Kiil, K.; et al. Origin and evolution of European community-acquired methicillin-resistant Staphylococcus aureus. mBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Labandeira-Rey, M.; Couzon, F.; Boisset, S.; Brown, E.L.; Bes, M.; Benito, Y.; Barbu, E.M.; Vazquez, V.; Höök, M.; Etienne, J.; et al. Staphylococcus aureus Panton-Valentine leukocidin causes necrotizing pneumonia. Science 2007, 315, 1130–1133. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.; Doye, A.; Rolando, M.; Flatau, G.; Munro, P.; Gounon, P.; Clément, R.; Pulcini, C.; Popoff, M.R.; Mettouchi, A.; et al. Induction of transient macroapertures in endothelial cells through RhoA inhibition by Staphylococcus aureus factors. J. Cell Biol. 2006, 173, 809–819. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Courjon, J.; Munro, P.; Benito, Y.; Visvikis, O.; Bouchiat, C.; Boyer, L.; Doye, A.; Lepidi, H.; Ghigo, E.; Lavigne, J.-P.; et al. EDIN-B Promotes the Translocation of Staphylococcus aureus to the Bloodstream in the Course of Pneumonia. Toxins 2015, 7, 4131-4142. https://doi.org/10.3390/toxins7104131
Courjon J, Munro P, Benito Y, Visvikis O, Bouchiat C, Boyer L, Doye A, Lepidi H, Ghigo E, Lavigne J-P, et al. EDIN-B Promotes the Translocation of Staphylococcus aureus to the Bloodstream in the Course of Pneumonia. Toxins. 2015; 7(10):4131-4142. https://doi.org/10.3390/toxins7104131
Chicago/Turabian StyleCourjon, Johan, Patrick Munro, Yvonne Benito, Orane Visvikis, Coralie Bouchiat, Laurent Boyer, Anne Doye, Hubert Lepidi, Eric Ghigo, Jean-Philippe Lavigne, and et al. 2015. "EDIN-B Promotes the Translocation of Staphylococcus aureus to the Bloodstream in the Course of Pneumonia" Toxins 7, no. 10: 4131-4142. https://doi.org/10.3390/toxins7104131
APA StyleCourjon, J., Munro, P., Benito, Y., Visvikis, O., Bouchiat, C., Boyer, L., Doye, A., Lepidi, H., Ghigo, E., Lavigne, J. -P., Vandenesch, F., & Lemichez, E. (2015). EDIN-B Promotes the Translocation of Staphylococcus aureus to the Bloodstream in the Course of Pneumonia. Toxins, 7(10), 4131-4142. https://doi.org/10.3390/toxins7104131