
Microfluidic Single-Cell Proteomics Assay Chip: Lung Cancer Cell Line Case Study

 ,
, 
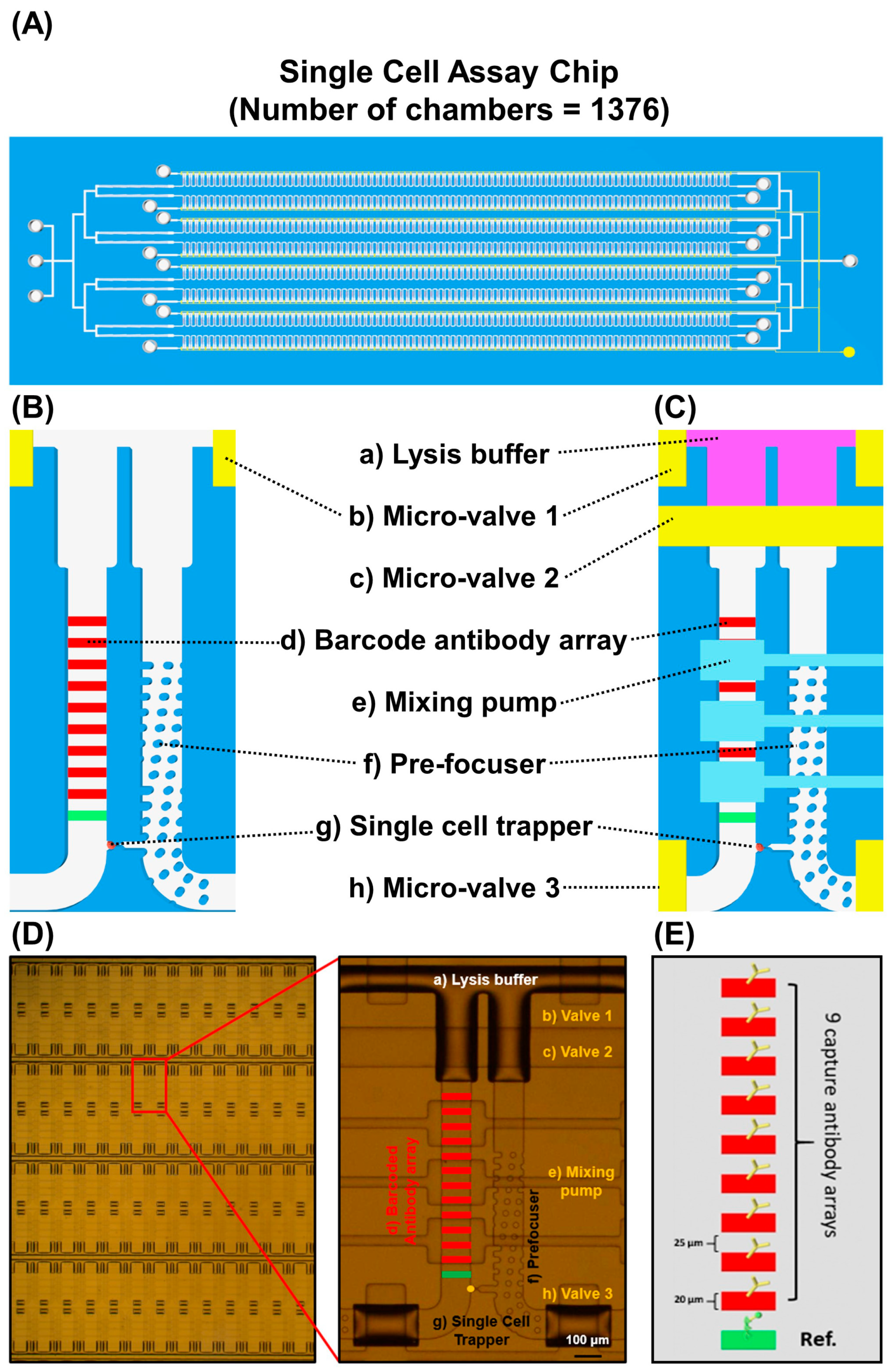{kind=link}
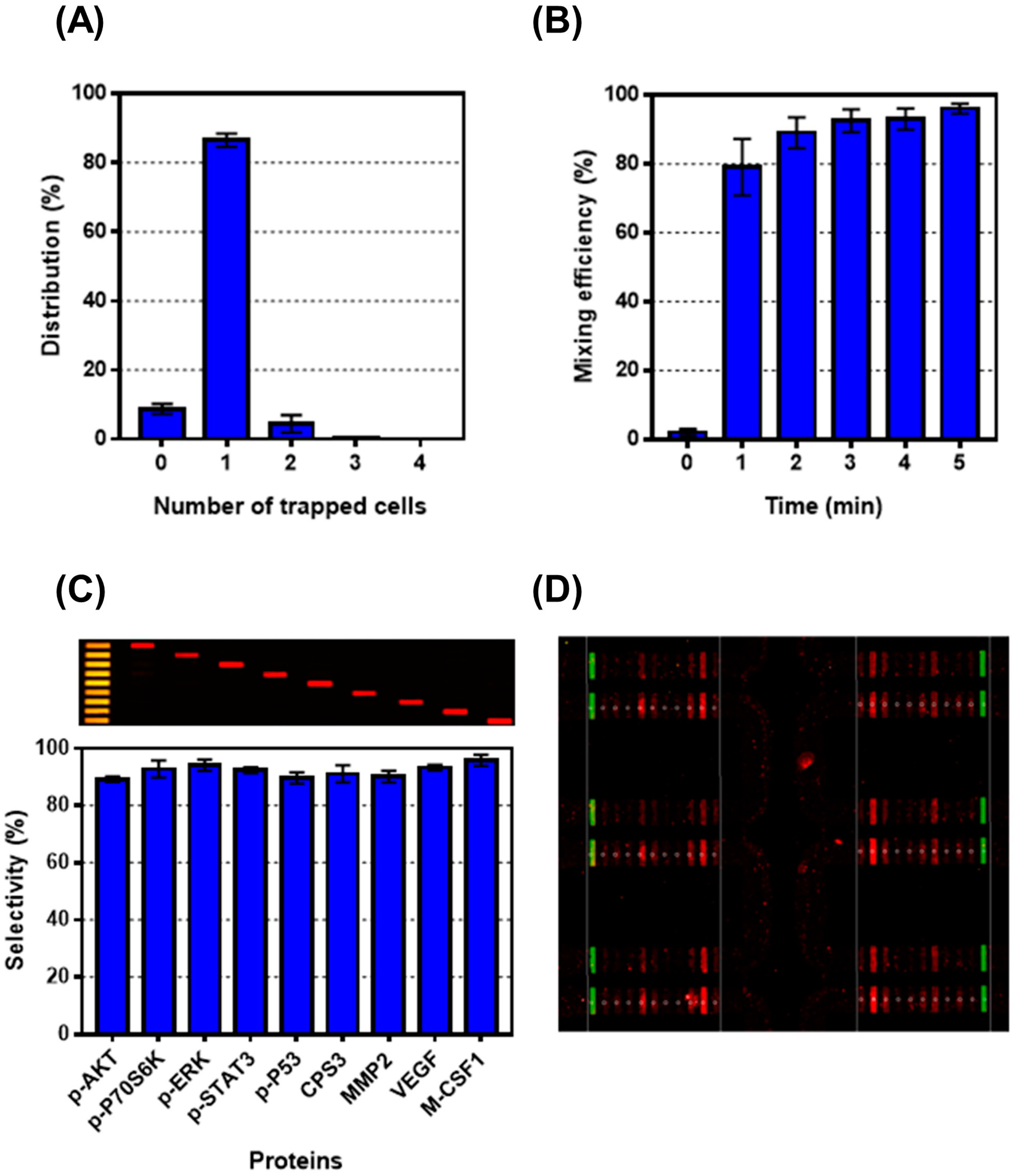{kind=link}
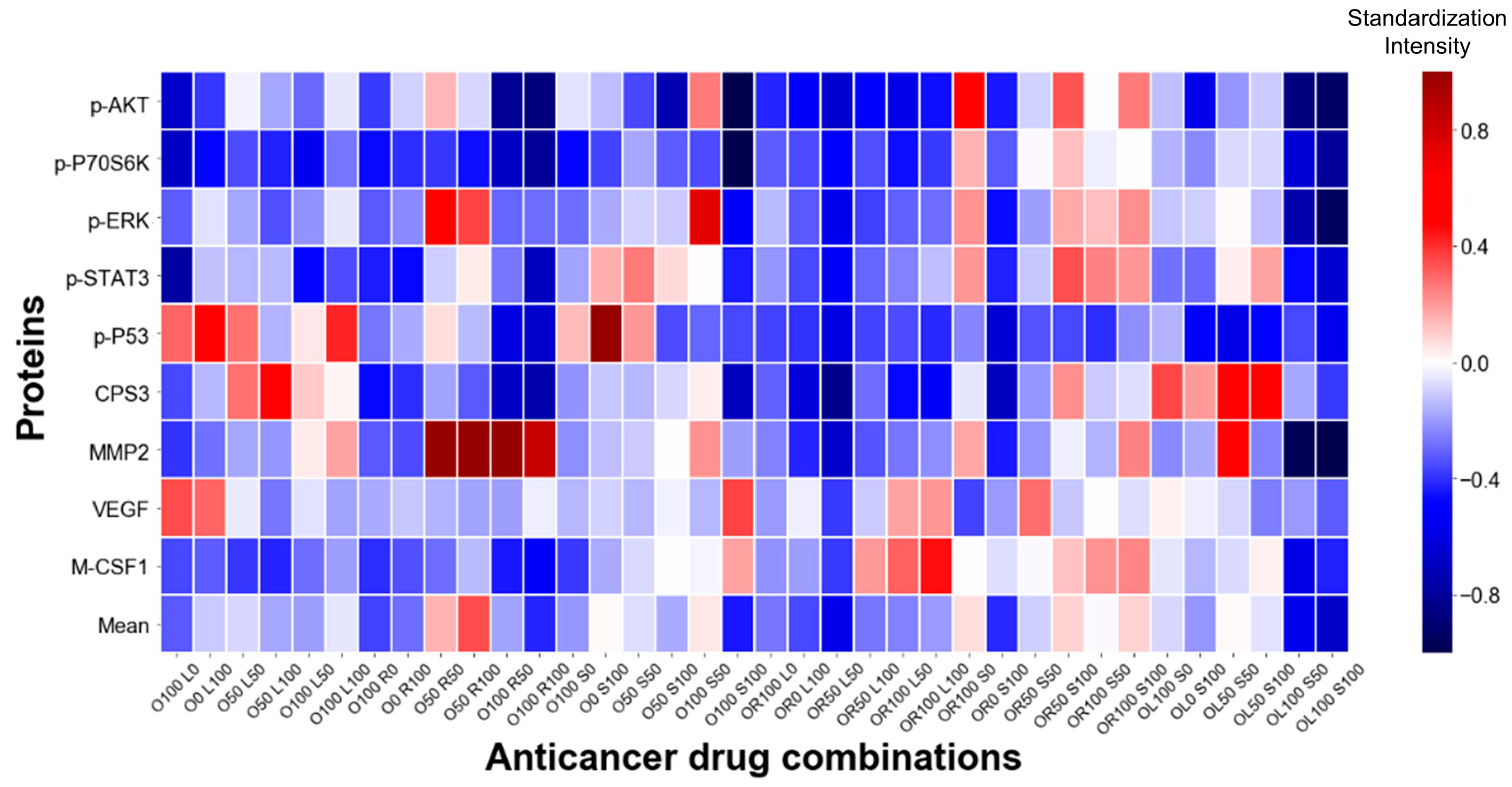{kind=link}
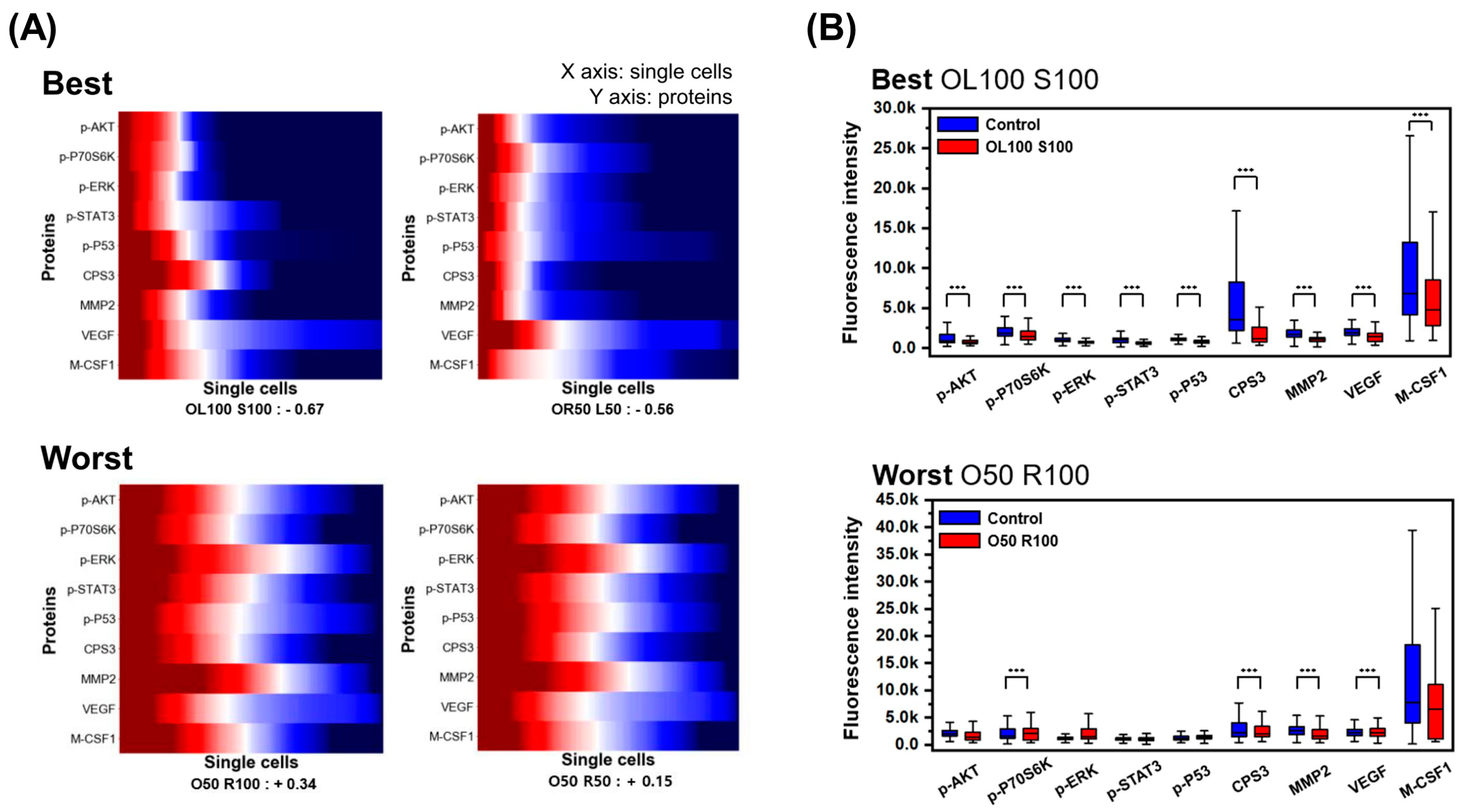{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Device Preparation
2.2. Preparation of the Multiplex Barcode Array
2.3. Cell Line and Reagents
2.4. MTT Assay and Drug IC50 Measurement
2.5. Single-Cell Assay
2.6. Single-Cell Data Analysis
2.7. Heterogeneity Evaluation and Analysis
2.8. ELISA Assay
3. Results
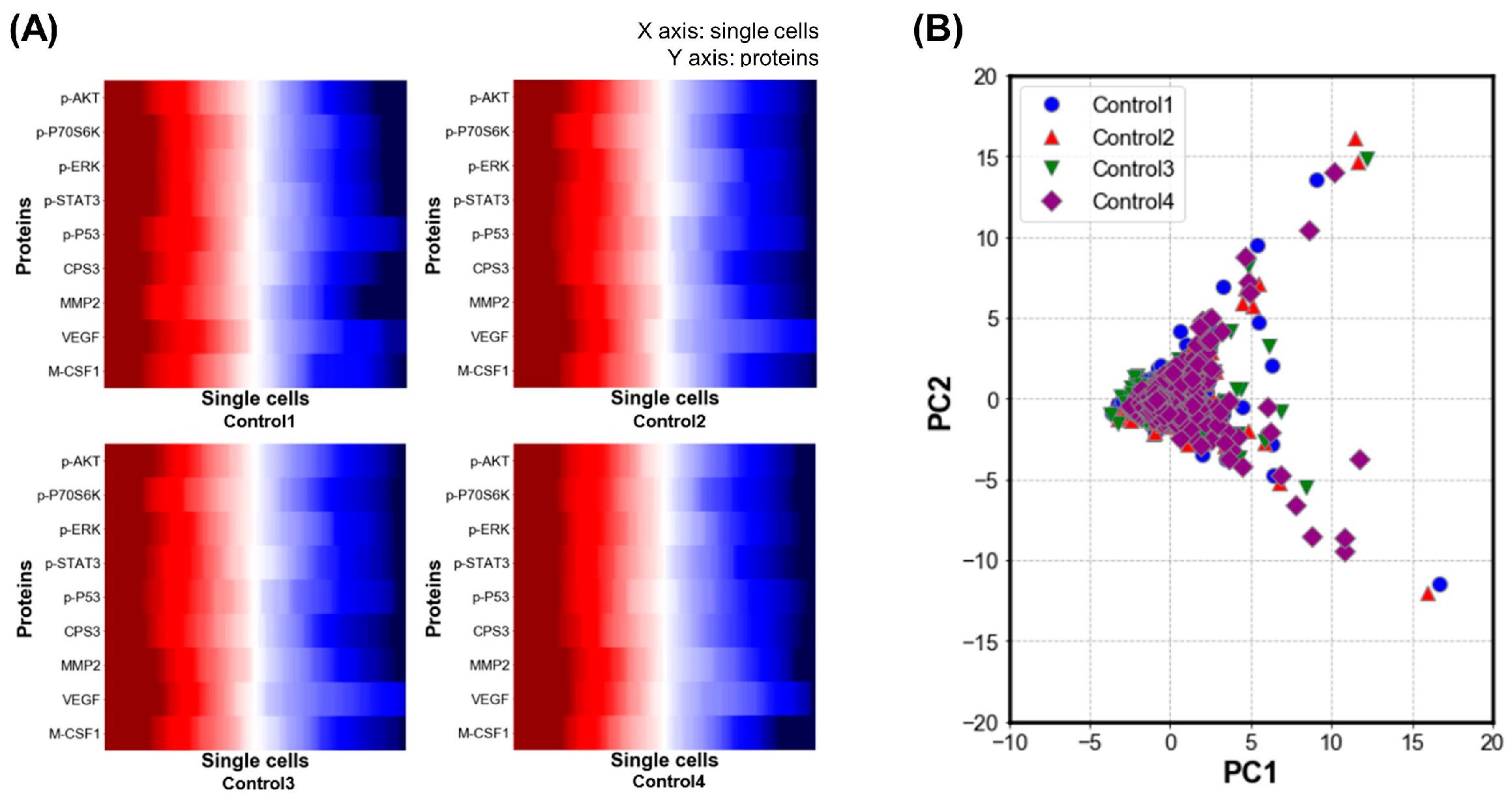
3.1. Validation of Single-Cell Proteomics Assay Chip Using H1650 Lung-Cancer Cell Line
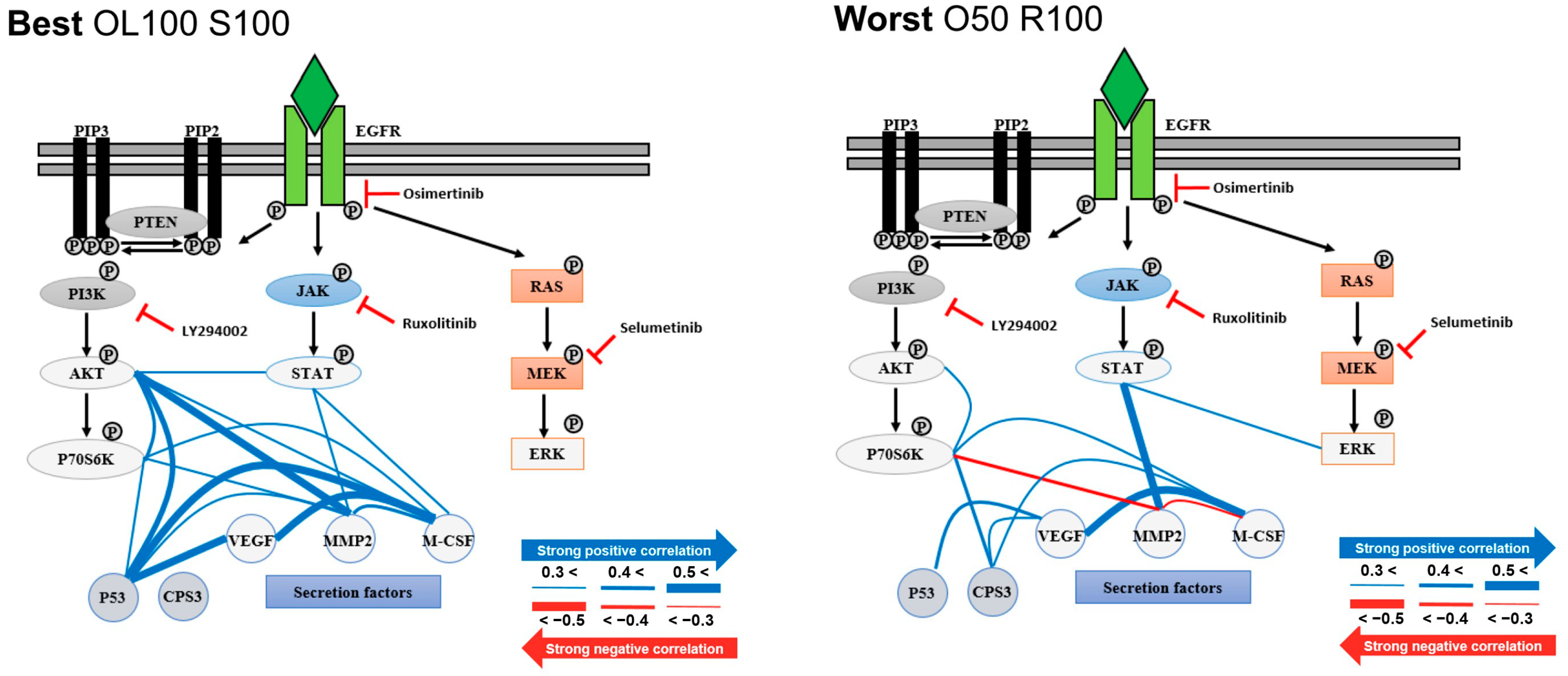
3.2. Single-Cell Analysis Using H1975 Lung-Cancer Cell Line
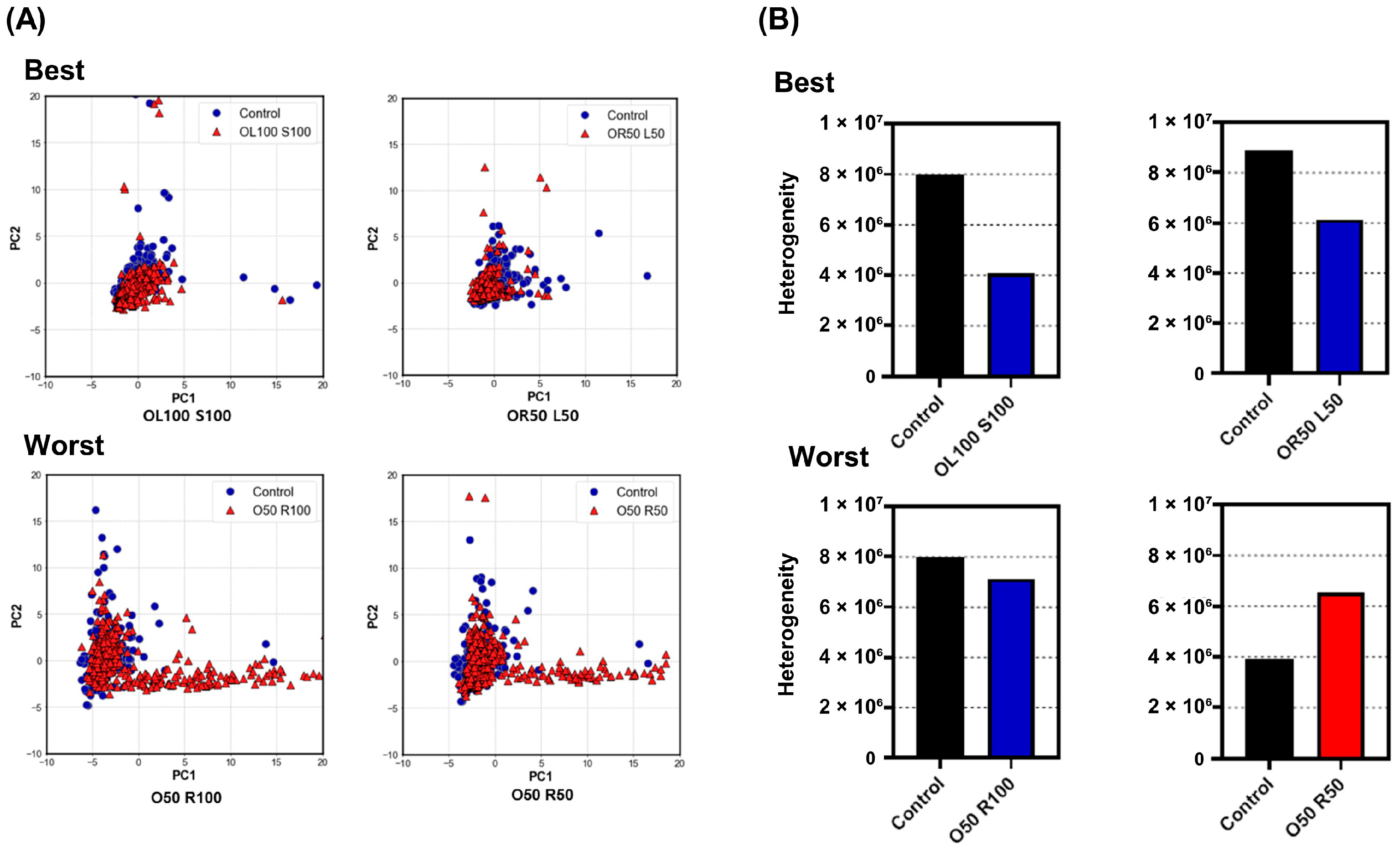
3.3. Evaluation of Heterogeneity
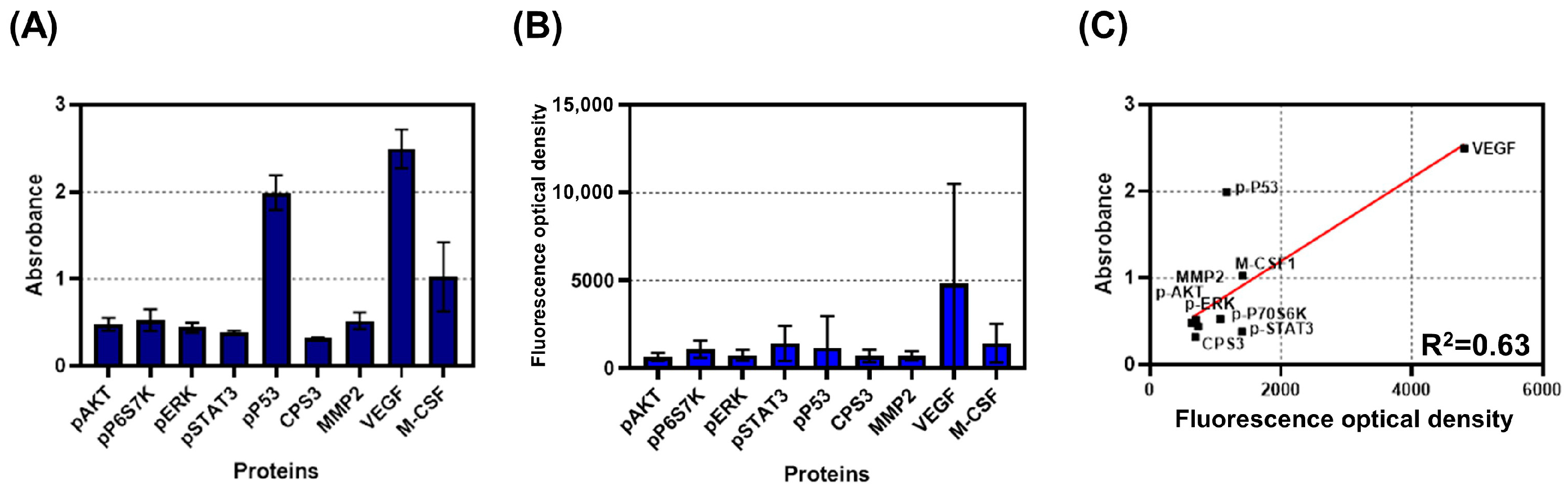
3.4. Comparison between Bulk and Single-Cell Assay
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Saunders, N.A.; Simpson, F.; Thompson, E.W.; Hill, M.M.; Endo-Munoz, L.; Leggatt, G.; Minchin, R.F.; Guminski, A. Role of intratumoural heterogeneity in cancer drug resistance: Molecular and clinical perspectives. EMBO Mol. Med. 2012, 4, 675–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; Swanton, C. Tumour heterogeneity and the evolution of polyclonal drug resistance. Mol. Oncol. 2014, 8, 1095–1111. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Burrell, R.A.; Endesfelder, D.; Novelli, M.R.; Swanton, C. Cancer chromosomal instability: Therapeutic and diagnostic challenges. EMBO Rep. 2012, 13, 528–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mroz, E.A.; Tward, A.D.; Hammon, R.J.; Ren, Y.; Rocco, J.W. Intra-tumor genetic heterogeneity and mortality in head and neck cancer: Analysis of data from the Cancer Genome Atlas. PLoS Med. 2015, 12, e1001786. [Google Scholar] [CrossRef] [Green Version]
- Lim, L.C.; Lim, Y.M. Proteome Heterogeneity in Colorectal Cancer. Proteomics 2018, 18, 1700169. [Google Scholar] [CrossRef]
- Greenbaum, A.; Martin, D.R.; Bocklage, T.; Lee, J.H.; Ness, S.A.; Rajput, A. Tumor Heterogeneity as a Predictor of Response to Neoadjuvant Chemotherapy in Locally Advanced Rectal Cancer. Clin. Colorectal Cancer 2019, 18, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Kikutake, C.; Yoshihara, M.; Sato, T.; Saito, D.; Suyama, M. Pan-cancer analysis of intratumor heterogeneity associated with patient prognosis using multidimensional measures. Oncotarget 2018, 9, 37689–37699. [Google Scholar] [CrossRef] [Green Version]
- Seoane, J.; De Mattos-Arruda, L. The challenge of intratumour heterogeneity in precision medicine. J. Intern. Med. 2014, 276, 41–51. [Google Scholar] [CrossRef]
- Shalek, A.K.; Benson, M. Single-cell analyses to tailor treatments. Sci. Transl. Med. 2017, 9, 408. [Google Scholar] [CrossRef] [Green Version]
- Zito Marino, F.; Bianco, R.; Accardo, M.; Ronchi, A.; Cozzolino, I.; Morgillo, F.; Rossi, G.; Franco, R. Molecular heterogeneity in lung cancer: From mechanisms of origin to clinical implications. Int. J. Med. Sci. 2019, 16, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.F.; Li, G.R.; Wang, R.J.; Yi, Y.T.; Yang, L.; Jiang, D.; Zhang, X.P.; Peng, Y. Application of next-generation sequencing in clinical oncology to advance personalized treatment of cancer. Chin. J. Cancer 2012, 31, 463–470. [Google Scholar] [CrossRef] [Green Version]
- Waters, K.M.; Liu, T.; Quesenberry, R.D.; Willse, A.R.; Bandyopadhyay, S.; Kathmann, L.E.; Weber, T.J.; Smith, R.D.; Wiley, H.S.; Thrall, B.D. Network analysis of epidermal growth factor signaling using integrated genomic, proteomic and phosphorylation data. PLoS ONE 2012, 7, e34515. [Google Scholar] [CrossRef] [Green Version]
- Boja, E.S.; Rodriguez, H. Proteogenomic convergence for understanding cancer pathways and networks. Clin. Proteom. 2014, 11, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.; Qin, L.; Wei, W.; Geng, F.; Fan, R.; Shin, Y.S.; Guo, D.; Hood, L.; Mischel, P.S.; Heath, J.R. Single-cell proteomic chip for profiling intracellular signaling pathways in single tumor cells. Proc. Natl. Acad. Sci. USA 2012, 109, 419–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navin, N.E. The first five years of single-cell cancer genomics and beyond. Genome Res. 2015, 25, 1499–1507. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Shin, Y.S.; Xue, M.; Matsutani, T.; Masui, K.; Yang, H.; Ikegami, S.; Gu, Y.; Herrmann, K.; Johnson, D.; et al. Single-Cell Phosphoproteomics Resolves Adaptive Signaling Dynamics and Informs Targeted Combination Therapy in Glioblastoma. Cancer Cell 2016, 29, 563–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hara, D.M.; Xu, Y.; Liang, Z.; Reddy, M.P.; Wu, D.Y.; Litwin, V. Recommendations for the validation of flow cytometric testing during drug development: II assays. J. Immunol. Methods 2011, 363, 120–134. [Google Scholar] [CrossRef]
- O’Brien, P.J.; Lee, M.; Spilker, M.E.; Zhang, C.C.; Yan, Z.; Nichols, T.C.; Li, W.; Johnson, C.H.; Patti, G.J.; Siuzdak, G. Monitoring metabolic responses to chemotherapy in single cells and tumors using nanostructure-initiator mass spectrometry (NIMS) imaging. Cancer Metab. 2013, 1, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Tham, D.; Wei, W.; Shin, Y.S.; Ma, C.; Ahmad, H.; Shi, Q.; Yu, J.; Levine, R.D.; Heath, J.R. Quantitating cell-cell interaction functions with applications to glioblastoma multiforme cancer cells. Nano Lett. 2012, 12, 6101–6106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Shi, Q.; Remacle, F.; Qin, L.; Shackelford, D.B.; Shin, Y.S.; Mischel, P.S.; Levine, R.D.; Heath, J.R. Hypoxia induces a phase transition within a kinase signaling network in cancer cells. Proc. Natl. Acad. Sci. USA 2013, 110, E1352–E1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kravchenko-Balasha, N.; Wang, J.; Remacle, F.; Levine, R.D.; Heath, J.R. Glioblastoma cellular architectures are predicted through the characterization of two-cell interactions. Proc. Natl. Acad. Sci. USA 2014, 111, 6521–6526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Masterman-Smith, M.D.; Graham, N.A.; Jiao, J.; Mottahedeh, J.; Laks, D.R.; Ohashi, M.; DeJesus, J.; Kamei, K.; Lee, K.B.; et al. A microfluidic platform for systems pathology: Multiparameter single-cell signaling measurements of clinical brain tumor specimens. Cancer Res. 2010, 70, 6128–6138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinkala, E.; Herr, A.E. Single-Cell Western Blotting. Methods Mol. Biol. 2015, 1346, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Xue, M.; Wei, W.; Su, Y.; Kim, J.; Shin, Y.S.; Mai, W.X.; Nathanson, D.A.; Heath, J.R. Chemical methods for the simultaneous quantitation of metabolites and proteins from single cells. J. Am. Chem. Soc. 2015, 137, 4066–4069. [Google Scholar] [CrossRef] [Green Version]
- Jin, D.; Deng, B.; Li, J.X.; Cai, W.; Tu, L.; Chen, J.; Wu, Q.; Wang, W.H. A microfluidic device enabling high-efficiency single cell trapping. Biomicrofluidics 2015, 9, 014101. [Google Scholar] [CrossRef] [Green Version]
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-small-cell lung cancer. Nat. Rev. Dis Primers 2015, 1, 15009. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.S.; Gilligan, D.; Pacey, S. Treatment approaches for EGFR-inhibitor-resistant patients with non-small-cell lung cancer. Lancet Oncol. 2015, 16, e447–e459. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Su, C.; Ren, S.; Cappuzzo, F.; Rocco, G.; Palmer, J.D.; van Zandwijk, N.; Blackhall, F.; Le, X.; Pennell, N.A.; et al. A consensus on the role of osimertinib in non-small cell lung cancer from the AME Lung Cancer Collaborative Group. J. Thorac. Dis. 2018, 10, 3909–3921. [Google Scholar] [CrossRef]
- Huang, L.; Fu, L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm. Sin. B 2015, 5, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Casaluce, F.; Sgambato, A.; Maione, P.; Sacco, P.C.; Santabarbara, G.; Gridelli, C. Selumetinib for the treatment of non-small cell lung cancer. Expert Opin. Investig. Drugs 2017, 26, 973–984. [Google Scholar] [CrossRef]
- Giaccone, G.; Sanborn, R.E.; Waqar, S.N.; Martinez-Marti, A.; Ponce, S.; Zhen, H.; Kennealey, G.; Erickson-Viitanen, S.; Schaefer, E. A Placebo-Controlled Phase II Study of Ruxolitinib in Combination with Pemetrexed and Cisplatin for First-Line Treatment of Patients with Advanced Nonsquamous Non-Small-Cell Lung Cancer and Systemic Inflammation. Clin. Lung Cancer 2018, 19, e567–e574. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lei, H.L.; Zhang, Y.F.; Xu, Y.G.; Shen, S.; Ge, Y.R.; Li, H.M.; Xie, J.M. Non-covalent modification of graphene oxide nanocomposites with chitosan/dextran and its application in drug delivery. RSC Adv. 2016, 6, 9328–9337. [Google Scholar] [CrossRef]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Campbell, K.R.; Lun, A.T.; Wills, Q.F. Scater: Pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 2017, 33, 1179–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bro, R.; Smilde, A.K. Principal component analysis. Anal. Methods 2014, 6, 2812–2831. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Murtagh, F.; Van Poucke, S.; Lin, S.; Lan, P. Hierarchical cluster analysis in clinical research with heterogeneous study population: Highlighting its visualization with R. Ann. Transl. Med. 2017, 5, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Vicent, S.; López-Picazo, J.M.; Toledo, G.; Lozano, M.D.; Torre, W.; Garcia-Corchón, C.; Quero, C.; Soria, J.C.; Martín-Algarra, S.; Manzano, R.G.; et al. ERK1/2 is activated in non-small-cell lung cancer and associated with advanced tumours. Br. J. Cancer 2004, 90, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, T.; Pirinen, R.; Bohm, J.; Johansson, R.; Kosma, V.M. Increased expression of matrix metalloproteinase-2 (MMP-2) predicts tumour recurrence and unfavourable outcome in non-small cell lung cancer. Histol. Histopathol. 2008, 23, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Grzywa, T.M.; Paskal, W.; Wlodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975. [Google Scholar] [CrossRef]
- Liu, J.; Dang, H.; Wang, X.W. The significance of intertumor and intratumor heterogeneity in liver cancer. Exp. Mol. Med. 2018, 50, e416. [Google Scholar] [CrossRef]
- Obulkasim, A.; Ylstra, B.; van Essen, H.F.; Benner, C.; Stenning, S.; Langley, R.; Allum, W.; Cunningham, D.; Inam, I.; Hewitt, L.C.; et al. Reduced genomic tumor heterogeneity after neoadjuvant chemotherapy is related to favorable outcome in patients with esophageal adenocarcinoma. Oncotarget 2016, 7, 44084–44095. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C. Defining cell types and states with single-cell genomics. Genome Res. 2015, 25, 1491–1498. [Google Scholar] [CrossRef] [Green Version]
- Yuan, G.C.; Cai, L.; Elowitz, M.; Enver, T.; Fan, G.; Guo, G.; Irizarry, R.; Kharchenko, P.; Kim, J.; Orkin, S.; et al. Challenges and emerging directions in single-cell analysis. Genome Biol. 2017, 18, 84. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, X.; Zhang, Y.; Liu, J. Advancing single-cell proteomics and metabolomics with microfluidic technologies. Analyst 2019, 144, 846–858. [Google Scholar] [CrossRef]
- Pandey, A.; Mann, M. Proteomics to study genes and genomes. Nature 2000, 405, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Chandramouli, K.; Qian, P.Y. Proteomics: Challenges, techniques and possibilities to overcome biological sample complexity. Hum. Genom. Proteom. 2009, 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Giner, F.; Gkountela, S.; Donato, C.; Alborelli, I.; Quagliata, L.; Ng, C.K.Y.; Piscuoglio, S.; Aceto, N. Cancer Diagnosis Using a Liquid Biopsy: Challenges and Expectations. Diagnostics 2018, 8, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Zhao, H. Liquid biopsy in tumors: Opportunities and challenges. Ann. Transl. Med. 2018, 6, S89. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, Y.; Son, M.; Nam, Y.R.; Choi, J.; Heath, J.R.; Yang, S. Microfluidic Single-Cell Proteomics Assay Chip: Lung Cancer Cell Line Case Study. Micromachines 2021, 12, 1147. https://doi.org/10.3390/mi12101147
Jung Y, Son M, Nam YR, Choi J, Heath JR, Yang S. Microfluidic Single-Cell Proteomics Assay Chip: Lung Cancer Cell Line Case Study. Micromachines. 2021; 12(10):1147. https://doi.org/10.3390/mi12101147
Chicago/Turabian StyleJung, Yugyung, Minkook Son, Yu Ri Nam, Jongchan Choi, James R. Heath, and Sung Yang. 2021. "Microfluidic Single-Cell Proteomics Assay Chip: Lung Cancer Cell Line Case Study" Micromachines 12, no. 10: 1147. https://doi.org/10.3390/mi12101147
APA StyleJung, Y., Son, M., Nam, Y. R., Choi, J., Heath, J. R., & Yang, S. (2021). Microfluidic Single-Cell Proteomics Assay Chip: Lung Cancer Cell Line Case Study. Micromachines, 12(10), 1147. https://doi.org/10.3390/mi12101147