Hypoxia-Induced Cisplatin Resistance in Non-Small Cell Lung Cancer Cells Is Mediated by HIF-1α and Mutant p53 and Can Be Overcome by Induction of Oxidative Stress

 , , , , , and
, , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
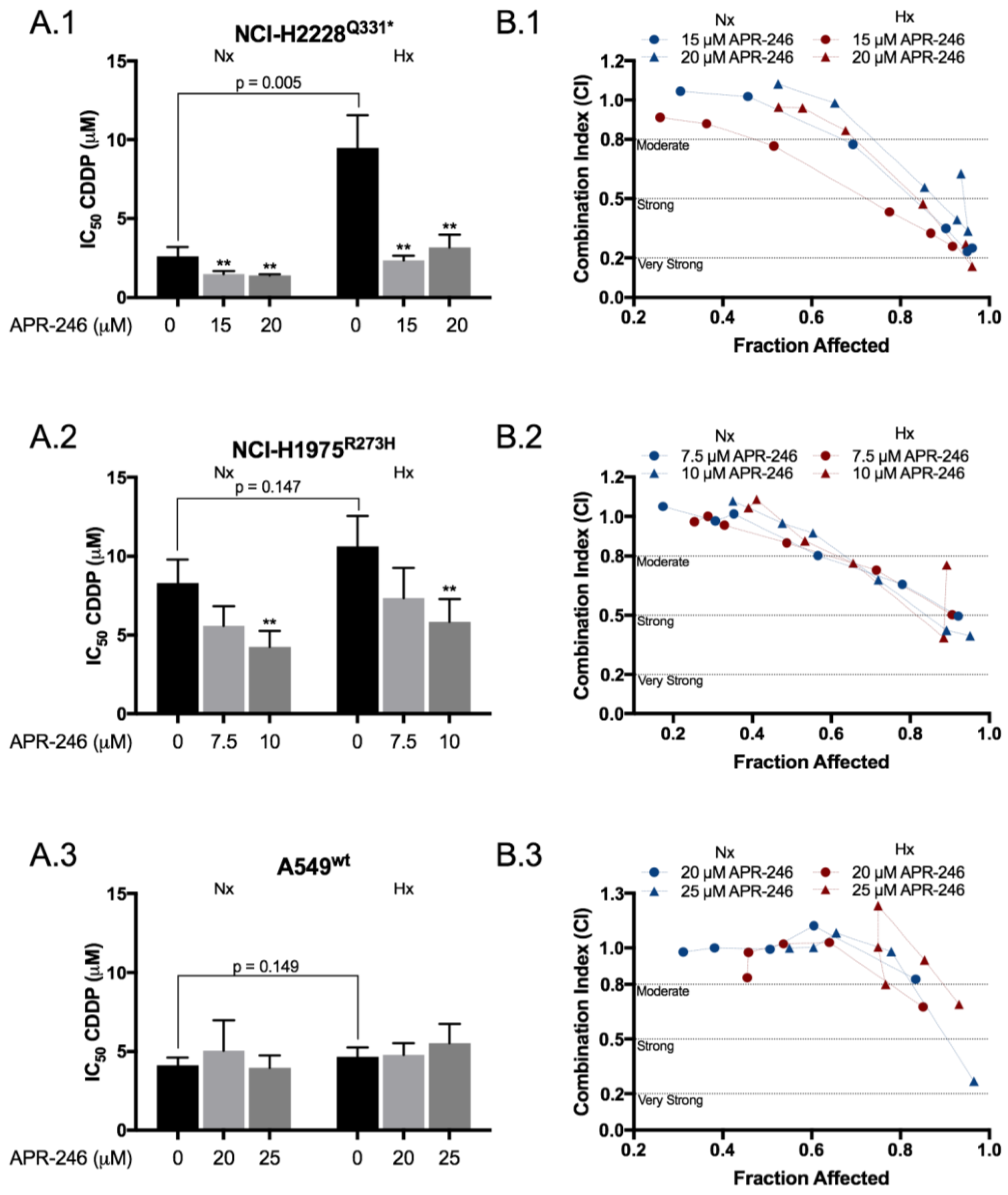
2.1. APR-246 Acted Synergistically with Cisplatin in a Mutant p53 Background under Normal and Reduced Oxygen Levels
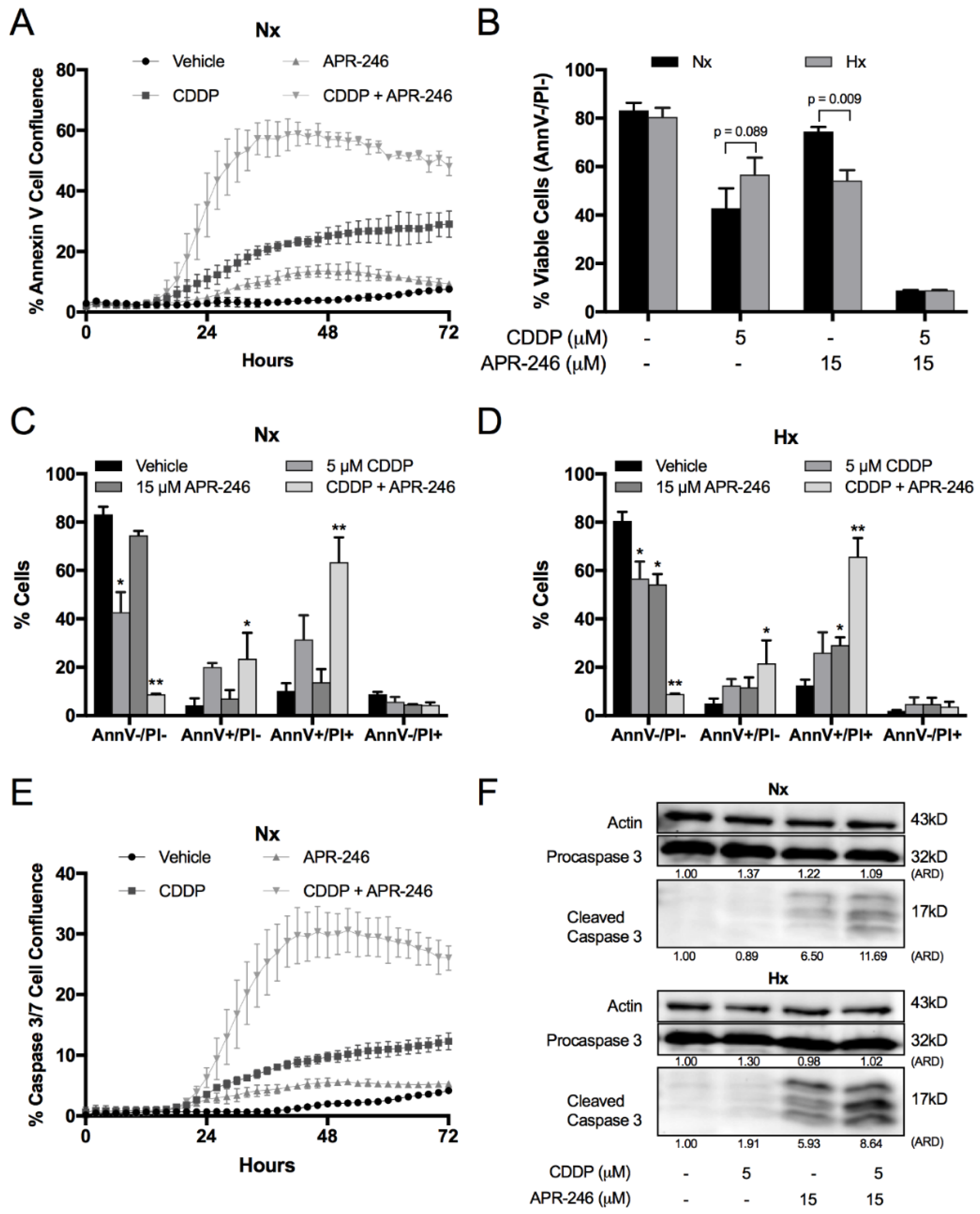
2.2. Hypoxia Affected the Apoptotic Response to CDDP and APR-246 Monotherapy, While Co-Treatment Resulted in an Equally Strong Apoptotic Response Compared to Normoxia in NCI-H2228Q331*
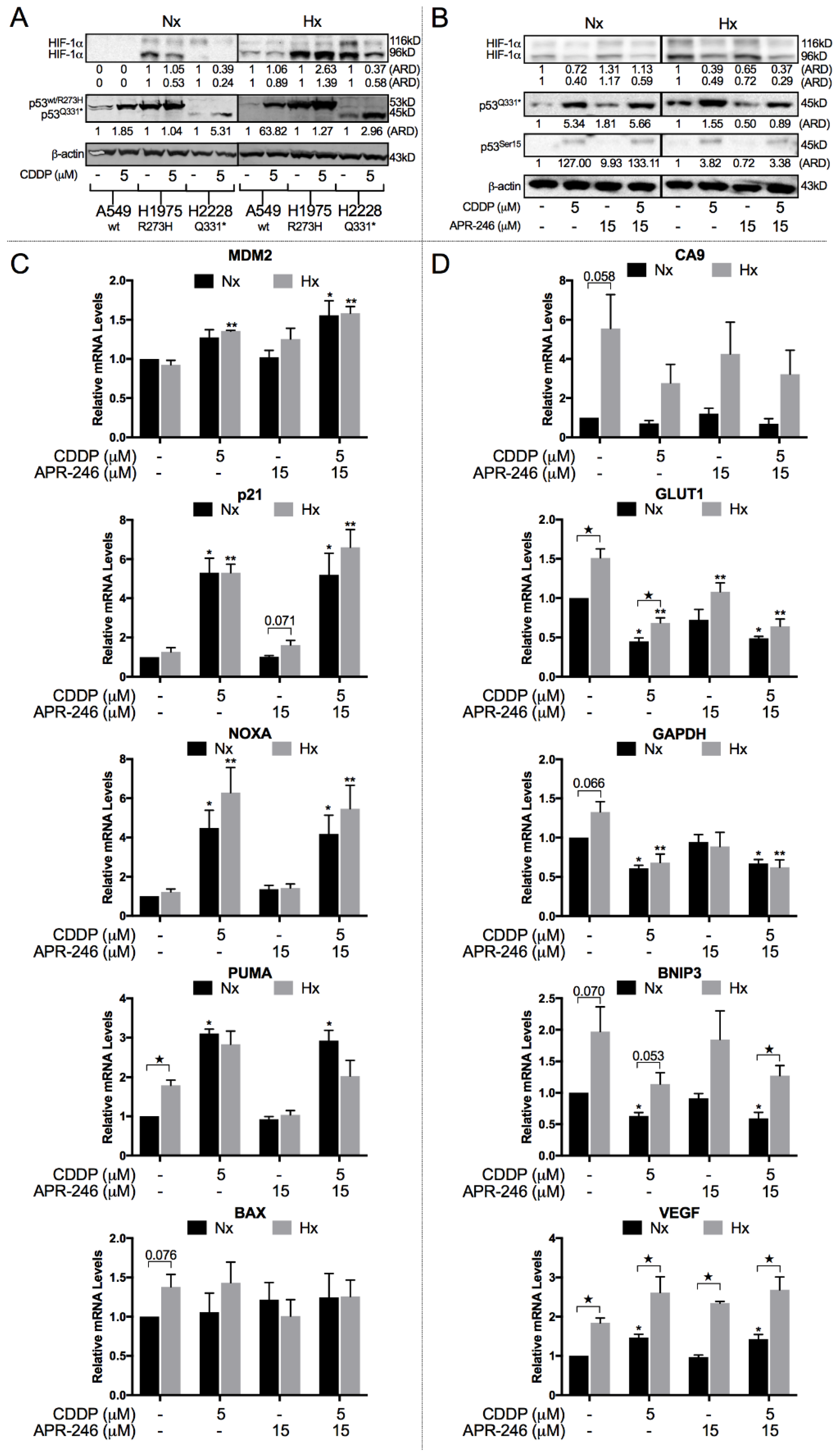
2.3. CDDP Induced a Shift from HIF-1α to p53Q331* Protein Expression and Transcription under Both Normoxic and Hypoxic Conditions
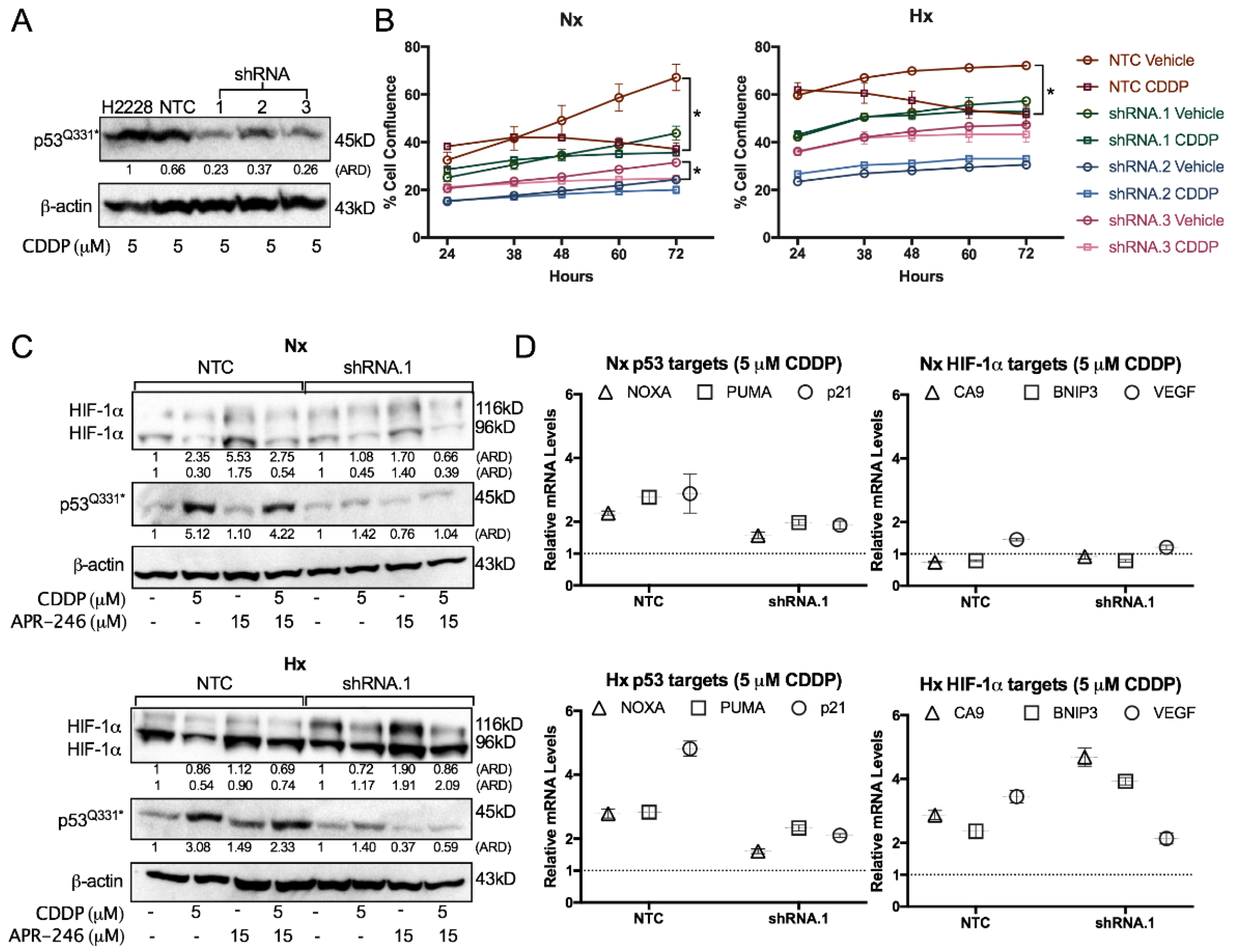
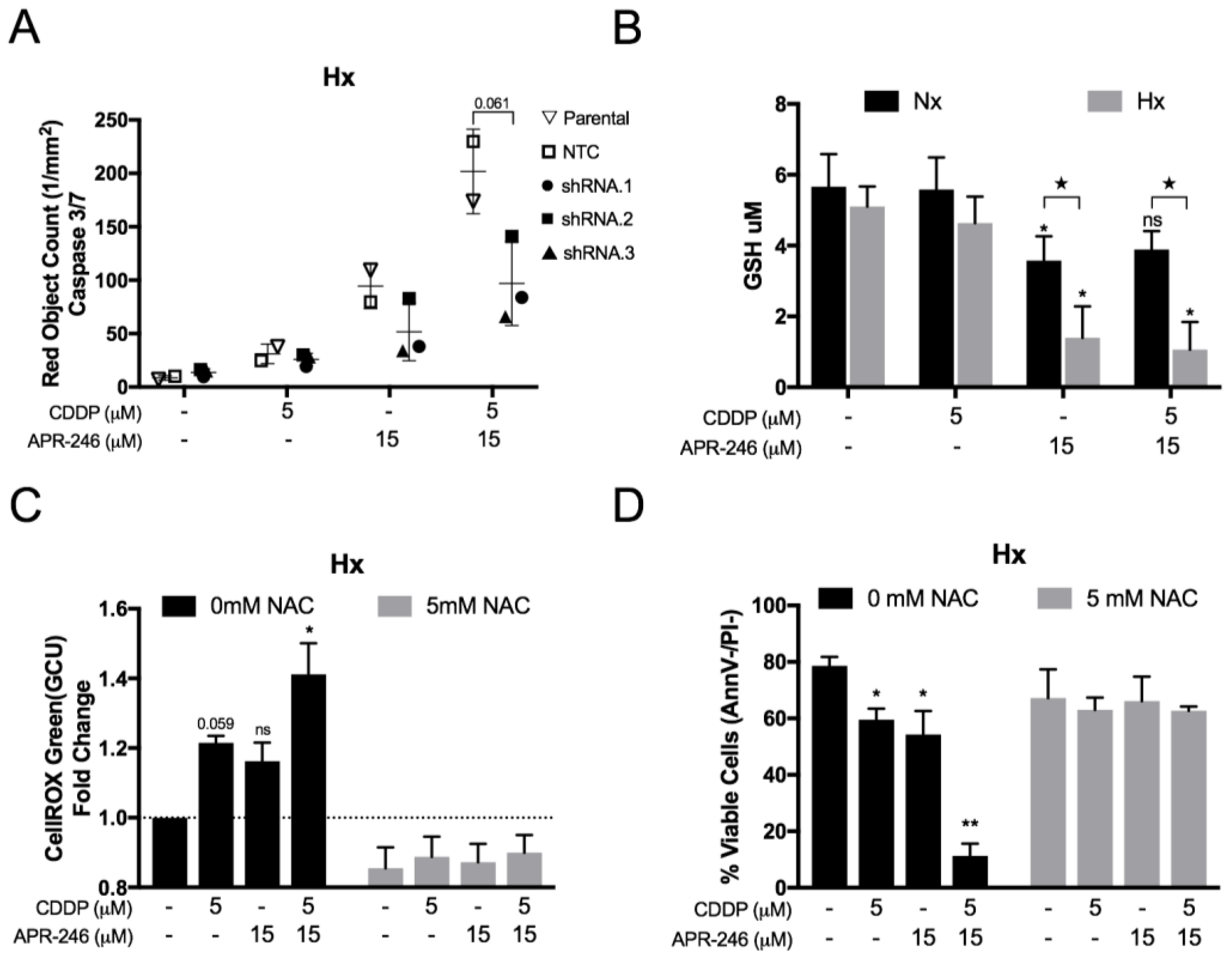
2.4. The Synergistic Interaction between APR-246 and CDDP was Both Mutant p53Q331*- and ROS-Dependent Following APR-246-Induced GSH Reduction under Hypoxic Conditions
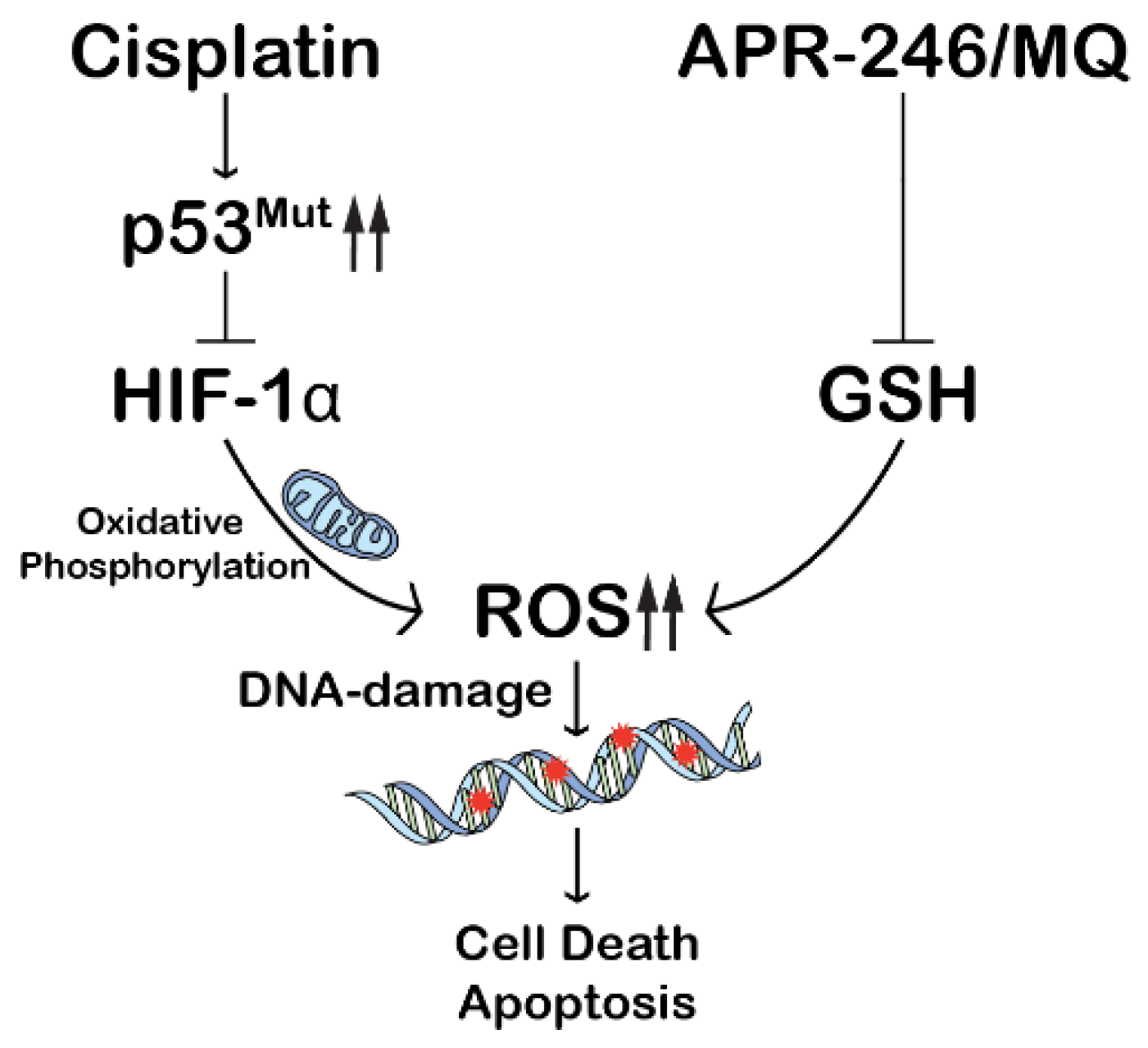
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Survival Assay and Synergism
4.3. Western Blot Analysis
4.4. Apoptotic/Cell Death Assays
4.5. Quantitative Reverse Transcription PCR (RT-qPCR)
4.6. Reactive Oxygen Species Detection
4.7. Total Glutathione Content
4.8. Transduction
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Deben, C.; Deschoolmeester, V.; Lardon, F.; Rolfo, C.; Pauwels, P. TP53 and MDM2 genetic alterations in non-small cell lung cancer: Evaluating their prognostic and predictive value. Crit. Rev. Oncol. Hematol. 2016, 99, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin resistance: A cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol. Rev. 2012, 64, 706–721. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1alpha pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-inducible factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J. Biol. Med. 2007, 80, 51–60. [Google Scholar] [PubMed]
- Sermeus, A.; Michiels, C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2011, 2, e164. [Google Scholar] [CrossRef] [PubMed]
- Hammond, E.M.; Giaccia, A.J. Hypoxia-inducible factor-1 and p53: Friends, acquaintances, or strangers? Clin. Cancer Res. 2006, 12, 5007–5009. [Google Scholar] [CrossRef] [PubMed]
- Kamat, C.D.; Green, D.E.; Warnke, L.; Thorpe, J.E.; Ceriello, A.; Ihnat, M.A. Mutant p53 facilitates pro-angiogenic, hyperproliferative phenotype in response to chronic relative hypoxia. Cancer Lett. 2007, 249, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Wohlkoenig, C.; Leithner, K.; Deutsch, A.; Hrzenjak, A.; Olschewski, A.; Olschewski, H. Hypoxia-induced cisplatin resistance is reversible and growth rate independent in lung cancer cells. Cancer Lett. 2011, 308, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Leithner, K.; Wohlkoenig, C.; Quehenberger, F.; Bertsch, A.; Olschewski, A.; Olschewski, H.; Hrzenjak, A. Panobinostat reduces hypoxia-induced cisplatin resistance of non-small cell lung carcinoma cells via HIF-1alpha destabilization. Mol. Cancer 2015, 14, 4. [Google Scholar] [CrossRef] [PubMed]
- Ai, Z.; Lu, Y.; Qiu, S.; Fan, Z. Overcoming cisplatin resistance of ovarian cancer cells by targeting HIF-1-regulated cancer metabolism. Cancer Lett. 2016, 373, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Koshikawa, N.; Maejima, C.; Miyazaki, K.; Nakagawara, A.; Takenaga, K. Hypoxia selects for high-metastatic lewis lung carcinoma cells overexpressing mcl-1 and exhibiting reduced apoptotic potential in solid tumors. Oncogene 2006, 25, 917–928. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.Y.; Tsai, M.H.; Osmanian, C.; Graeber, T.G.; Lee, J.E.; Giffard, R.G.; DiPaolo, J.A.; Peehl, D.M.; Giaccia, A.J. Selection of human cervical epithelial cells that possess reduced apoptotic potential to low-oxygen conditions. Cancer Res. 1997, 57, 4200–4204. [Google Scholar] [PubMed]
- Sabapathy, K.; Lane, D.P. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. Apr-246 reactivates mutant p53 by targeting cysteines 124 and 277. BioRxiv 2017. [Google Scholar] [CrossRef]
- Perdrix, A.; Najem, A.; Saussez, S.; Awada, A.; Journe, F.; Ghanem, G.; Krayem, M. PRIMA-1 and PRIMA-1(met) (APR-246): From mutant/wild type p53 reactivation to unexpected mechanisms underlying their potent anti-tumor effect in combinatorial therapies. Cancers 2017, 9, 172. [Google Scholar] [CrossRef] [PubMed]
- Rieber, M.; Strasberg-Rieber, M. Hypoxia, Mn-SOD and H2O2 regulate p53 reactivation and PRIMA-1 toxicity irrespective of p53 status in human breast cancer cells. Biochem. Pharmacol. 2012, 84, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. Prima-1(met) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef] [PubMed]
- Mohell, N.; Alfredsson, J.; Fransson, A.; Uustalu, M.; Bystrom, S.; Gullbo, J.; Hallberg, A.; Bykov, V.J.; Bjorklund, U.; Wiman, K.G. Apr-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 2015, 6, e1794. [Google Scholar] [CrossRef] [PubMed]
- Fransson, A.; Glaessgen, D.; Alfredsson, J.; Wiman, K.G.; Bajalica-Lagercrantz, S.; Mohell, N. Strong synergy with APR-246 and DNA-damaging drugs in primary cancer cells from patients with TP53 mutant high-grade serous ovarian cancer. J. Ovarian Res. 2016, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Jiang, Z.F.; Ding, P.S.; Shao, L.J.; Liu, R.Y. Hypoxia-induced autophagy mediates cisplatin resistance in lung cancer cells. Sci. Rep. 2015, 5, 12291. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Melino, G. The p53 family and the hypoxia-inducible factors (HIFs): Determinants of cancer progression. Trends Biochem. Sci. 2015, 40, 425–434. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar]
- Zhao, T.; Zhu, Y.; Morinibu, A.; Kobayashi, M.; Shinomiya, K.; Itasaka, S.; Yoshimura, M.; Guo, G.; Hiraoka, M.; Harada, H. HIF-1-mediated metabolic reprogramming reduces ROS levels and facilitates the metastatic colonization of cancers in lungs. Sci. Rep. 2014, 4, 3793. [Google Scholar] [CrossRef] [PubMed]
- Solaini, G.; Baracca, A.; Lenaz, G.; Sgarbi, G. Hypoxia and mitochondrial oxidative metabolism. Biochim. Biophys. Acta 2010, 1797, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.N.; Yang, W.K.; Kim, J.; Kim, H.S.; Kim, E.J.; Yun, H.; Park, H.; Kim, S.S.; Choe, W.; Kang, I.; et al. Reactive oxygen species stabilize hypoxia-inducible factor-1 alpha protein and stimulate transcriptional activity via amp-activated protein kinase in du145 human prostate cancer cells. Carcinogenesis 2008, 29, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: New developments in an old debate. J. Cell Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef] [PubMed]
- The ros scavenger nac reduced hif-1α levels under hypoxia in response to cisplatin treatment, Christophe Deben. University of Antwerp: Wilrijk, Belgium, Unpublished data. 2018.
- Tessoulin, B.; Descamps, G.; Moreau, P.; Maiga, S.; Lode, L.; Godon, C.; Marionneau-Lambot, S.; Oullier, T.; Le Gouill, S.; Amiot, M.; et al. Prima-1met induces myeloma cell death independent of p53 by impairing the gsh/ros balance. Blood 2014, 124, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Soderqvist, M.; Segerback, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Pauwels, B.; Korst, A.E.; de Pooter, C.M.; Pattyn, G.G.; Lambrechts, H.A.; Baay, M.F.; Lardon, F.; Vermorken, J.B. Comparison of the sulforhodamine b assay and the clonogenic assay for in vitro chemoradiation studies. Cancer Chemother. Pharmacol. 2003, 51, 221–226. [Google Scholar] [PubMed]
- Jonsson, E.; Fridborg, H.; Nygren, P.; Larsson, R. Synergistic interactions of combinations of topotecan with standard drugs in primary cultures of human tumor cells from patients. Eur. J. Clin. Pharmacol. 1998, 54, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Valeriote, F.; Lin, H. Synergistic interaction of anticancer agents: A cellular perspective. Cancer Chemother. Rep. 1975, 59, 895–900. [Google Scholar] [PubMed]
- Pauwels, B.; Korst, A.E.; Andriessen, V.; Baay, M.F.; Pattyn, G.G.; Lambrechts, H.A.; Pooter, C.M.; Lardon, F.; Vermorken, J.B. Unraveling the mechanism of radiosensitization by gemcitabine: The role of tp53. Radiat. Res. 2005, 164, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Deben, C.; Wouters, A.; Op de Beeck, K.; van Den Bossche, J.; Jacobs, J.; Zwaenepoel, K.; Peeters, M.; Van Meerbeeck, J.; Lardon, F.; Rolfo, C.; et al. The MDM2-inhibitor nutlin-3 synergizes with cisplatin to induce p53 dependent tumor cell apoptosis in non-small cell lung cancer. Oncotarget 2015, 6, 22666–22679. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deben, C.; Deschoolmeester, V.; De Waele, J.; Jacobs, J.; Van den Bossche, J.; Wouters, A.; Peeters, M.; Rolfo, C.; Smits, E.; Lardon, F.; et al. Hypoxia-Induced Cisplatin Resistance in Non-Small Cell Lung Cancer Cells Is Mediated by HIF-1α and Mutant p53 and Can Be Overcome by Induction of Oxidative Stress. Cancers 2018, 10, 126. https://doi.org/10.3390/cancers10040126
Deben C, Deschoolmeester V, De Waele J, Jacobs J, Van den Bossche J, Wouters A, Peeters M, Rolfo C, Smits E, Lardon F, et al. Hypoxia-Induced Cisplatin Resistance in Non-Small Cell Lung Cancer Cells Is Mediated by HIF-1α and Mutant p53 and Can Be Overcome by Induction of Oxidative Stress. Cancers. 2018; 10(4):126. https://doi.org/10.3390/cancers10040126
Chicago/Turabian StyleDeben, Christophe, Vanessa Deschoolmeester, Jorrit De Waele, Julie Jacobs, Jolien Van den Bossche, An Wouters, Marc Peeters, Christian Rolfo, Evelien Smits, Filip Lardon, and et al. 2018. "Hypoxia-Induced Cisplatin Resistance in Non-Small Cell Lung Cancer Cells Is Mediated by HIF-1α and Mutant p53 and Can Be Overcome by Induction of Oxidative Stress" Cancers 10, no. 4: 126. https://doi.org/10.3390/cancers10040126
APA StyleDeben, C., Deschoolmeester, V., De Waele, J., Jacobs, J., Van den Bossche, J., Wouters, A., Peeters, M., Rolfo, C., Smits, E., Lardon, F., & Pauwels, P. (2018). Hypoxia-Induced Cisplatin Resistance in Non-Small Cell Lung Cancer Cells Is Mediated by HIF-1α and Mutant p53 and Can Be Overcome by Induction of Oxidative Stress. Cancers, 10(4), 126. https://doi.org/10.3390/cancers10040126