Insights of Crosstalk between p53 Protein and the MKK3/MKK6/p38 MAPK Signaling Pathway in Cancer


Abstract
:1. Introduction
2. TP53: Apoptosis and More
3. MKK3/MKK6/P38 Signaling in Cancer: Friend or Foe
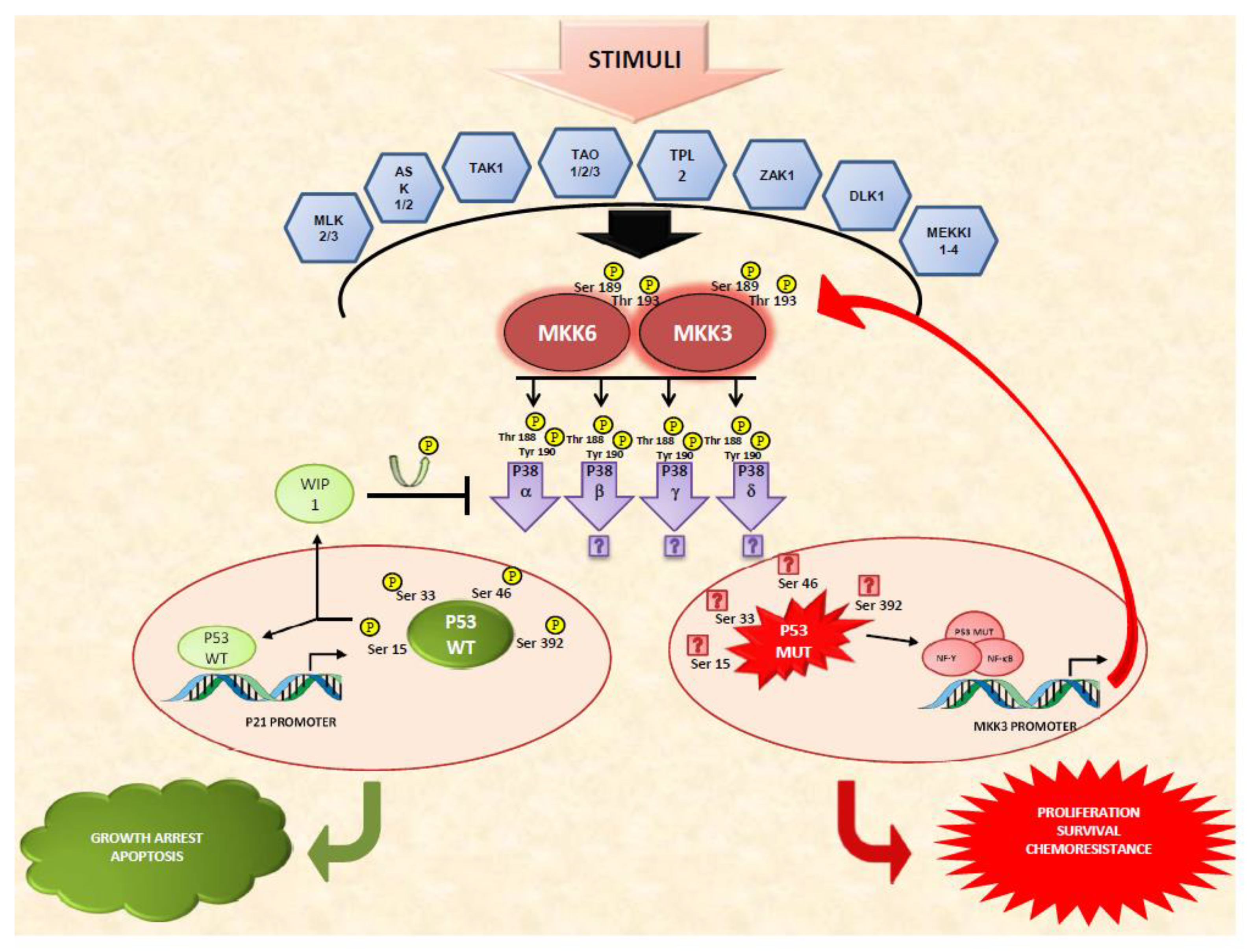
4. Two Pathways Intersect: What Is the Outcome
5. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Corre, I.; Paris, F.; Huot, J. The p38 pathway, a major pleiotropic cascade that transduces stress and metastatic signals in endothelial cells. Oncotarget 2017, 8, 55684–55714. [Google Scholar] [CrossRef] [PubMed]
- Keshet, Y.; Seger, R. The map kinase signaling cascades: A system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 2010, 661, 3–38. [Google Scholar] [PubMed]
- Bulavin, D.V.; Saito, S.; Hollander, M.C.; Sakaguchi, K.; Anderson, C.W.; Appella, E.; Fornace, A.J. Phosphorylation of human p53 by p38 kinase coordinates n-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J. 1999, 18, 6845–6854. [Google Scholar] [CrossRef] [PubMed]
- Saba-El-Leil, M.K.; Frémin, C.; Meloche, S. Redundancy in the world of map kinases: All for one. Front. Cell Dev. Biol. 2016, 4, 67. [Google Scholar] [CrossRef] [PubMed]
- Beckta, J.M.; Ahmad, S.F.; Yang, H.; Valerie, K. Revisiting p53 for cancer-specific chemo- and radiotherapy: Ten years after. Cell Cycle 2014, 13, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2017, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Mello, S.S.; Attardi, L.D. Deciphering p53 signaling in tumor suppression. Curr. Opin. Cell Biol. 2017, 51, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Hager, K.M.; Gu, W. Understanding the non-canonical pathways involved in p53-mediated tumor suppression. Carcinogenesis 2014, 35, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Engelberg, D. Stress-activated protein kinases-tumor suppressors or tumor initiators? Semin. Cancer Biol. 2004, 14, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Blank, M. Targeting p38 map kinase signaling in cancer through post-translational modifications. Cancer Lett. 2017, 384, 19–26. [Google Scholar] [CrossRef] [PubMed]
- García-Cano, J.; Roche, O.; Cimas, F.J.; Pascual-Serra, R.; Ortega-Muelas, M.; Fernández-Aroca, D.M.; Sánchez-Prieto, R. P38MAPK and chemotherapy: We always need to hear both sides of the story. Front. Cell Dev. Biol. 2016, 4, 69. [Google Scholar] [CrossRef] [PubMed]
- Baldari, S.; Ubertini, V.; Garufi, A.; D’Orazi, G.; Bossi, G. Targeting MKK3 as a novel anticancer strategy: Molecular mechanisms and therapeutical implications. Cell Death Dis. 2015, 6, e1621. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, A.; Starace, G.; Norelli, G.; Piaggio, G.; Sacchi, A.; Bossi, G. Mutant p53-induced up-regulation of mitogen-activated protein kinase kinase 3 contributes to gain of function. J. Biol. Chem. 2010, 285, 14160–14169. [Google Scholar] [CrossRef] [PubMed]
- Bossi, G. MKK as oncotarget. Aging (Albany NY) 2016, 8, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Bossi, G.; Marampon, F.; Maor-Aloni, R.; Zani, B.; Rotter, V.; Oren, M.; Strano, S.; Blandino, G.; Sacchi, A. Conditional RNA interference in vivo to study mutant p53 oncogenic gain of function on tumor malignancy. Cell Cycle 2008, 7, 1870–1879. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Zhang, Y.; Lozano, G. Mutant p53: Multiple mechanisms define biologic activity in cancer. Front. Oncol. 2015, 5, 249. [Google Scholar] [CrossRef] [PubMed]
- Bossi, G.; Lapi, E.; Strano, S.; Rinaldo, C.; Blandino, G.; Sacchi, A. Mutant p53 gain of function: Reduction of tumor malignancy of human cancer cell lines through abrogation of mutant p53 expression. Oncogene 2006, 25, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.; Wiman, K.G. Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett. 2014, 588, 2622–2627. [Google Scholar] [CrossRef] [PubMed]
- Valente, L.J.; Gray, D.H.; Michalak, E.M.; Pinon-Hofbauer, J.; Egle, A.; Scott, C.L.; Janic, A.; Strasser, A. P53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep. 2013, 3, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Hickman, J.H.; Wang, S.J.; Gu, W. Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle 2015, 14, 2881–2885. [Google Scholar] [CrossRef] [PubMed]
- Ubertini, V.; Norelli, G.; D’Arcangelo, D.; Gurtner, A.; Cesareo, E.; Baldari, S.; Gentileschi, M.P.; Piaggio, G.; Nisticò, P.; Soddu, S.; et al. Mutant p53 gains new function in promoting inflammatory signals by repression of the secreted interleukin-1 receptor antagonist. Oncogene 2015, 34, 2493–2504. [Google Scholar] [CrossRef] [PubMed]
- Cuenda, A.; Sanz-Ezquerro, J.J. P38γ and p38δ: From spectators to key physiological players. Trends Biochem. Sci. 2017, 42, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Escós, A.; Risco, A.; Alsina-Beauchamp, D.; Cuenda, A. P38γ and p38δ mitogen activated protein kinases (MAPKs), new stars in the MAPK galaxy. Front. Cell Dev. Biol. 2016, 4, 31. [Google Scholar] [CrossRef] [PubMed]
- Cerezo-Guisado, M.I.; del Reino, P.; Remy, G.; Kuma, Y.; Arthur, J.S.; Gallego-Ortega, D.; Cuenda, A. Evidence of p38γ and p38δ involvement in cell transformation processes. Carcinogenesis 2011, 32, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Beenstock, J.; Ben-Yehuda, S.; Melamed, D.; Admon, A.; Livnah, O.; Ahn, N.G.; Engelberg, D. The p38β mitogen-activated protein kinase possesses an intrinsic autophosphorylation activity, generated by a short region composed of the α-G helix and MAPK insert. J. Biol. Chem. 2014, 289, 23546–23556. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.F.; Martin, E.D.; Chaikuad, A.; Bassi, R.; Clark, J.; Martino, L.; Verma, S.; Sicard, P.; Tata, R.; Atkinson, R.A.; et al. Mechanism and consequence of the autoactivation of p38α mitogen-activated protein kinase promoted by tab1. Nat. Struct. Mol. Biol. 2013, 20, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Mittelstadt, P.R.; Salvador, J.M.; Fornace, A.J.; Ashwell, J.D. Activating p38 MAPK: New tricks for an old kinase. Cell Cycle 2005, 4, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Brancho, D.; Tanaka, N.; Jaeschke, A.; Ventura, J.J.; Kelkar, N.; Tanaka, Y.; Kyuuma, M.; Takeshita, T.; Flavell, R.A.; Davis, R.J. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003, 17, 1969–1978. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Zhao, Y.; Zhang, J.; Li, L.; Zou, L.; Yao, Y.; Xu, Y. β-Elemene inhibits proliferation of human glioblastoma cells and causes cell-cycle G0/G1 arrest via mutually compensatory activation of MKK3 and MKK6. Int. J. Oncol. 2011, 38, 419–426. [Google Scholar] [PubMed]
- Boyle, D.L.; Hammaker, D.; Edgar, M.; Zaiss, M.M.; Teufel, S.; David, J.P.; Schett, G.; Firestein, G.S. Differential roles of MAPK kinases MKK3 and MKK6 in osteoclastogenesis and bone loss. PLoS ONE 2014, 9, e84818. [Google Scholar] [CrossRef]
- Galan-Moya, E.M.; de la Cruz-Morcillo, M.A.; Llanos Valero, M.; Callejas-Valera, J.L.; Melgar-Rojas, P.; Hernadez Losa, J.; Salcedo, M.; Fernández-Aramburo, A.; Ramon y Cajal, S.; Sánchez-Prieto, R. Balance between MKK6 and MKK3 mediates p38 MAPK associated resistance to cisplatin in nsclc. PLoS ONE 2011, 6, e28406. [Google Scholar] [CrossRef] [PubMed]
- Remy, G.; Risco, A.M.; Iñesta-Vaquera, F.A.; González-Terán, B.; Sabio, G.; Davis, R.J.; Cuenda, A. Differential activation of p38MAPK isoforms by MKK6 and MKK3. Cell Signal. 2010, 22, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.; Hong, L.; Liao, R.; Deng, Q.; Han, J.; Sun, P. P38alpha and p38gamma mediate oncogenic Ras-induced senescence through differential mechanisms. J. Biol. Chem. 2009, 284, 11237–11246. [Google Scholar] [CrossRef] [PubMed]
- Lovett, F.A.; Cosgrove, R.A.; Gonzalez, I.; Pell, J.M. Essential role for p38alpha MAPK but not p38gamma MAPK in Igf2 expression and myoblast differentiation. Endocrinology 2010, 151, 4368–4380. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Pohl, N.M.; Loesch, M.; Hou, S.; Li, R.; Qin, J.Z.; Cuenda, A.; Chen, G. P38alpha antagonizes p38gamma activity through c-Jun-dependent ubiquitin-proteasome pathways in regulating Ras transformation and stress response. J. Biol. Chem. 2007, 282, 31398–31408. [Google Scholar] [CrossRef] [PubMed]
- Pramanik, R.; Qi, X.; Borowicz, S.; Choubey, D.; Schultz, R.M.; Han, J.; Chen, G. P38 isoforms have opposite effects on ap-1-dependent transcription through regulation of c-Jun. The determinant roles of the isoforms in the p38 MAPK signal specificity. J. Biol. Chem. 2003, 278, 4831–4839. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Ferraris, J.D.; Dmitrieva, N.I.; Liu, Y.; Burg, M.B. Mkp-1 inhibits high NaCl-induced activation of p38 but does not inhibit the activation of TonRBP/OREBP: Opposite roles of p38alpha and p38delta. Proc. Natl. Acad. Sci. USA 2008, 105, 5620–5625. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Haluska, P.; Tolcher, A.W.; Erlichman, C.; Papadopoulos, K.P.; Lensing, J.L.; Beeram, M.; Molina, J.R.; Rasco, D.W.; Arcos, R.R.; et al. A first-in-human phase I study of the oral p38 MAPK inhibitor, ralimetinib (LY2228820 Dimesylate), in patients with advanced cancer. Clin. Cancer Res. 2016, 22, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; Ala-Aho, R.; Jokilehto, T.; Peltonen, J.; Kallajoki, M.; Grenman, R.; Jaakkola, P.; Westermarck, J.; Kähäri, V.M. P38alpha and p38delta mitogen-activated protein kinase isoforms regulate invasion and growth of head and neck squamous carcinoma cells. Oncogene 2007, 26, 5267–5279. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Behera, R.; Lohite, K.; Karnik, S.; Kundu, G.C. P38 kinase is crucial for osteopontin-induced furin expression that supports cervical cancer progression. Cancer Res. 2010, 70, 10381–10391. [Google Scholar] [CrossRef] [PubMed]
- Song, W.J.; Dong, Y.; Luo, C.; Chen, Y.Y. P38MAPK family isoform p38α and activating transcription factor 2 are associated with the malignant phenotypes and poor prognosis of patients with ovarian adenocarcinoma. Pathol. Res. Pract. 2017, 213, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Del Reino, P.; Alsina-Beauchamp, D.; Escós, A.; Cerezo-Guisado, M.I.; Risco, A.; Aparicio, N.; Zur, R.; Fernandez-Estévez, M.; Collantes, E.; Montans, J.; et al. Pro-oncogenic role of alternative p38 mitogen-activated protein kinases p38γ and p38δ, linking inflammation and cancer in colitis-associated colon cancer. Cancer Res. 2014, 74, 6150–6160. [Google Scholar] [CrossRef] [PubMed]
- Kuma, Y.; Sabio, G.; Bain, J.; Shpiro, N.; Márquez, R.; Cuenda, A. BIRB796 inhibits all p38 MAPK isoforms in vitro and in vivo. J. Biol. Chem. 2005, 280, 19472–19479. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, Z.; Stebbins, J.L.; Zhang, X.; Hoffman, R.; Moore, A.; Pellecchia, M. A fragment-based approach for the discovery of isoform-specific p38alpha inhibitors. ACS Chem. Biol. 2007, 2, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.; Kobayashi, T.; Lawson, J.D.; Saitoh, M.; Shimokawa, K.; Bigi, S.V.; Hixon, M.S.; Smith, C.R.; Tatamiya, T.; Goto, M.; et al. Fragment-based drug discovery of potent and selective MKK3/6 inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 1086–1089. [Google Scholar] [CrossRef] [PubMed]
- Huth, H.W.; Albarnaz, J.D.; Torres, A.A.; Bonjardim, C.A.; Ropert, C. MEK2 controls the activation of MKK3/MKK6-p38 axis involved in the MDA-MB-231 breast cancer cell survival: Correlation with cyclin D1 expression. Cell Signal. 2016, 28, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- MacNeil, A.J.; Jiao, S.C.; McEachern, L.A.; Yang, Y.J.; Dennis, A.; Yu, H.; Xu, Z.; Marshall, J.S.; Lin, T.J. MAPK kinase 3 is a tumor suppressor with reduced copy number in breast cancer. Cancer Res. 2014, 74, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Parray, A.A.; Baba, R.A.; Bhat, H.F.; Wani, L.; Mokhdomi, T.A.; Mushtaq, U.; Bhat, S.S.; Kirmani, D.; Kuchay, S.; Wani, M.M.; et al. MKK6 is upregulated in human esophageal, stomach, and colon cancers. Cancer Invest. 2014, 32, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Lotan, T.L.; Lyon, M.; Huo, D.; Taxy, J.B.; Brendler, C.; Foster, B.A.; Stadler, W.; Rinker-Schaeffer, C.W. Up-regulation of MKK4, MKK6 and MKK7 during prostate cancer progression: An important role for SAPK signalling in prostatic neoplasia. J. Pathol. 2007, 212, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Shin, I.; Kim, S.; Song, H.; Kim, H.R.; Moon, A. H-Ras-specific activation of RAC-MKK3/6-p38 pathway: Its critical role in invasion and migration of breast epithelial cells. J. Biol. Chem. 2005, 280, 14675–14683. [Google Scholar] [CrossRef] [PubMed]
- Hickson, J.A.; Huo, D.; Vander Griend, D.J.; Lin, A.; Rinker-Schaeffer, C.W.; Yamada, S.D. The p38 kinases MKK4 and MKK6 suppress metastatic colonization in human ovarian carcinoma. Cancer Res. 2006, 66, 2264–2270. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Kang, J.; Wang, X.; Jiang, W.; Liao, H.; Yuan, J. TAT-OSBP-1-MKK6(e), a novel TAT-fusion protein with high selectivity for human ovarian cancer, exhibits anti-tumor activity. Med. Oncol. 2015, 32, 118. [Google Scholar] [CrossRef] [PubMed]
- Cerezo-Guisado, M.I.; Zur, R.; Lorenzo, M.J.; Risco, A.; Martín-Serrano, M.A.; Alvarez-Barrientos, A.; Cuenda, A.; Centeno, F. Implication of Akt, ERK1/2 and alternative p38MAPK signalling pathways in human colon cancer cell apoptosis induced by green tea EGCG. Food Chem. Toxicol. 2015, 84, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Rovida, E.; Stecca, B. Mitogen-activated protein kinases and Hedgehog-GLI signaling in cancer: A crosstalk providing therapeutic opportunities? Semin. Cancer Biol. 2015, 35, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.A.; Gonzalez-Pecchi, V.; Khuri, L.F.; Niu, Q.; Wang, Y.; Xu, Y.; Bai, Y.; Mo, X.; Prochownik, E.V.; Johns, M.A.; et al. OncoPPi-informed discovery of mitogen-activated protein kinase kinase 3 as a novel binding partner of c-Myc. Oncogene 2017, 36, 5852–5860. [Google Scholar] [CrossRef] [PubMed]
- Serra, C.; Palacios, D.; Mozzetta, C.; Forcales, S.V.; Morantte, I.; Ripani, M.; Jones, D.R.; Du, K.; Jhala, U.S.; Simone, C.; et al. Functional interdependence at the chromatin level between the MKK6/p38 and IGF1/PI3k/AKT pathways during muscle differentiation. Mol. Cell 2007, 28, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Stepniak, E.; Hui, L.; Leibbrandt, A.; Katada, T.; Nishina, H.; Wagner, E.F.; Penninger, J.M. Antagonistic control of cell fates by JNK and p38-MAPK signaling. Cell Death Differ. 2008, 15, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPKpathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Loesch, M.; Zhi, H.Y.; Hou, S.W.; Qi, X.M.; Li, R.S.; Basir, Z.; Iftner, T.; Cuenda, A.; Chen, G. P38gamma MAPK cooperates with c-Jun in trans-activating matrix metalloproteinase 9. J. Biol. Chem. 2010, 285, 15149–15158. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.; Chen, M.; Lv, D.; Luo, N.; Su, W.; Xiang, R.; Sun, P. Induction of p38δ expression plays an essential role in oncogenic Ras-induced senescence. Mol. Cell. Biol. 2013, 33, 3780–3794. [Google Scholar] [CrossRef] [PubMed]
- Takekawa, M.; Adachi, M.; Nakahata, A.; Nakayama, I.; Itoh, F.; Tsukuda, H.; Taya, Y.; Imai, K. P53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 2000, 19, 6517–6526. [Google Scholar] [CrossRef] [PubMed]
- Kishi, H.; Nakagawa, K.; Matsumoto, M.; Suga, M.; Ando, M.; Taya, Y.; Yamaizumi, M. Osmotic shock induces G1 arrest through p53 phosphorylation at Ser33 by activated p38MAPK without phosphorylation at Ser15 and Ser20. J. Biol. Chem. 2001, 276, 39115–39122. [Google Scholar] [CrossRef] [PubMed]
- Lafarga, V.; Cuadrado, A.; Lopez de Silanes, I.; Bengoechea, R.; Fernandez-Capetillo, O.; Nebreda, A.R. P38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G(1)/s checkpoint. Mol. Cell. Biol. 2009, 29, 4341–4351. [Google Scholar] [CrossRef] [PubMed]
- Hrstka, R.; Bouchalova, P.; Michalova, E.; Matoulkova, E.; Muller, P.; Coates, P.J.; Vojtesek, B. AGR2 oncoprotein inhibits p38 mapk and p53 activation through a DUSP10-mediated regulatory pathway. Mol. Oncol. 2016, 10, 652–662. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz-Morcillo, M.A.; Valero, M.L.; Callejas-Valera, J.L.; Arias-González, L.; Melgar-Rojas, P.; Galán-Moya, E.M.; García-Gil, E.; García-Cano, J.; Sánchez-Prieto, R. P38MSPK is a major determinant of the balance between apoptosis and autophagy triggered by 5-fluorouracil: Implication in resistance. Oncogene 2012, 31, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.J.; Chua, M.S.; So, S.K. Suppression of ATAD2 inhibits hepatocellular carcinoma progression through activation of p53- and p38-mediated apoptotic signaling. Oncotarget 2015, 6, 41722–41735. [Google Scholar] [CrossRef] [PubMed]
- Cannell, I.G.; Merrick, K.A.; Morandell, S.; Zhu, C.Q.; Braun, C.J.; Grant, R.A.; Cameron, E.R.; Tsao, M.S.; Hemann, M.T.; Yaffe, M.B. A pleiotropic RNA-binding protein controls distinct cell cycle checkpoints to drive resistance of p53-defective tumors to chemotherapy. Cancer Cell 2015, 28, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Morandell, S.; Reinhardt, H.C.; Cannell, I.G.; Kim, J.S.; Ruf, D.M.; Mitra, T.; Couvillon, A.D.; Jacks, T.; Yaffe, M.B. A reversible gene-targeting strategy identifies synthetic lethal interactions between MK2 and p53 in the DNA damage response in vivo. Cell Rep. 2013, 5, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Goloudina, A.R.; Tanoue, K.; Hammann, A.; Fourmaux, E.; Le Guezennec, X.; Bulavin, D.V.; Mazur, S.J.; Appella, E.; Garrido, C.; Demidov, O.N. Wip1 promotes RUNX2-dependent apoptosis in p53-negative tumors and protects normal tissues during treatment with anticancer agents. Proc. Natl. Acad. Sci. USA 2012, 109, E68–E75. [Google Scholar] [CrossRef] [PubMed]
- Goloudina, A.R.; Kochetkova, E.Y.; Pospelova, T.V.; Demidov, O.N. Wip1 phosphatase: Between p53 and MAPK kinases pathways. Oncotarget 2016, 7, 31563–31571. [Google Scholar] [CrossRef] [PubMed]
- Guma, M.; Hammaker, D.; Topolewski, K.; Corr, M.; Boyle, D.L.; Karin, M.; Firestein, G.S. Antiinflammatory functions of p38 in mouse models of rheumatoid arthritis: Advantages of targeting upstream kinases MKK-3 or MKK-6. Arthritis Rheumatol. 2012, 64, 2887–2895. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.M.; Menendez, D.; Fessler, M.B. A new inflammatory role for p53 in human macrophages. Cell Cycle 2014, 13, 2983–2984. [Google Scholar] [CrossRef] [PubMed]
- Perdrix, A.; Najem, A.; Saussez, S.; Awada, A.; Journe, F.; Ghanem, G.; Krayem, M. Prima-1 and prima-1. Cancers (Basel) 2017, 9, 172. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Study | Agent | Secondary Agent(s) | Target | Disease | Phase | Status |
|---|---|---|---|---|---|---|
| NCT01463631 | LY3007113 | N/A | p38 | Metastatic cancer | I | Completed |
| NCT02364206 | LY2228820 | TMZ, Radiotherapy | p38 | Glioblastoma | II | Active |
| NCT01663857 | LY2228820 | Carboplatin, Gemcitabine | p38 | Ovarian cancer | II | Active |
| NCT02322853 | LY2228820 | Tamoxifen | p38 | Breast cancer | II | Terminated |
| NCT01393990 | LY2228820 | N/A | p38 | Advanced cancer | I | Completed |
| NCT02860780 | LY2228820 | Prexasertib | p38 | Colorectal cancer, NSCLC | I | Completed |
| NCT00095680 | SCIO-469 | Bortezomib | p38 | Multiple Myeloma | II | Completed |
| NCT00087867 | SCIO-469 | Bortezomib | p38 | Multiple Myeloma | II | Completed |
| NCT00113893 | SCIO-469 | N/A | p38 | Myelodysplastic syndrome | II | Completed |
| NCT01496495 | ARRY-614 | N/A | p38/Tie2 | Myelodysplastic syndrome | I | Completed |
| NCT00916227 | ARRY-614 | N/A | p38/Tie2 | Myelodysplastic syndrome | I | Completed |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stramucci, L.; Pranteda, A.; Bossi, G. Insights of Crosstalk between p53 Protein and the MKK3/MKK6/p38 MAPK Signaling Pathway in Cancer. Cancers 2018, 10, 131. https://doi.org/10.3390/cancers10050131
Stramucci L, Pranteda A, Bossi G. Insights of Crosstalk between p53 Protein and the MKK3/MKK6/p38 MAPK Signaling Pathway in Cancer. Cancers. 2018; 10(5):131. https://doi.org/10.3390/cancers10050131
Chicago/Turabian StyleStramucci, Lorenzo, Angelina Pranteda, and Gianluca Bossi. 2018. "Insights of Crosstalk between p53 Protein and the MKK3/MKK6/p38 MAPK Signaling Pathway in Cancer" Cancers 10, no. 5: 131. https://doi.org/10.3390/cancers10050131
APA StyleStramucci, L., Pranteda, A., & Bossi, G. (2018). Insights of Crosstalk between p53 Protein and the MKK3/MKK6/p38 MAPK Signaling Pathway in Cancer. Cancers, 10(5), 131. https://doi.org/10.3390/cancers10050131