The EBV-Encoded Oncoprotein, LMP1, Induces an Epithelial-to-Mesenchymal Transition (EMT) via Its CTAR1 Domain through Integrin-Mediated ERK-MAPK Signalling

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Results
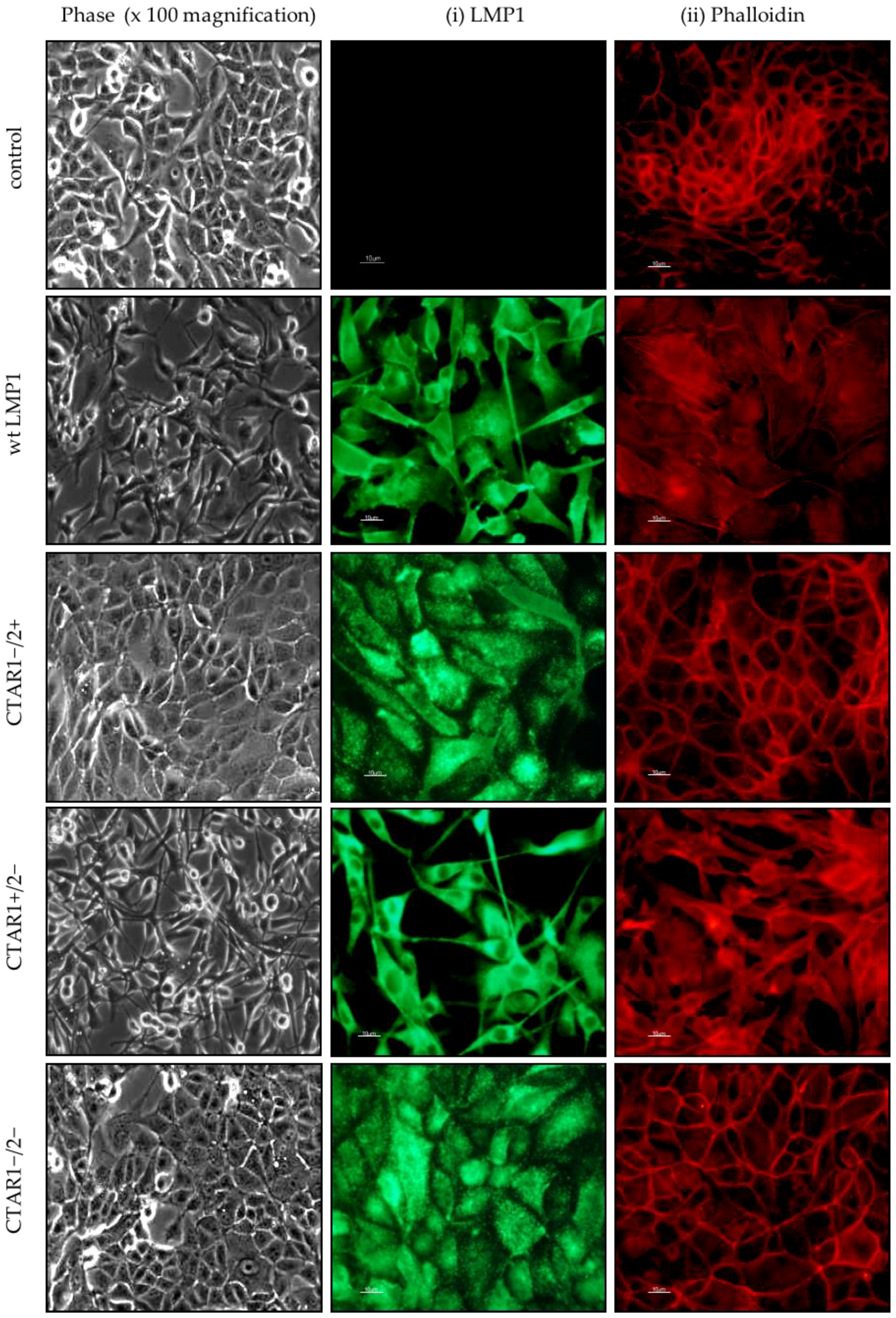
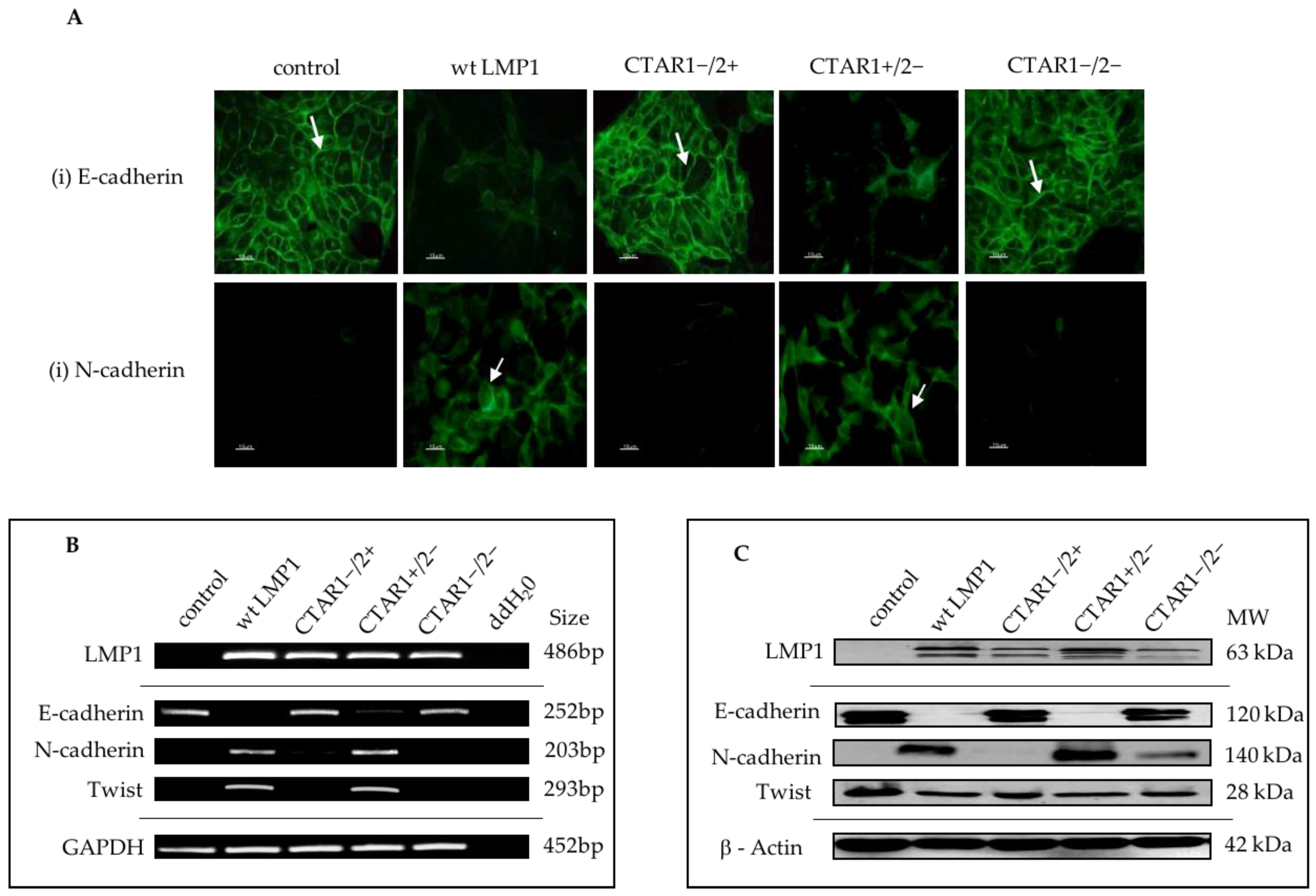
2.1. LMP1 Induces EMT in MDCK Cells via CTAR1
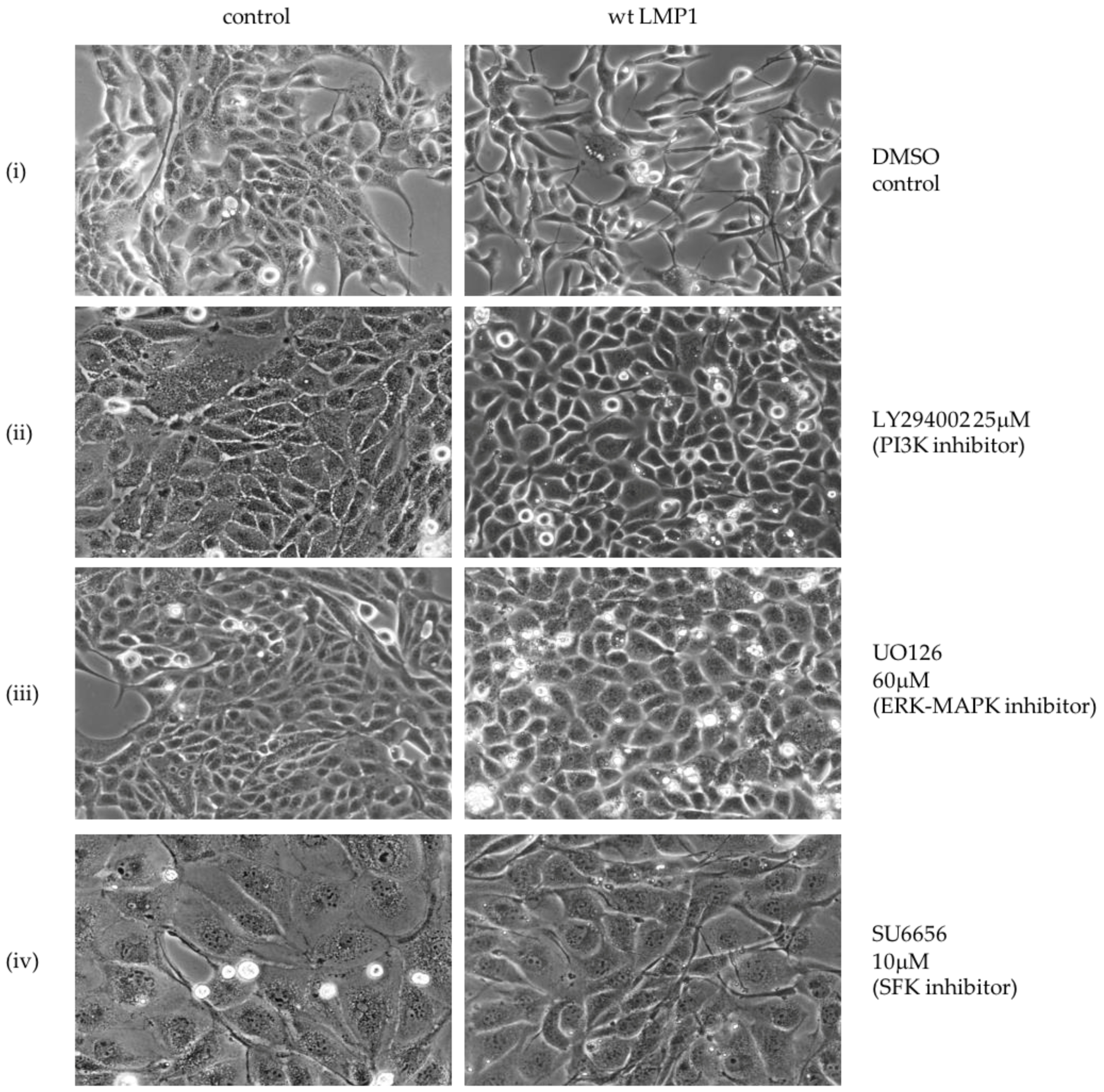
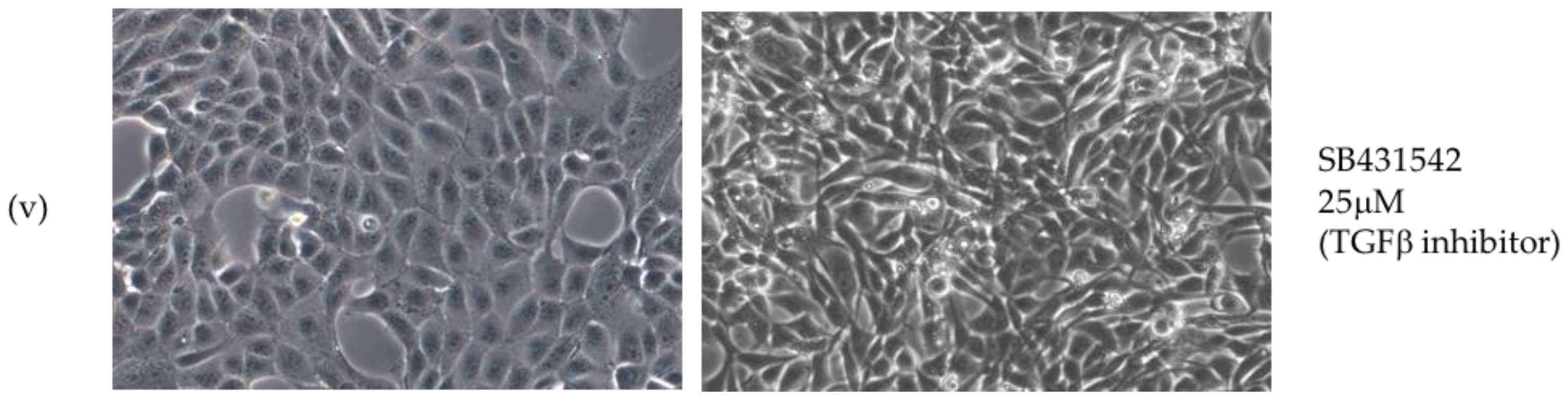
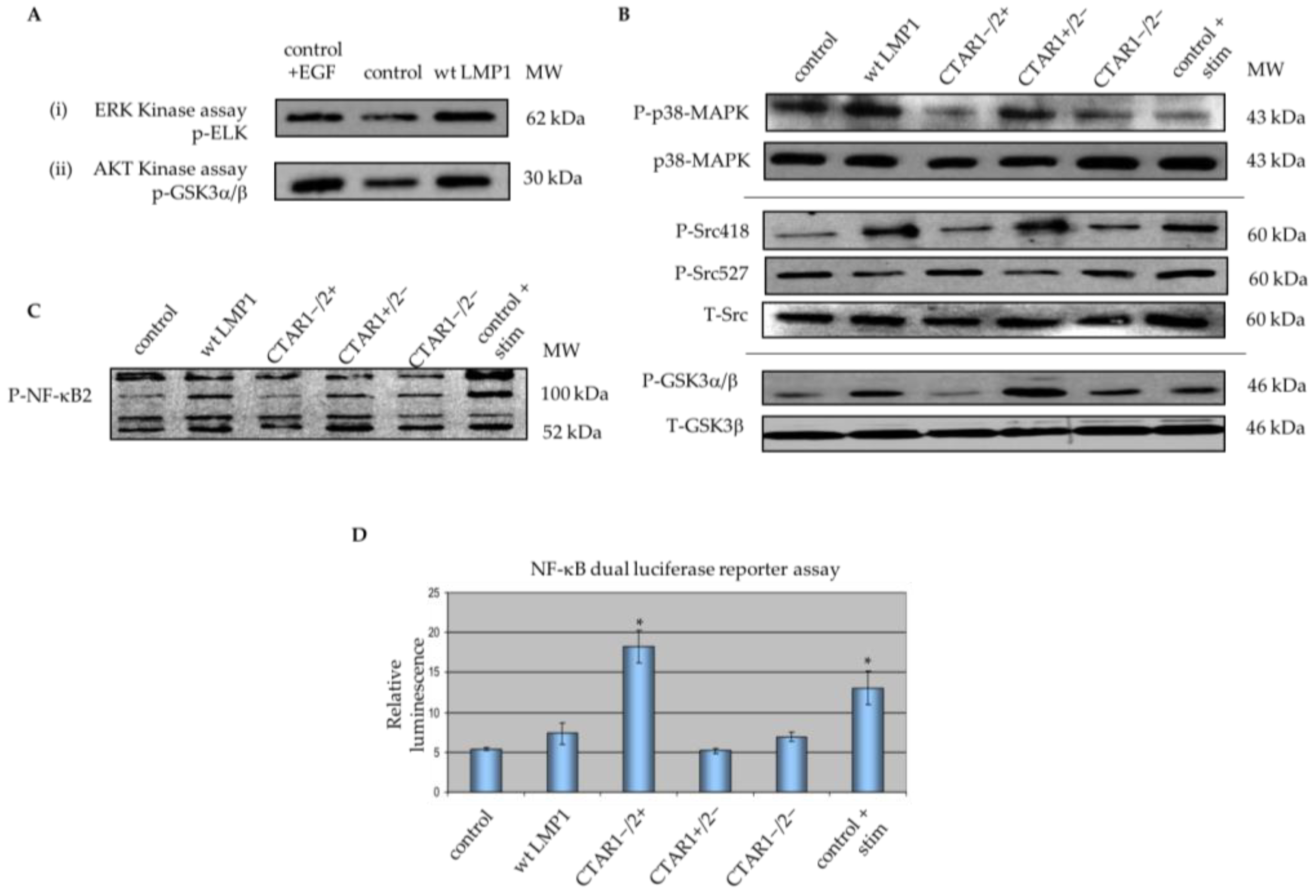
2.2. Critical Roles for ERK-MAPK, PI3-Kinase, Src Family Kinases (SFKs) But Not TGFβ Signalling Pathways in LMP1-Mediated EMT
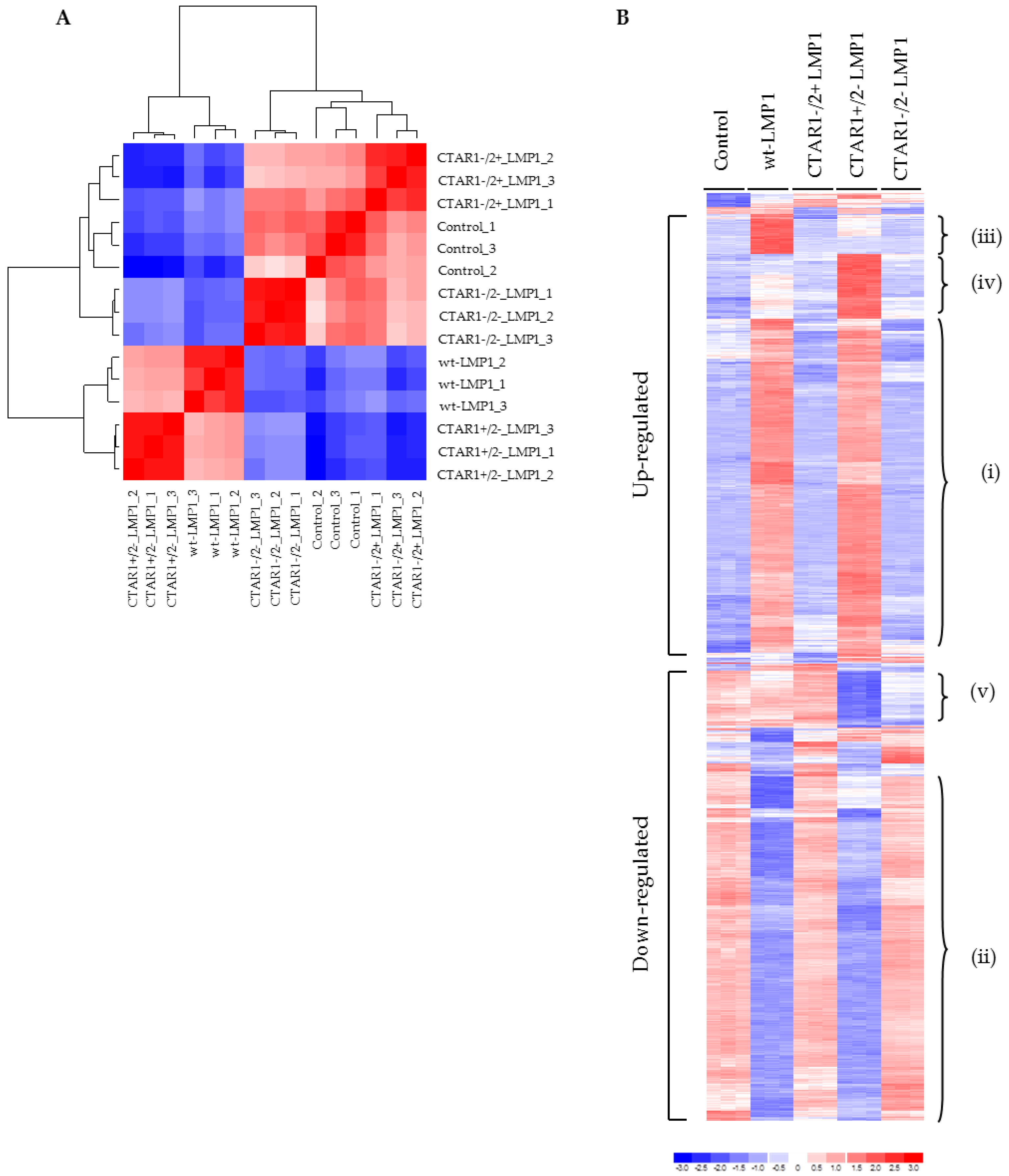
2.3. Global Gene Expression Analysis Reveals a Distinct Role for CTAR1 in Deregulating Genes from Key Signalling Pathways Implicated in EMT
2.4. Stable Expression of LMP1 in MDCK Cells Is Accompanied by the Complete Loss of Epithelial Markers and Upregulation of Mesenchymal Markers
2.5. LMP1 Deregulates Various Components of MAPK Signalling Implicated in EMT
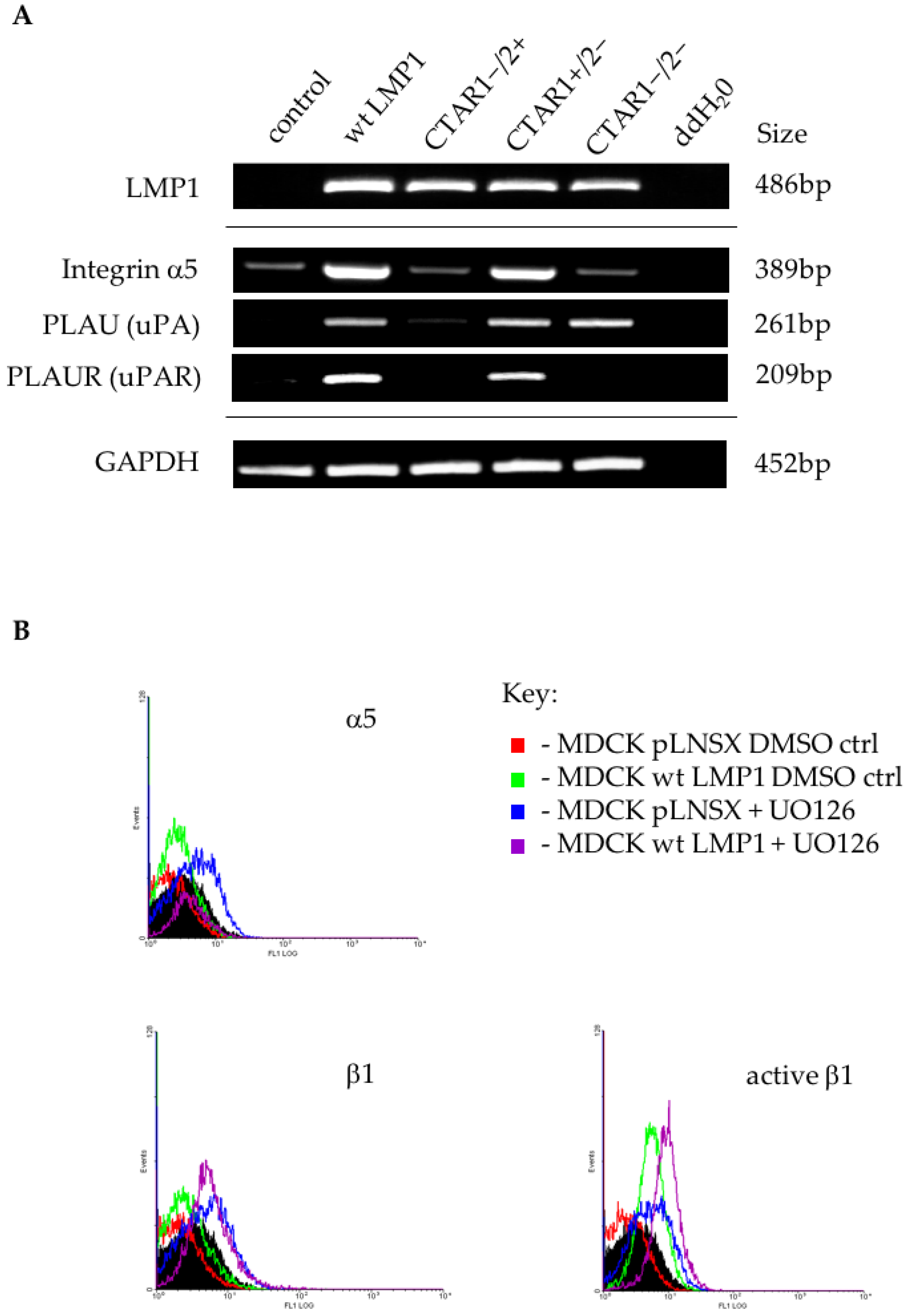
2.6. LMP1 Deregulates the Expression of Multiple Genes in the Integrin Signalling Pathway Implicated in the Generation of an EMT
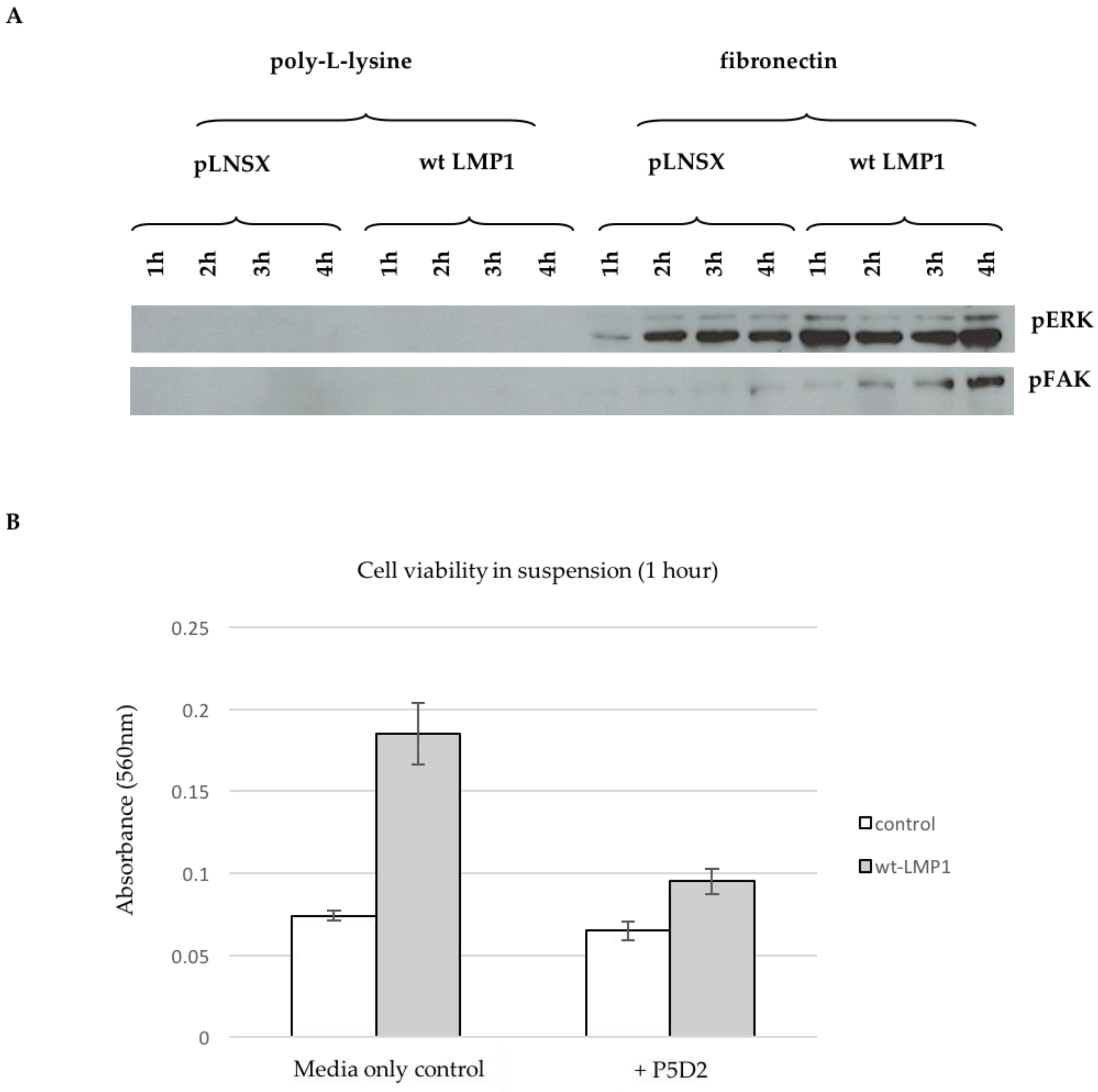
2.7. Ligand-Induced β1 Integrin Signalling Facilitates LMP1-Mediated ERK and FAK Phosphorylation, and Protects Epithelial Cells from Anoikis
3. Discussion
3.1. LMP1 Induces EMT in MDCK Cells via CTAR1 with Critical Roles for ERK-MAPK, PI3-Kinase, Src Family Kinases (SFKs) But Not TGFβ Signalling Pathways
3.2. Stable Expression of LMP1 in MDCK Cells is Accompanied by the Complete Loss of Epithelial Markers and Upregulation of Mesenchymal Markers
3.3. Microarray Analysis Identifies Deregulated Genes from Key Signalling Pathways Implicated in EMT, Including MAPK and Integrin Signalling Components
3.4. Ligand-Induced β1 Integrin Signalling Facilitates LMP1-Mediated ERK and FAK Phosphorylation, and Protects Epithelial Cells from Anoikis
3.5. EMT and NPC
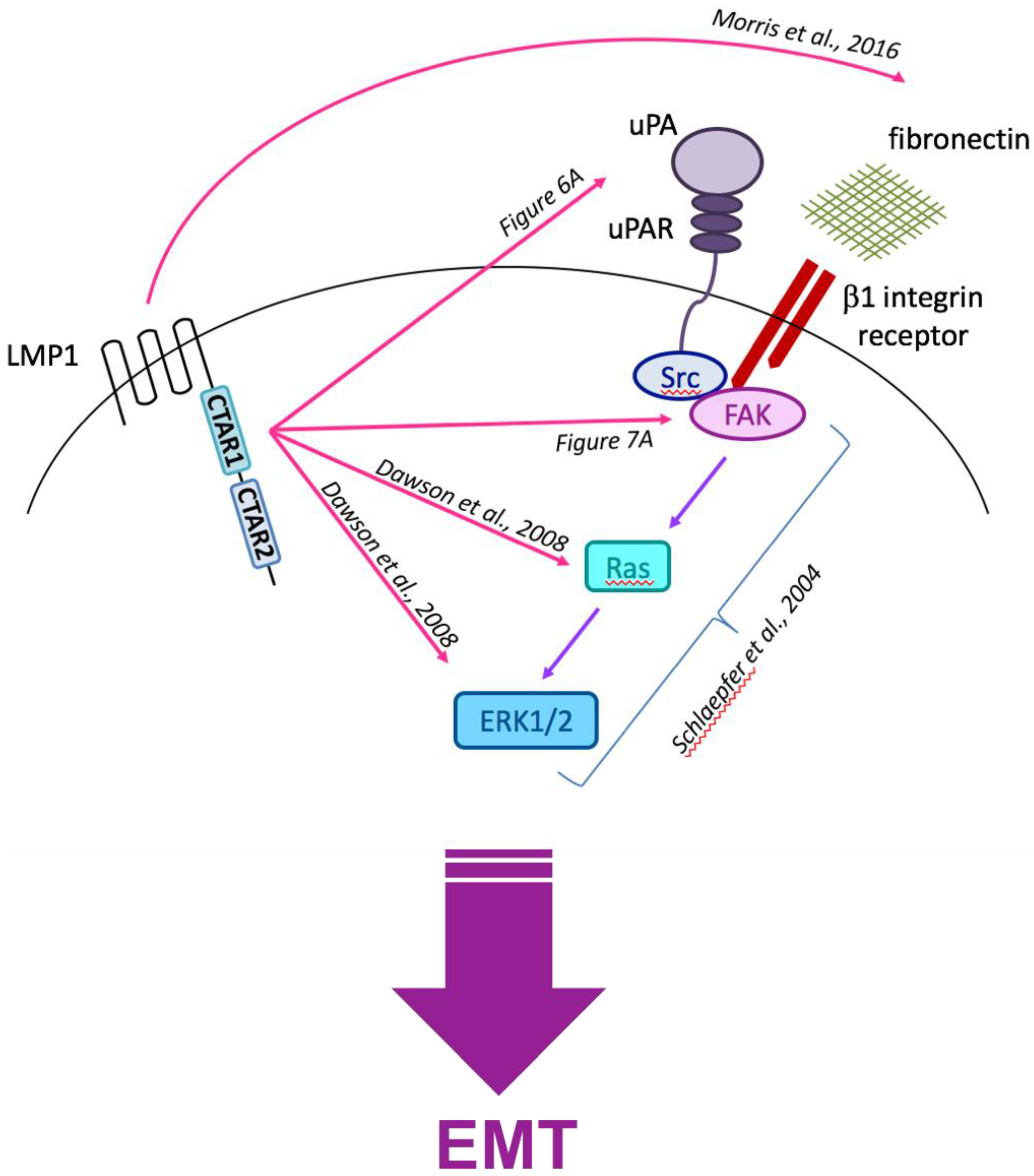
3.6. Current Working Hypothesis: LMP1-Mediated EMT Signals Converge at the Cell Surface with Integrin–Ligand Interactions
4. Materials and Methods
4.1. Cell Lines
4.2. Treatment with Selective Pharmacological Inhibitors
4.3. Transfections
4.4. Immunofluorescence Microscopy
4.5. Western Blotting
4.6. Reverse-Transcriptase Polymerase Chain Reaction (RT-PCR)
4.7. Non-Radioactive Kinase Assays
4.8. MTT Assay
4.9. Gene Expression Profiling
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Morris, M.A. Epstein-Barr Virus. In eLS; John Wiley & Sons, Ltd.: Chichester, UK, 2017; pp. 1–11. [Google Scholar]
- Young, L.S. Epstein-Barr virus at 50-future perspectives. Chin. J. Cancer 2014, 33, 527–528. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liebowitz, D.; Kieff, E. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell 1985, 43 Pt 2, 831–840. [Google Scholar] [CrossRef]
- Tao, Q.; Young, L.S.; Woodman, C.B.J.; Murray, P.G. Epstein-Barr virus (EBV) and its associated human cancers—Genetics, epigenetics, pathobiology and novel therapeutics. Front. Biosci. 2006, 11, 2672–2713. [Google Scholar] [CrossRef] [PubMed]
- Pathmanathan, R.; Prasad, U.; Chandrika, G.; Sadler, R.; Flynn, K.; Raab-Traub, N. Undifferentiated, nonkeratinizing, and squamous cell carcinoma of the nasopharynx. Variants of Epstein-Barr virus-infected neoplasia. Am. J. Pathol. 1995, 146, 1355–1367. [Google Scholar] [PubMed]
- Kieser, A.; Sterz, K.R. The latent membrane protein 1 (LMP1). Curr. Top. Microbiol. Immunol. 2015, 391, 119–149. [Google Scholar] [PubMed]
- Morris, M.A.; Dawson, C.W.; Laverick, L.; Davis, A.M.; Dudman, J.P.; Raveenthiraraj, S.; Ahmad, Z.; Yap, L.F.; Young, L.S. The Epstein-Barr virus encoded LMP1 oncoprotein modulates cell adhesion via regulation of activin A/TGFβ and β1 integrin signalling. Sci. Rep. 2016, 6, 19533. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, T.; Yang, J.; Kondo, S.; Yoshizaki, T.; Joab, I.; Furukawa, M.; Pagano, J.S. Twist and epithelial-mesenchymal transition are induced by the EBV oncoprotein latent membrane protein 1 and are associated with metastatic nasopharyngeal carcinoma. Cancer Res. 2007, 67, 1970–1978. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, T.; Yoshizaki, T.; Kondo, S.; Furukawa, M.; Kaizaki, Y.; Pagano, J.S. Epstein-Barr Virus latent membrane protein 1 induces Snail and epithelial-mesenchymal transition in metastatic nasopharyngeal carcinoma. Br. J. Cancer 2011, 104, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.R.; Yoshizaki, T.; Miyamori, H.; Hasegawa, K.; Horikawa, T.; Furukawa, M.; Harada, S.; Seiki, M.; Sato, H. Transformation of Madin-Darby canine kidney (MDCK) epithelial cells by Epstein-Barr virus latent membrane protein 1 (LMP1) induces expression of Ets1 and invasive growth. Oncogene 2000, 19, 1764–1771. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, V.; Lemieux, B.; Sawan, B.; Lichtensztejn, D.; Lichtensztejn, Z.; Wellinger, R.; Mai, S.; Knecht, H. LMP1 mediates multinuclearity through downregulation of shelterin proteins and formation of telomeric aggregates. Blood 2015, 125, 2101–2110. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Massagué, J. Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 2004, 118, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Kraut, N.; Beug, H. Molecular requirements for epithelial–mesenchymal transition during tumor progression. Curr. Opin. Cell Biol. 2005, 17, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Muthusamy, B.P.; Saeteurn, K.Y. Signaling pathway cooperation in TGF-β-induced epithelial–mesenchymal transition. Curr. Opin. Cell Biol. 2014, 31, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Kang, Y. Multilayer control of the EMT master regulators. Oncogene 2014, 33, 1755–1763. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K. Transforming growth factor-beta signaling in epithelial-mesenchymal transition and progression of cancer. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Quintanilla, M.; Cano, A. Transforming Growth Factor β-1 Induces Snail Transcription Factor in Epithelial Cell Lines. J. Biol. Chem. 2003, 278, 21113–21123. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E.; Pierschbacher, M.D. New perspectives in cell adhesion: RGD and integrins. Science 1987, 238, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Gil, D.; Ciołczyk-Wierzbicka, D.; Dulińska-Litewka, J.; Zwawa, K.; McCubrey, J.A.; Laidler, P. The mechanism of contribution of integrin linked kinase (ILK) to epithelial-mesenchymal transition (EMT). Adv. Enzym. Regul. 2011, 51, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Cicchini, C.; Laudadio, I.; Citarella, F.; Corazzari, M.; Steindler, C.; Conigliaro, A.; Fantoni, A.; Amicone, L.; Tripodi, M. TGFbeta-induced EMT requires focal adhesion kinase (FAK) signaling. Exp. Cell Res. 2008, 314, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Schober, M.; Fuchs, E. Tumor-initiating stem cells of squamous cell carcinomas and their control by TGF-β and integrin/focal adhesion kinase (FAK) signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 10544–10549. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.C.; Oloumi, A.; Mills, J.; Dobreva, I.; Maidan, M.; Gray, V.; Wederell, E.D.; Bally, M.B.; Foster, L.J.; Dedhar, S. Rictor and Integrin-Linked Kinase Interact and Regulate Akt Phosphorylation and Cancer Cell Survival. Cancer Res. 2008, 68, 1618–1624. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Law, B.K.; Chytil, A.M.; Brown, K.A.; Aakre, M.E.; Moses, H.L. Activation of the Erk Pathway Is Required for TGF-β1-Induced EMT In Vitro. Neoplasia 2004, 6, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kuiatse, I.; Lee, A.V.; Pan, J.; Giuliano, A.; Cui, X. Sustained c-Jun-NH2-kinase activity promotes epithelial-mesenchymal transition, invasion, and survival of breast cancer cells by regulating extracellular signal-regulated kinase activation. Mol. Cancer Res. 2010, 8, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-N.; Tsai, C.-L.; Tse, K.-P.; Chang, H.-Y.; Chang, Y.-S. The Epstein-Barr virus oncogene product, latent membrane protein 1, induces the downregulation of E-cadherin gene expression via activation of DNA methyltransferases. Proc. Natl. Acad. Sci. USA 2002, 99, 10084–10089. [Google Scholar] [CrossRef] [PubMed]
- Wasil, L.R.; Shair, K.H.Y. Epstein-Barr virus LMP1 induces focal adhesions and epithelial cell migration through effects on integrin-α5 and N-cadherin. Oncogenesis 2015, 4, e171. [Google Scholar] [CrossRef] [PubMed]
- Sides, M.D.; Klingsberg, R.C.; Shan, B.; Gordon, K.A.; Nguyen, H.T.; Lin, Z.; Takahashi, T.; Flemington, E.K.; Lasky, J.A. The Epstein-Barr virus latent membrane protein 1 and transforming growth factor—β1 synergistically induce epithelial—Mesenchymal transition in lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 2011, 44, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Shair, K.H.Y.; Schnegg, C.I.; Raab-Traub, N. EBV Latent Membrane Protein 1 Effects on Plakoglobin, Cell Growth, and Migration. Cancer Res. 2008, 68, 6997–7005. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Wakisaka, N.; Muramatsu, M.; Zen, Y.; Endo, K.; Murono, S.; Sugimoto, H.; Yamaoka, S.; Pagano, J.S.; Yoshizaki, T. Epstein-Barr Virus Latent Membrane Protein 1 Induces Cancer Stem/Progenitor-Like Cells in Nasopharyngeal Epithelial Cell Lines. J. Virol. 2011, 85, 11255–11264. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.L.; Zhang, J.; Liu, L.Z.; Zhou, Q.; Du, S.J.; Xin, S.Y.; Ning, Z.P.; Yang, J.; Yu, H.B.; Yue, W.X.; et al. Cadherin 6 is activated by Epstein-Barr virus LMP1 to mediate EMT and metastasis as an interplay node of multiple pathways in nasopharyngeal carcinoma. Oncogenesis 2017, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.A.; Dawson, C.W.; Wei, W.; O’Neil, J.D.; Stewart, S.E.; Jia, J.; Bell, A.I.; Young, L.S.; Arrand, J.R. Epstein-Barr virus-encoded LMP1 induces a hyperproliferative and inflammatory gene expression programme in cultured keratinocytes. J. Gen. Virol. 2008, 89, 2806–2820. [Google Scholar] [CrossRef] [PubMed]
- Dawson, C.W.; Laverick, L.; Morris, M.A.; Tramoutanis, G.; Young, L.S. Epstein-Barr virus-encoded LMP1 regulates epithelial cell motility and invasion via the ERK-MAPK pathway. J. Virol. 2008, 82, 3654–3664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, H.-J.; Moon, A. Epithelial-mesenchymal Transition and Cell Invasion. Toxicol. Res. 2010, 26, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.-T.; Peng, J.-P.; Chang, H.-C.; Hung, W.-C. RECK is a target of Epstein-Barr virus latent membrane protein 1. Oncogene 2003, 22, 8263–8270. [Google Scholar] [CrossRef] [PubMed]
- Dawson, C.W.; Tramountanis, G.; Eliopoulos, A.G.; Young, L.S. Epstein-Barr virus latent membrane protein 1 (LMP1) activates the phosphatidylinositol 3-kinase/Akt pathway to promote cell survival and induce actin filament remodeling. J. Biol. Chem. 2003, 278, 3694–3704. [Google Scholar] [CrossRef] [PubMed]
- Khanna, C.; Lindblad-Toh, K.; Vail, D.; London, C.; Bergman, P.; Barber, L.; Breen, M.; Kitchell, B.; McNeil, E.; Modiano, J.F.; et al. The dog as a cancer model. Nat. Biotechnol. 2006, 24, 1065–1066. [Google Scholar] [CrossRef] [PubMed]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [PubMed]
- Breitling, R.; Armengaud, P.; Amtmann, A.; Herzyk, P. Rank products: A simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett. 2004, 573, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Breitling, R.; Herzyk, P. Rank-based methods as a non-parametric alternative of the T-statistic for the analysis of biological microarray data. J. Bioinform. Comput. Biol. 2005, 3, 1171–1189. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Schramek, H.; Feifel, E.; Marschitz, I.; Golochtchapova, N.; Gstraunthaler, G.; Montesano, R. Loss of active MEK1-ERK1/2 restores epithelial phenotype and morphogenesis in transdifferentiated MDCK cells. Am. J. Physiol. Cell Physiol. 2003, 285, C652–C661. [Google Scholar] [CrossRef] [PubMed]
- Huen, D.S.; Henderson, S.A.; Croom-Carter, D.; Rowe, M. The Epstein-Barr virus latent membrane protein-1 (LMP1) mediates activation of NF-kappa B and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene 1995, 10, 549–560. [Google Scholar] [PubMed]
- Montuori, N.; Pesapane, A.; Rossi, F.W.; Giudice, V.; De Paulis, A.; Selleri, C.; Ragno, P. Urokinase type plasminogen activator receptor (uPAR) as a new therapeutic target in cancer. Transl. Med. UniSa 2016, 15, 15–21. [Google Scholar] [PubMed]
- Madsen, C.; Sidenius, N. The interaction between urokinase receptor and vitronectin in cell adhesion and signalling. Eur. J. Cell Biol. 2008, 87, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Widmaier, M.; Rognoni, E.; Radovanac, K.; Azimifar, S.B.; Fassler, R. Integrin-linked kinase at a glance. J. Cell Sci. 2012, 125, 1839–1843. [Google Scholar] [CrossRef] [PubMed]
- Legate, K.R.; Montañez, E.; Kudlacek, O.; Füssler, R. ILK, PINCH and parvin: The tIPP of integrin signalling. Nat. Rev. Mol. Cell Biol. 2006, 7, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.J.; Lee, S.W.; Lin, L.C.; Chen, T.J.; Chang, I.W.; Hsu, H.P.; Chang, K.Y.; Huang, H.Y.; Li, C.F. Fibronectin overexpression is associated with latent membrane protein 1 expression and has independent prognostic value for nasopharyngeal carcinoma. Tumour Biol. 2014, 35, 1703–1712. [Google Scholar] [CrossRef] [PubMed]
- Watt, F.M. New EMBO Member’s Review: Role of integrins in regulating epidermal adhesion, growth and differentiation. EMBO J. 2002, 21, 3919–3926. [Google Scholar] [CrossRef] [PubMed]
- Benoit, Y.D.; Groulx, J.-F.; Gagné, D.; Beaulieu, J.-F. RGD-Dependent Epithelial Cell-Matrix Interactions in the Human Intestinal Crypt. J. Signal Transduct. 2012, 2012, 248759. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.T. Focal adhesion kinase: The first ten years. J. Cell Sci. 2003, 116 Pt 8, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Sawai, H.; Okada, Y.; Funahashi, H.; Matsuo, Y.; Takahashi, H.; Takeyama, H.; Manabe, T. Activation of focal adhesion kinase enhances the adhesion and invasion of pancreatic cancer cells via extracellular signal-regulated kinase-1/2 signaling pathway activation. Mol. Cancer 2005, 4, 37. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.J.; Brunton, V.G.; Frame, M.C. Adhesion-linked kinases in cancer; emphasis on src, focal adhesion kinase and PI 3-kinase. Eur. J. Cancer 2000, 36, 1595–1606. [Google Scholar] [CrossRef]
- Schlaepfer, D.D.; Mitra, S.K.; Ilic, D. Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochim. Biophys. Acta Mol. Cell Res. 2004, 1692, 77–102. [Google Scholar] [CrossRef] [PubMed]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Vuori, K.; Reed, J.C.; Ruoslahti, E. The alpha 5 beta 1 integrin supports survival of cells on fibronectin and up-regulates Bcl-2 expression. Proc. Natl. Acad. Sci. USA 1995, 92, 6161–6165. [Google Scholar] [CrossRef] [PubMed]
- Matter, M.L.; Zhang, Z.; Nordstedt, C.; Ruoslahti, E. The alpha5beta1 integrin mediates elimination of amyloid-beta peptide and protects against apoptosis. J. Cell Biol. 1998, 141, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.K.F.; Huang, D.P.; Lo, K.W.; Chui, Y.L.; Li, H.M.; Pang, J.C.S.; Tsao, S.W. Phenotypic alterations induced by the Hong Kong-prevalent Epstein-Barr virus-encoded LMP1 variant (2117-LMP1) in nasopharyngeal epithelial cells. Int. J. Cancer 2004, 109, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-β signaling. Cell Res. 2009, 19, 128. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Layseca, P.; Streuli, C.H. Signalling pathways linking integrins with cell cycle progression. Matrix Biol. 2014, 34, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Lowell, C.A. Src-family and Syk Kinases in Activating and Inhibitory Pathways in Innate Immune Cells: Signaling Cross Talk. Cold Spring Harb. Perspect. Biol. 2011, 3, a002352. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, R.; Mallets, E.; Gomez-Cambronero, J. The transcription factors Slug (SNAI2) and Snail (SNAI1) regulate phospholipase D (PLD) promoter in opposite ways towards cancer cell invasion. Mol. Oncol. 2016, 10, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bueno, G.; Cubillo, E.; Sarrió, D.; Peinado, H.; Rodríguez-Pinilla, S.M.; Villa, S.; Bolós, V.; Jordá, M.; Fabra, A.; Portillo, F.; et al. Genetic Profiling of Epithelial Cells Expressing E-Cadherin Repressors Reveals a Distinct Role for Snail, Slug, and E47 Factors in Epithelial-Mesenchymal Transition. Cancer Res. 2006, 66, 9543–9556. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wade, P.; Mandell, K.J.; Akyildiz, A.; Parkos, C.A.; Mrsny, R.J.; Nusrat, A. Raf 1 represses expression of the tight junction protein occludin via activation of the zinc-finger transcription factor slug. Oncogene 2007, 26, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Janda, E.; Nevolo, M.; Lehmann, K.; Downward, J.; Beug, H.; Grieco, M. Raf plus TGFbeta-dependent EMT is initiated by endocytosis and lysosomal degradation of E-cadherin. Oncogene 2006, 25, 7117–7130. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, P.A.; Kjøller, L.; Christensen, L.; Duffy, M.J. The urokinase-type plasminogen activator system in cancer metastasis: A review. Int. J. Cancer 1997, 72, 1–22. [Google Scholar] [CrossRef]
- Turner, F.E.; Broad, S.; Khanim, F.L.; Jeanes, A.; Talma, S.; Hughes, S.; Tselepis, C.; Hotchin, N.A. Slug regulates integrin expression and cell proliferation in human epidermal keratinocytes. J. Biol. Chem. 2006, 281, 21321–21331. [Google Scholar] [CrossRef] [PubMed]
- Birkenmeier, T.M.; McQuillan, J.J.; Boedeker, E.D.; Argraves, W.S.; Ruoslahti, E.; Dean, D.C. The alpha 5 beta 1 fibronectin receptor. Characterization of the alpha 5 gene promoter. J. Biol. Chem. 1991, 266, 20544–20549. [Google Scholar] [PubMed]
- Liu, Z.C.; Wang, H.S.; Zhang, G.; Liu, H.; Chen, X.H.; Zhang, F.; Chen, D.Y.; Cai, S.H.; Du, J. AKT/GSK-3β regulates stability and transcription of snail which is crucial for bFGF-induced epithelial-mesenchymal transition of prostate cancer cells. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 3096–3105. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Y.; Chung, G.T.; Lui, V.W.; To, K.F.; Ma, B.B.; Chow, C.; John, K.; Woo, S.; Yip, K.Y.; Seo, J.; et al. Exome and genome sequencing of nasopharynx cancer identifies NF-κB pathway activating mutations. Nat. Commun. 2017, 8, 14121. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.C.; Meng, X.; Hazawa, M.; Nagata, Y.; Varela, A.M.; Xu, L.; Sato, Y.; Liu, L.Z.; Ding, L.W.; Sharma, A.; et al. The genomic landscape of nasopharyngeal carcinoma. Nat. Genet. 2014, 46, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Niemhom, S.; Kitazawa, S.; Kitazawa, R.; Maeda, S.; Leopairat, J. Hypermethylation of epithelial-cadherin gene promoter is associated with Epstein-Barr virus in nasopharyngeal carcinoma. Cancer Detect. Prev. 2008, 32, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Li, W.F.; Zhang, Y.; Huang, X.B.; Du, X.J.; Tang, L.L.; Chen, L.; Peng, H.; Guo, R.; Sun, Y.; Ma, J. Prognostic value of plasma Epstein-Barr virus DNA level during posttreatment follow-up in the patients with nasopharyngeal carcinoma having undergone intensity-modulated radiotherapy. Chin. J. Cancer 2017, 36, 87. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Madin, S.H.; Darby, N.B. Established kidney cell lines of normal adult bovine and ovine origin. Proc. Soc. Exp. Biol. Med. 1958, 98, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Gene | Fold-Change in CTAR+/2− LMP1 |
|---|---|---|
| EMT phenotype | E-cadherin | −50.5 |
| N-cadherin | 7.3 | |
| Desmocollin 2 | −7.2 | |
| Desmoglein 3 | −33.0 | |
| Plakoglobin | −5.3 | |
| Plakophilin 3 | −7.4 | |
| Occludin | −26.4 | |
| Periplakin | −30.2 | |
| Claudin 3 | −10.0 | |
| ZEB1 | 199.3 | |
| ZEB2 | 12.0 | |
| Snail homologue 2 | 2.7 | |
| Podoplanin | 9.8 | |
| MAPK signalling | MAPK1/3 | 2.2 |
| RASA1 | 4.0 | |
| RRAS2 | 3.3 | |
| MAP3K4 | 2.4 | |
| IL-1α | 2.2 | |
| AKT3 | 3.8 | |
| Integrin | α5 | 9.1 |
| α6 | −3.2 | |
| β1 | 2.4 | |
| β4 | −3.8 | |
| Fibronectin | 22.8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morris, M.A.; Laverick, L.; Wei, W.; Davis, A.M.; O’Neill, S.; Wood, L.; Wright, J.; Dawson, C.W.; Young, L.S. The EBV-Encoded Oncoprotein, LMP1, Induces an Epithelial-to-Mesenchymal Transition (EMT) via Its CTAR1 Domain through Integrin-Mediated ERK-MAPK Signalling. Cancers 2018, 10, 130. https://doi.org/10.3390/cancers10050130
Morris MA, Laverick L, Wei W, Davis AM, O’Neill S, Wood L, Wright J, Dawson CW, Young LS. The EBV-Encoded Oncoprotein, LMP1, Induces an Epithelial-to-Mesenchymal Transition (EMT) via Its CTAR1 Domain through Integrin-Mediated ERK-MAPK Signalling. Cancers. 2018; 10(5):130. https://doi.org/10.3390/cancers10050130
Chicago/Turabian StyleMorris, Mhairi A., Louise Laverick, Wenbin Wei, Alexandra M. Davis, Samantha O’Neill, Liam Wood, Jack Wright, Christopher W. Dawson, and Lawrence S. Young. 2018. "The EBV-Encoded Oncoprotein, LMP1, Induces an Epithelial-to-Mesenchymal Transition (EMT) via Its CTAR1 Domain through Integrin-Mediated ERK-MAPK Signalling" Cancers 10, no. 5: 130. https://doi.org/10.3390/cancers10050130
APA StyleMorris, M. A., Laverick, L., Wei, W., Davis, A. M., O’Neill, S., Wood, L., Wright, J., Dawson, C. W., & Young, L. S. (2018). The EBV-Encoded Oncoprotein, LMP1, Induces an Epithelial-to-Mesenchymal Transition (EMT) via Its CTAR1 Domain through Integrin-Mediated ERK-MAPK Signalling. Cancers, 10(5), 130. https://doi.org/10.3390/cancers10050130