FOXD1 and Gal-3 Form a Positive Regulatory Loop to Regulate Lung Cancer Aggressiveness




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. High FOXD1 and Gal-3 Expression are Associated with the Poor Prognosis in Lung Cancer
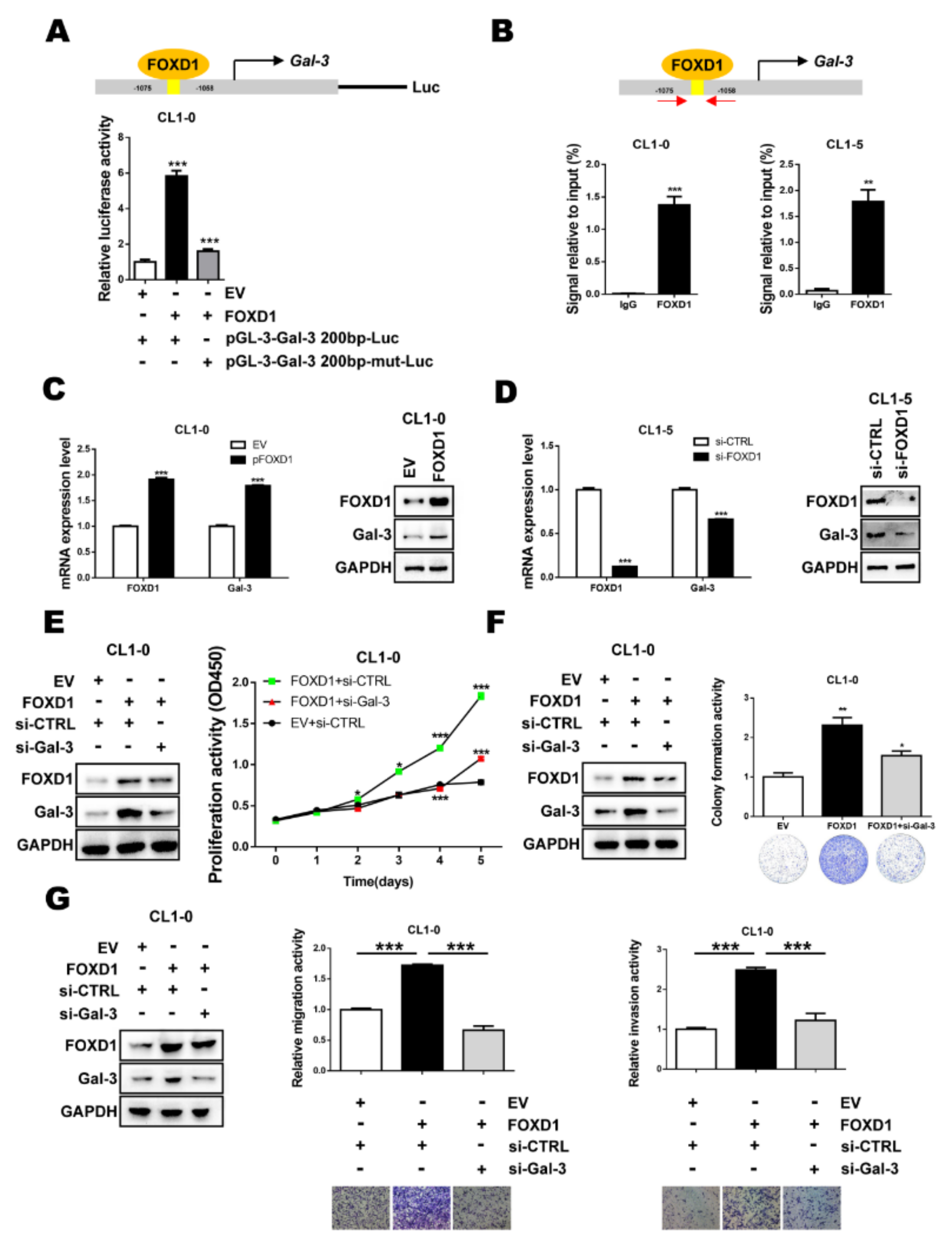
2.2. FOXD1 is a Transcription Factor of Gal-3
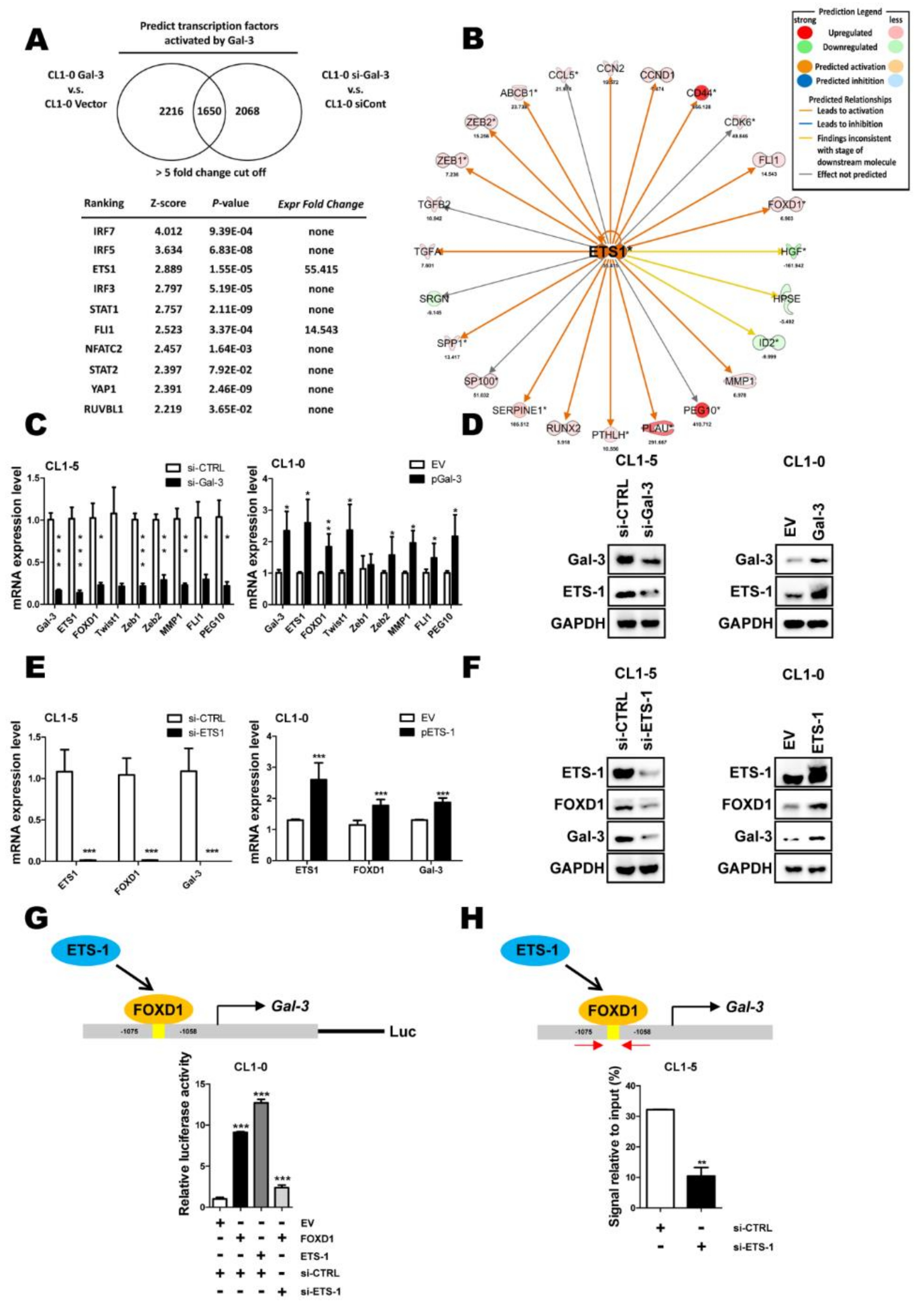
2.3. ETS-1 Is Involved in Gal-3-Mediated FOXD1 Expression
2.4. Gal-3 Regulates FOXD1 Expression through ITGβ1 Signaling
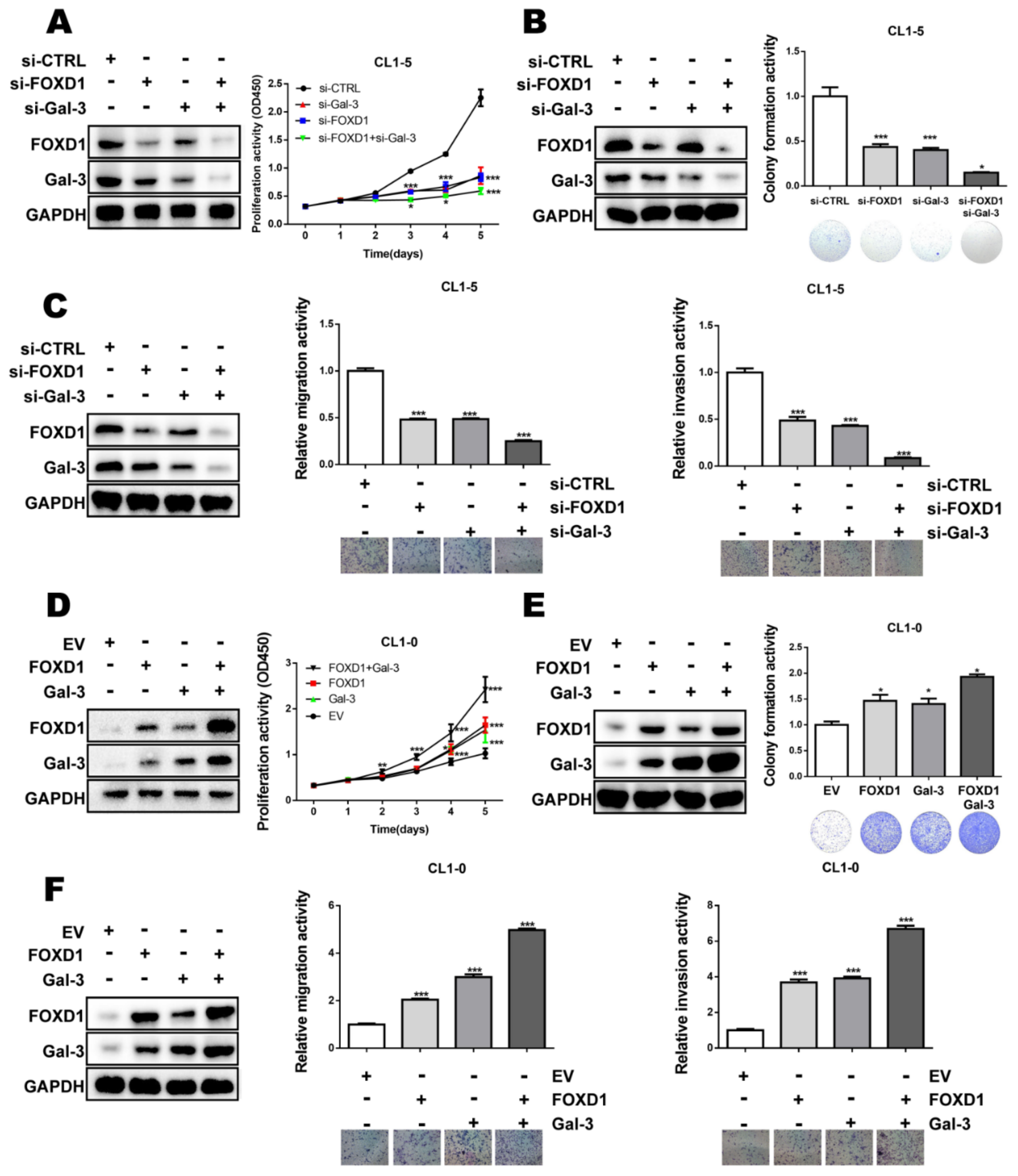
2.5. FOXD1 and Gal-3 form a Positive Regulatory Loop to Promote Lung Cancer Cell Growth and Motility
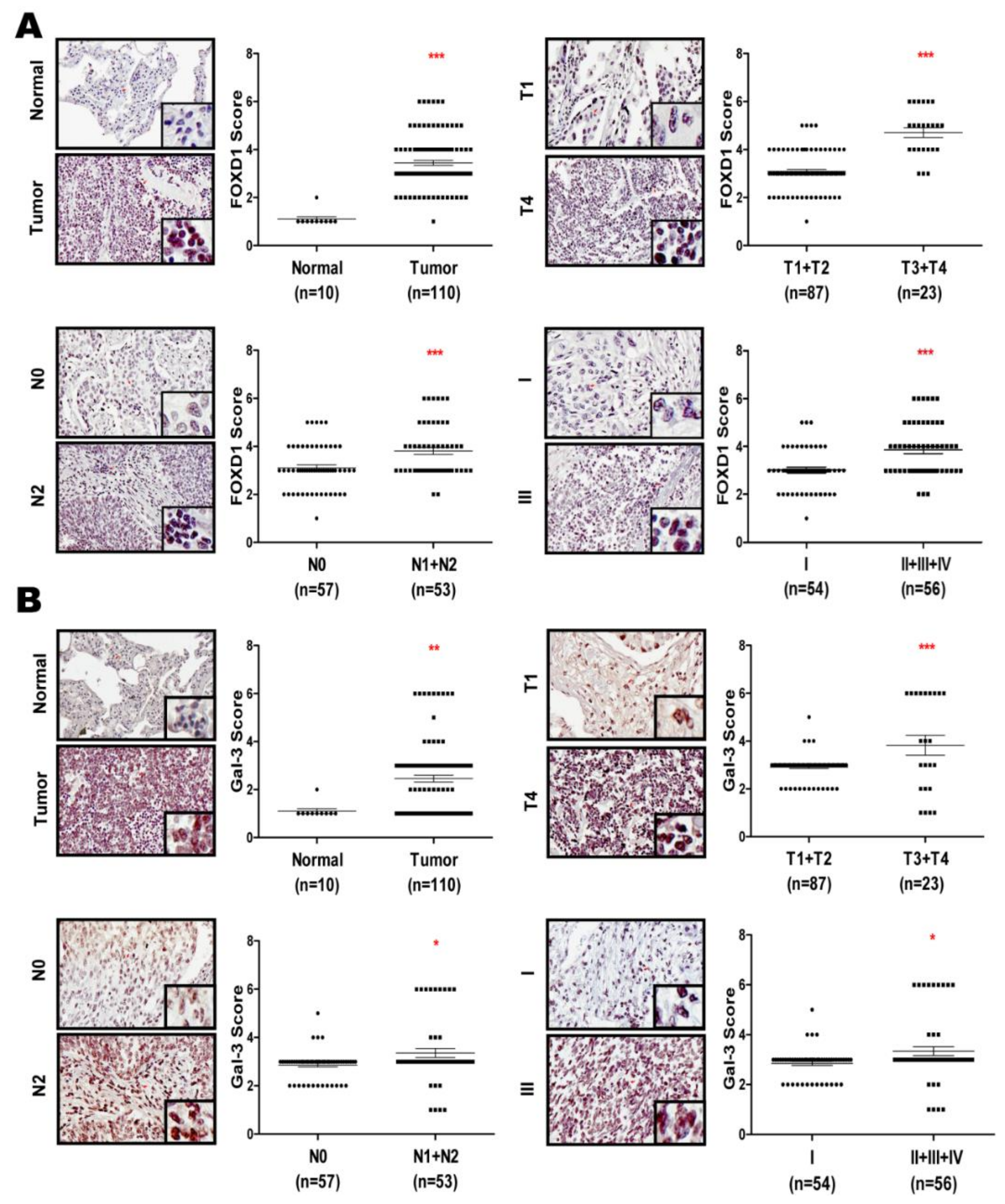
2.6. FOXD1 and Gal-3 are Positively Correlated in Human Lung Cancer Tissues
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Transfection
4.3. mRNA-Based qPCR Assay
4.4. Immunoblotting and Immunoprecipitation
4.5. Immunohistochemistry Analysis
4.6. Colonogenic Formation Assay
4.7. Extraction of Cytoplasma/Nucleus and Membrane Raft Proteins
4.8. Chromatin Immunoprecipitation Assay
4.9. Promoter Assay
4.10. Proliferation Assay
4.11. Transwell Migration and Invasion Assay
4.12. Immunofluorescence Staining
4.13. In-Silicon mRNA Microarrays
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ETS-1: | Proto-oncogene 1 |
| FOXD1: | Forkhead box D1 |
| FOX: | Forkhead box |
| Gal-3: | Galectin-3/LGALS3 |
| GPS: | Group-based prediction system |
| HR: | Hazard ratio |
| ITGβ1: | Integrin-β1 |
| IPA: | Ingenuity Pathway Analysis |
| NSCLC: | Non-small cell lung cancer |
| NSL: | Nuclear localization signals |
| OS: | Overall survival |
| PFS: | Progression-free survival |
| PRS: | Post-recurrence survival rate |
| TF: | Transcription factor |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Sun, S.; Schiller, J.H.; Spinola, M.; Minna, J.D. New molecularly targeted therapies for lung cancer. J. Clin. Investig. 2007, 117, 2740–2750. [Google Scholar] [CrossRef]
- Simeone, J.C.; Nordstrom, B.L.; Patel, K.; Mann, H.; Klein, A.B.; Horne, L. Treatment patterns and overall survival in metastatic urothelial carcinoma in a real-world, US setting. Cancer Epidemiol. 2019, 60, 121–127. [Google Scholar] [CrossRef]
- Sekihara, K.; Hishida, T.; Yoshida, J.; Oki, T.; Omori, T.; Katsumata, S.; Ueda, T.; Miyoshi, T.; Goto, M.; Nakasone, S.; et al. Long-term survival outcome after postoperative recurrence of non-small-cell lung cancer: Who is ’cured’ from postoperative recurrence? Eur. J. Cardio Thorac. 2017, 52, 522–528. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Tamiya, A.; Isa, S.I.; Nakahama, K.; Okishio, K.; Shiroyama, T.; Suzuki, H.; Inoue, T.; Tamiya, M.; Hirashima, T.; et al. Predictive Factors for Poor Progression-free Survival in Patients with Non-small Cell Lung Cancer Treated with Nivolumab. Anticancer Res. 2017, 37, 5857–5862. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.; Jambon, S.; Depauw, S.; David-Cordonnier, M.H. Targeting Transcription Factors for Cancer Treatment. Molecules 2018, 23, 1479. [Google Scholar] [CrossRef] [PubMed]
- Levinson, R.S.; Batourina, E.; Choi, C.; Vorontchikhina, M.; Kitajewski, J.; Mendelsohn, C.L. Foxd1-dependent signals control cellularity in the renal capsule, a structure required for normal renal development. Development 2005, 132, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Koga, M.; Matsuda, M.; Kawamura, T.; Sogo, T.; Shigeno, A.; Nishida, E.; Ebisuya, M. Foxd1 is a mediator and indicator of the cell reprogramming process. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.E.; Sequeira-Lopez, M.L.; Gomez, R.A. RBP-J in FOXD1+ renal stromal progenitors is crucial for the proper development and assembly of the kidney vasculature and glomerular mesangial cells. Am. J. Physiol. Ren. Physiol. 2014, 306, F249–F258. [Google Scholar] [CrossRef] [PubMed]
- Song, R.; Lopez, M.; Yosypiv, I.V. Foxd1 is an upstream regulator of the renin-angiotensin system during metanephric kidney development. Pediatr Res. 2017, 82, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Quintero-Ronderos, P.; Laissue, P. The multisystemic functions of FOXD1 in development and disease. J. Mol. Med. 2018, 96, 725–739. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.F.; Zhao, J.Y.; Yue, H.; Hu, K.S.; Shen, H.; Guo, Z.G.; Su, X.J. FOXD1 promotes breast cancer proliferation and chemotherapeutic drug resistance by targeting p27. Biochem. Biophys. Res. Commun. 2015, 456, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Wang, J.; Waghmare, I.; Sartini, S.; Coviello, V.; Zhang, Z.; Kim, S.H.; Mohyeldin, A.; Pavlyukov, M.S.; Minata, M.; et al. FOXD1-ALDH1A3 Signaling Is a Determinant for the Self-Renewal and Tumorigenicity of Mesenchymal Glioma Stem Cells. Cancer Res. 2016, 76, 7219–7230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.M.; Wang, T.; Wang, S.; Xiong, Y.L.; Zhang, R.; Zhang, X.; Zhao, J.; Yang, A.G.; Wang, L.; Jia, L.T. Nkx2-2as Suppression Contributes to the Pathogenesis of Sonic Hedgehog Medulloblastoma. Cancer Res. 2018, 78, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, S.; Soejima, K.; Yasuda, H.; Yoda, S.; Satomi, R.; Ikemura, S.; Terai, H.; Sato, T.; Yamaguchi, N.; Hamamoto, J.; et al. FOXD1 Expression Is Associated with Poor Prognosis in Non-small Cell Lung Cancer. Anticancer Res. 2015, 35, 261–268. [Google Scholar]
- Li, D.; Fan, S.; Yu, F.; Zhu, X.; Song, Y.; Ye, M.; Fan, L.; Lv, Z. FOXD1 Promotes Cell Growth and Metastasis by Activation of Vimentin in NSCLC. Cell Physiol. Biochem. 2018, 51, 2716–2731. [Google Scholar] [CrossRef]
- Wu, H.Z.; Larribere, L.; Sun, Q.; Novak, D.; Sachindra, S.; Granados, K.; Umansky, V.; Utikal, J. Loss of neural crest-associated gene FOXD1 impairs melanoma invasion and migration via RAC1B downregulation. Int. J. Cancer 2018, 143, 2962–2972. [Google Scholar] [CrossRef]
- Hughes, R.C. Secretion of the galectin family of mammalian carbohydrate-binding proteins. Biochim. Biophys. Acta Gen. Subj. 1999, 1473, 172–185. [Google Scholar] [CrossRef]
- Ahmed, H.; AlSadek, D.M.M. Galectin-3 as a Potential Target to Prevent Cancer Metastasis. Clin. Med. Insights Oncol. 2015, 9, 113–121. [Google Scholar] [CrossRef]
- Cardoso, A.C.F.; Andrade, L.N.D.; Bustos, S.O.; Chammas, R. Galectin-3 Determines Tumor Cell Adaptive Strategies in Stressed Tumor Microenvironments. Front. Oncol. 2016, 6, 127. [Google Scholar] [CrossRef]
- Chung, L.Y.; Tang, S.J.; Wu, Y.C.; Sun, G.H.; Liu, H.Y.; Sun, K.H. Galectin-3 augments tumor initiating property and tumorigenicity of lung cancer through interaction with beta-catenin. Oncotarget 2015, 6, 4936–4952. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, S.; Oka, N.; Raz, A. On the role of galectin-3 in cancer apoptosis. Apoptosis 2005, 10, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Akahani, S.; NangiaMakker, P.; Inohara, H.; Kim, H.R.C.; Raz, A. Galectin-3: A novel antiapoptotic molecule with a functional BH1 (NWGR) domain of Bcl-2 family. Cancer Res. 1997, 57, 5272–5276. [Google Scholar] [PubMed]
- Nakahara, S.; Raz, A. Regulation of cancer-related gene expression by galectin-3 and the molecular mechanism of its nuclear import pathway. Cancer Metastasis Rev. 2007, 26, 605–610. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, P.; Qin, Y.; Cong, Q.; Shao, C.; Du, Z.; Ni, X.; Li, P.; Ding, K. RN1, a novel galectin-3 inhibitor, inhibits pancreatic cancer cell growth in vitro and in vivo via blocking galectin-3 associated signaling pathways. Oncogene 2017, 36, 1297–1308. [Google Scholar] [CrossRef]
- Dong, R.; Zhang, M.; Hu, Q.Y.; Zheng, S.; Soh, A.; Zheng, Y.J.; Yuan, H. Galectin-3 as a novel biomarker for disease diagnosis and a target for therapy. Int. J. Mol. Med. 2018, 41, 599–614. [Google Scholar] [CrossRef]
- Margadant, C.; van den Bout, I.; van Boxtel, A.L.; Thijssen, V.L.; Sonnenberg, A. Epigenetic Regulation of Galectin-3 Expression by beta 1 Integrins Promotes Cell Adhesion and Migration. J. Biol. Chem. 2012, 287, 44684–44693. [Google Scholar] [CrossRef]
- Ramsey, J.; Butnor, K.; Peng, Z.H.; Leclair, T.; van der Velden, J.; Stein, G.; Lian, J.; Kinsey, C.M. Loss of RUNX1 is associated with aggressive lung adenocarcinomas. J. Cell. Physiol. 2018, 233, 3487–3497. [Google Scholar] [CrossRef]
- Seguin, L.; Kato, S.; Franovic, A.; Camargo, M.F.; Lesperance, J.; Elliott, K.C.; Yebra, M.; Mielgo, A.; Lowy, A.M.; Husain, H.; et al. An integrin beta(3)-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 2014, 16, 457. [Google Scholar] [CrossRef]
- Wang, L.; Guo, X.L. Molecular regulation of galectin-3 expression and therapeutic implication in cancer progression. Biomed. Pharmacother. 2016, 78, 165–171. [Google Scholar] [CrossRef]
- Fukushi, J.; Makagiansar, I.T.; Stallcup, W.B. NG2 proteoglycan promotes endothelial cell motility and angiogenesis via engagement of galectin-3 and alpha 3 beta 1 integrin. Mol. Biol. Cell 2004, 15, 3580–3590. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.C.; Wang, W.; Zhu, B.J.; Wang, X.D. Lung Cancer Heterogeneity and New Strategies for Drug Therapy. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Cochran, B.H. Extracellular signal-regulated kinase binds to TFII-I and regulates its activation of the c-fos promoter. Mol. Cell. Biol. 2000, 20, 1140–1148. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Liu, L.; Qiu, X.; Jiang, L.; Huang, B.; Li, H.; Li, Z.; Luo, W.; Wang, E. CCL21/CCR7 promotes G2/M phase progression via the ERK pathway in human non-small cell lung cancer cells. PLoS ONE 2011, 6, e21119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.M.; Ji, B.A.; Ramachandran, V.; Wang, H.M.; Hafley, M.; Logsdon, C.; Bresalier, R.S. Overexpressed Galectin-3 in Pancreatic Cancer Induces Cell Proliferation and Invasion by Binding Ras and Activating Ras Signaling. PloS ONE 2012, 7, e42699. [Google Scholar] [CrossRef] [Green Version]
- Ho, M.Y.; Liang, C.M.; Liang, S.M. MIG-7 and phosphorylated prohibitin coordinately regulate lung cancer invasion/metastasis. Oncotarget 2015, 6, 381–393. [Google Scholar] [CrossRef] [Green Version]
- Dumic, J.; Dabelic, S.; Flogel, M. Galectin-3: An open-ended story. Biochim. Biophys. Acta Gen. Subj. 2006, 1760, 616–635. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.-H.; Chang, Y.-C.; Hsiao, M.; Liang, S.-M. FOXD1 and Gal-3 Form a Positive Regulatory Loop to Regulate Lung Cancer Aggressiveness. Cancers 2019, 11, 1897. https://doi.org/10.3390/cancers11121897
Li C-H, Chang Y-C, Hsiao M, Liang S-M. FOXD1 and Gal-3 Form a Positive Regulatory Loop to Regulate Lung Cancer Aggressiveness. Cancers. 2019; 11(12):1897. https://doi.org/10.3390/cancers11121897
Chicago/Turabian StyleLi, Chien-Hsiu, Yu-Chan Chang, Michael Hsiao, and Shu-Mei Liang. 2019. "FOXD1 and Gal-3 Form a Positive Regulatory Loop to Regulate Lung Cancer Aggressiveness" Cancers 11, no. 12: 1897. https://doi.org/10.3390/cancers11121897
APA StyleLi, C. -H., Chang, Y. -C., Hsiao, M., & Liang, S. -M. (2019). FOXD1 and Gal-3 Form a Positive Regulatory Loop to Regulate Lung Cancer Aggressiveness. Cancers, 11(12), 1897. https://doi.org/10.3390/cancers11121897