Antimicrobial Peptide TP4 Induces ROS-Mediated Necrosis by Triggering Mitochondrial Dysfunction in Wild-Type and Mutant p53 Glioblastoma Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
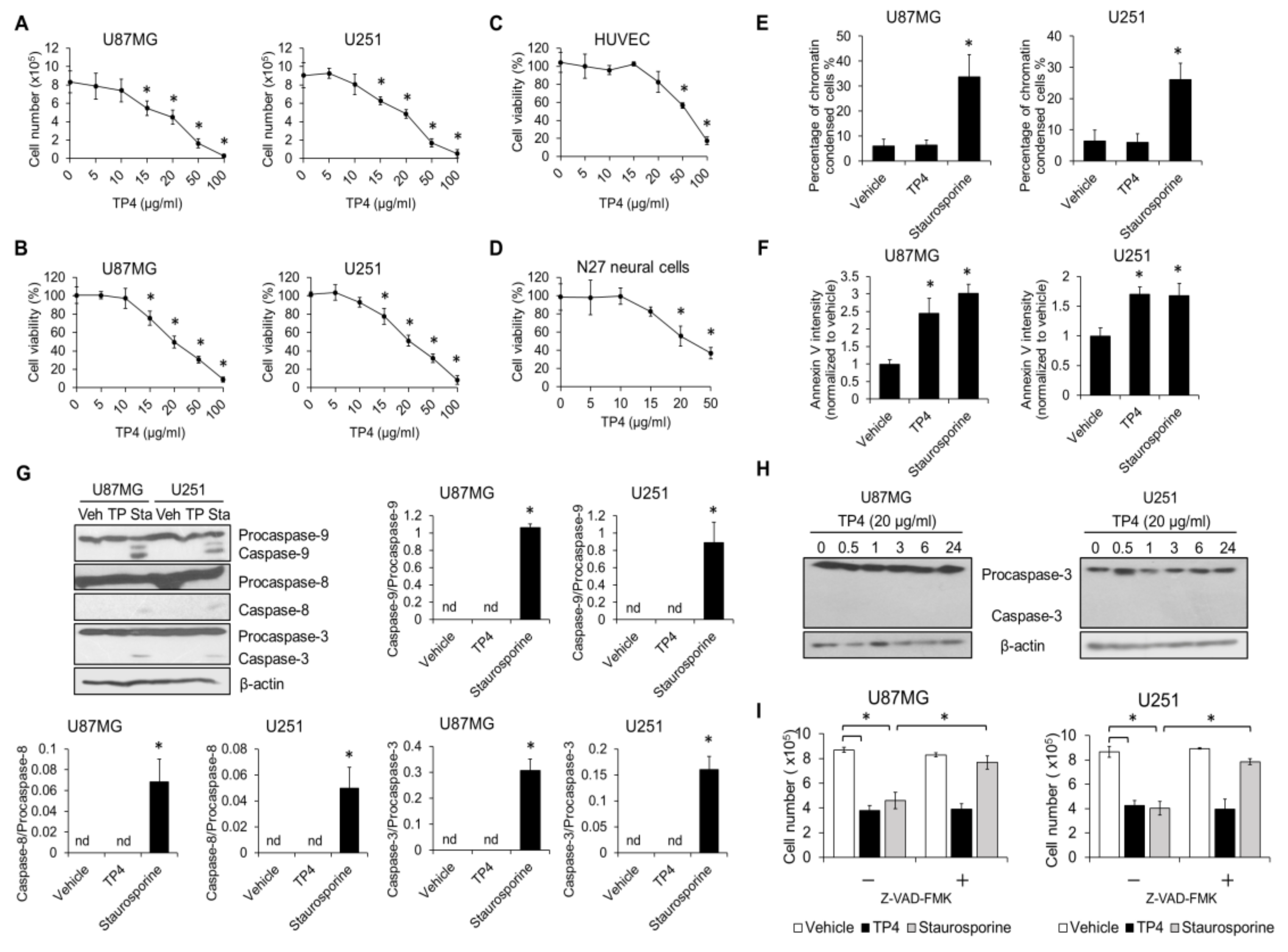
2.1. TP4 Induces Death in Glioblastoma Cell Lines through a p53-Independent Mechanism
2.2. TP4 Induces Caspase-Independent Cytotoxicity in Glioblastoma Cells
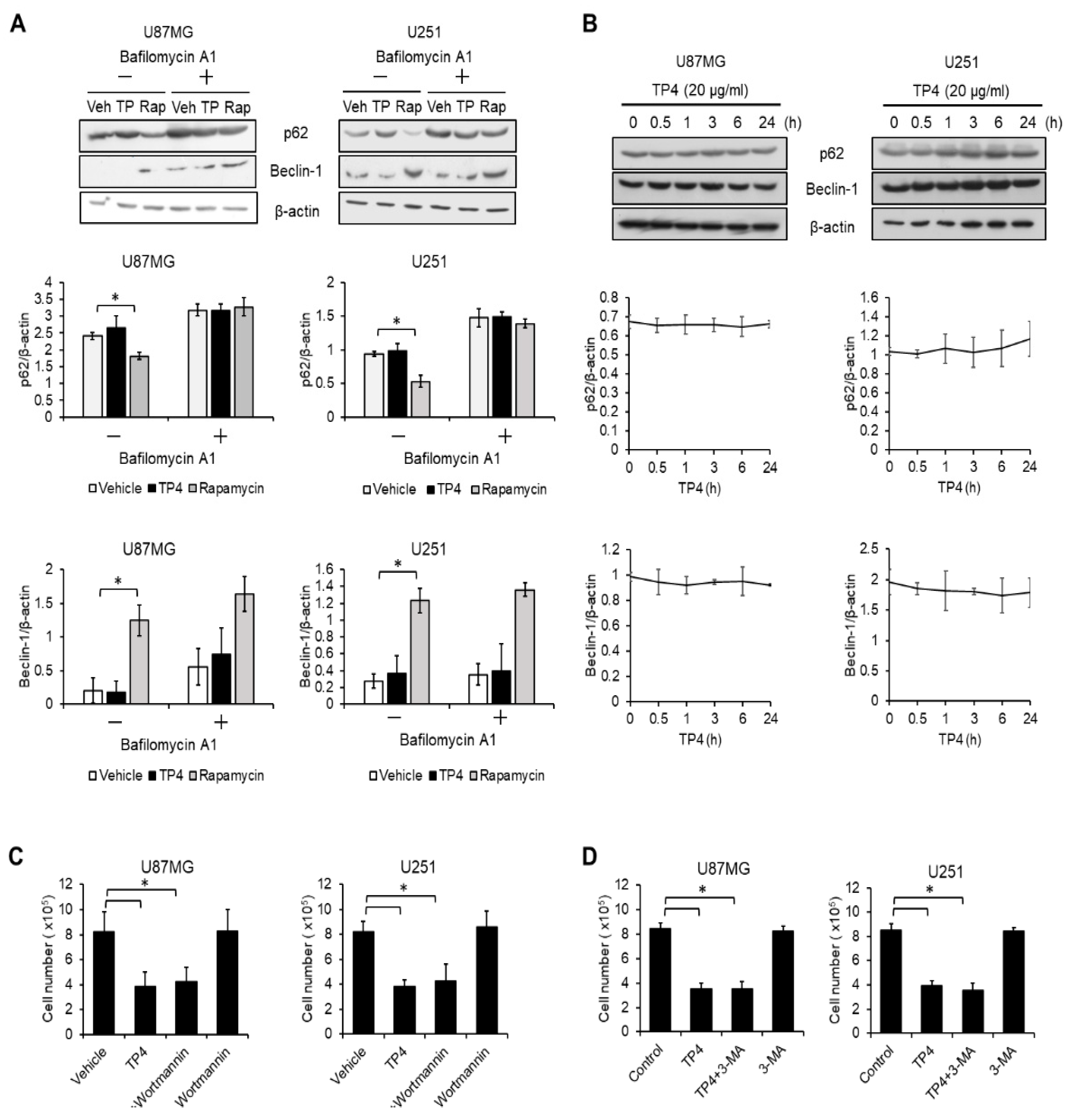
2.3. Autophagy Is not Activated by TP4 in Glioblastoma Cell Lines
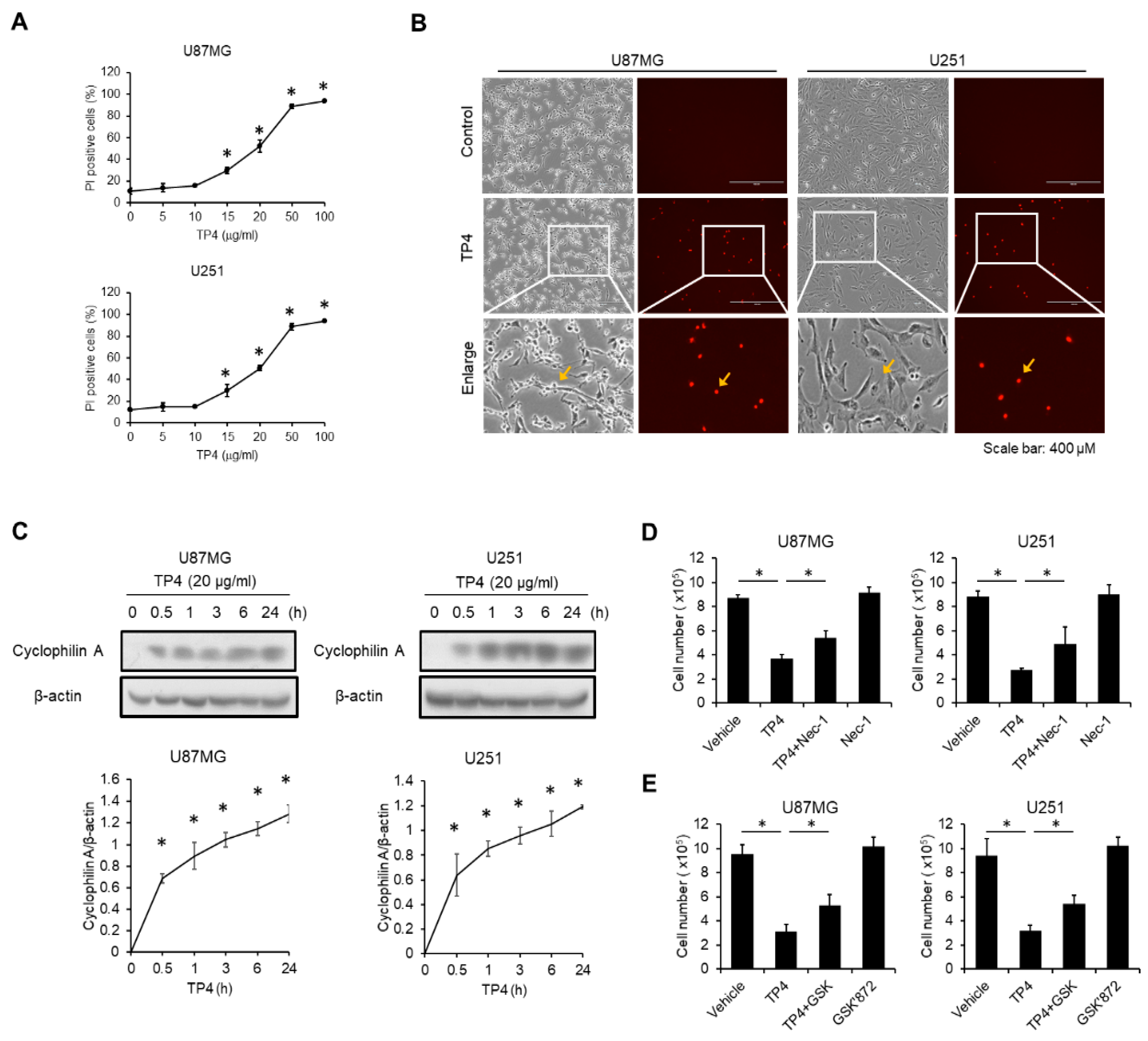
2.4. TP4 Induces Necrotic Cell Death in Glioblastoma Cell Lines
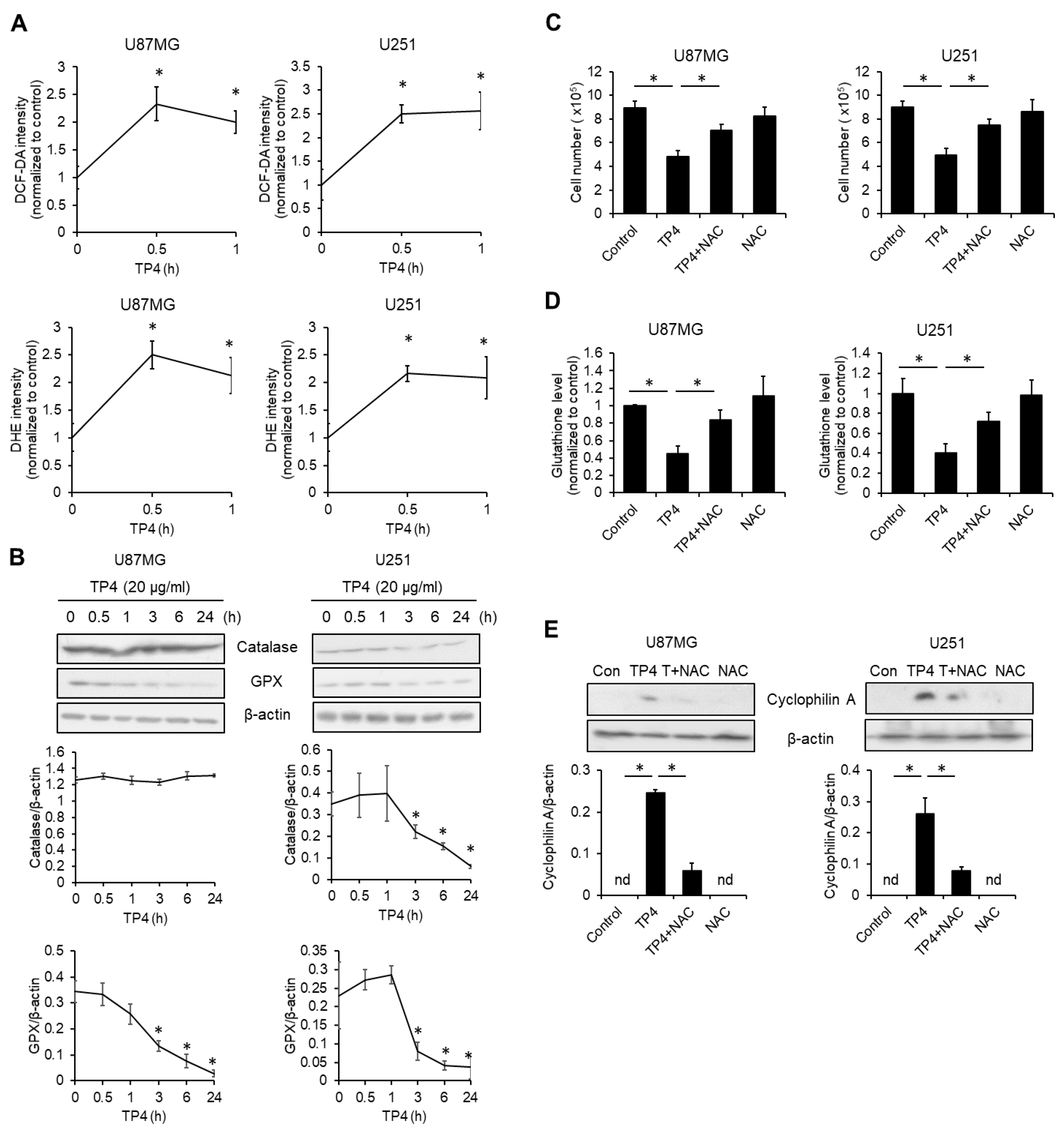
2.5. TP4 Elevates Intracellular Reactive Oxygen Species (ROS)
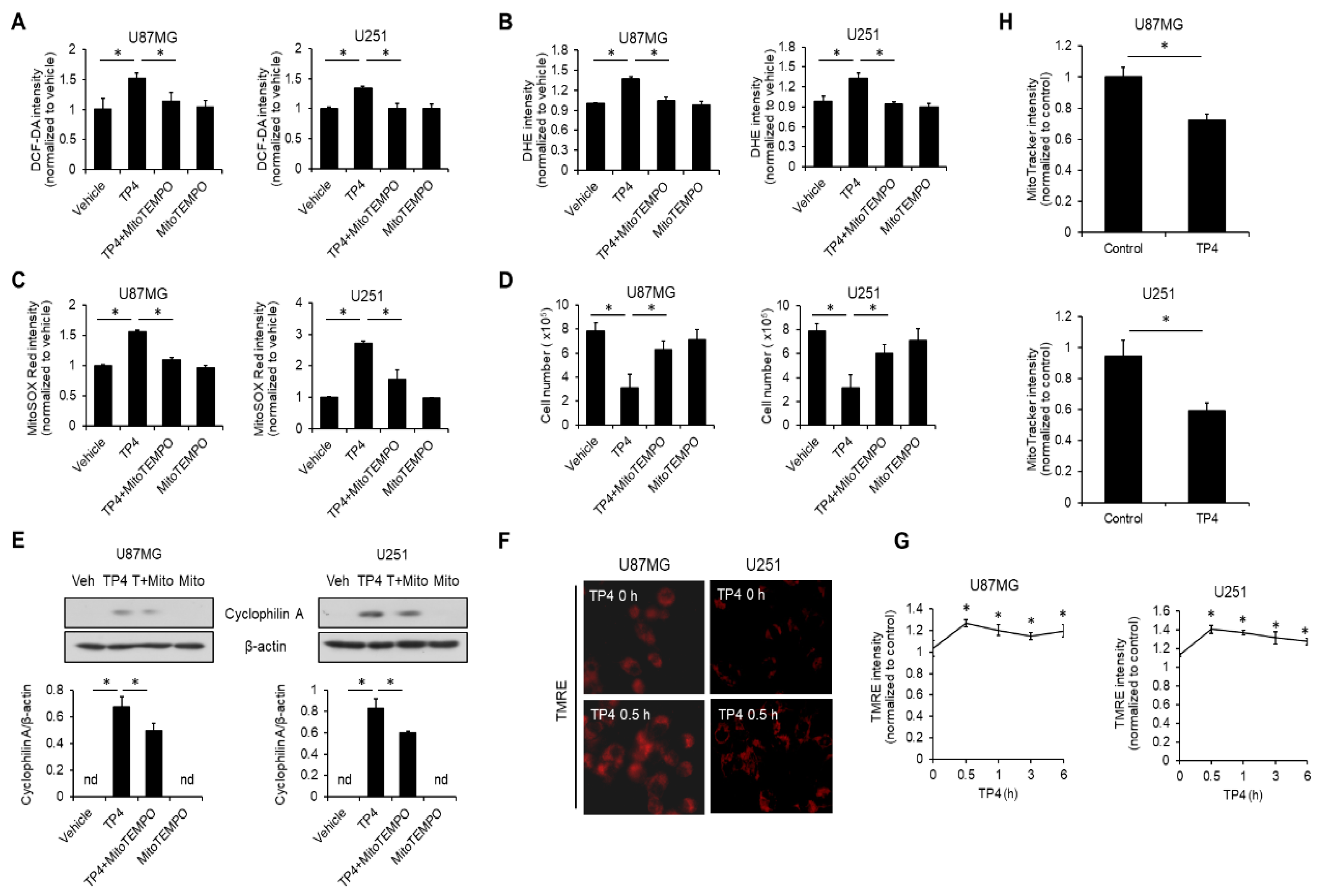
2.6. TP4 Impairs Mitochondria, Resulting in ROS Accumulation
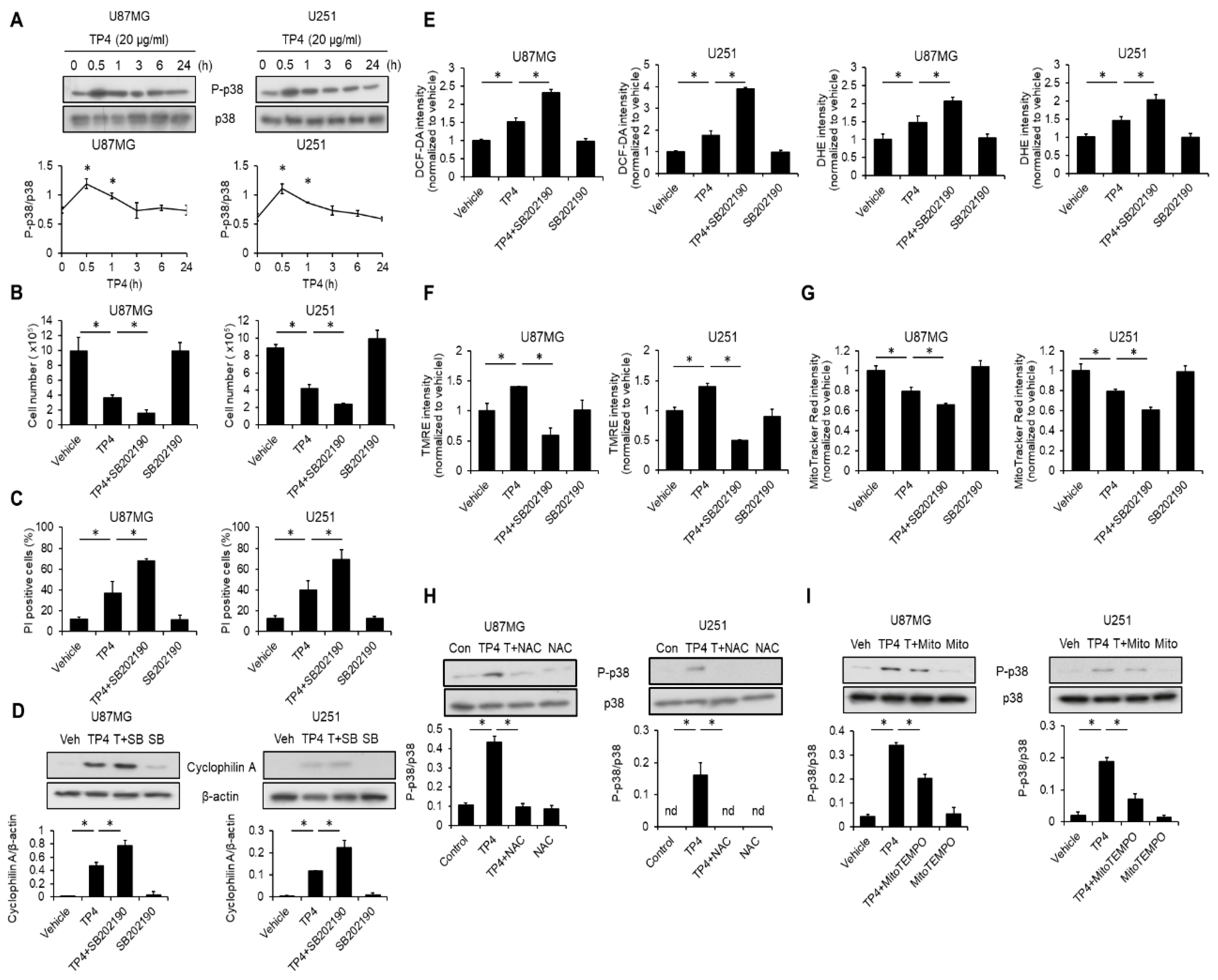
2.7. TP4 Induces a p38-Mediated Protective Effect
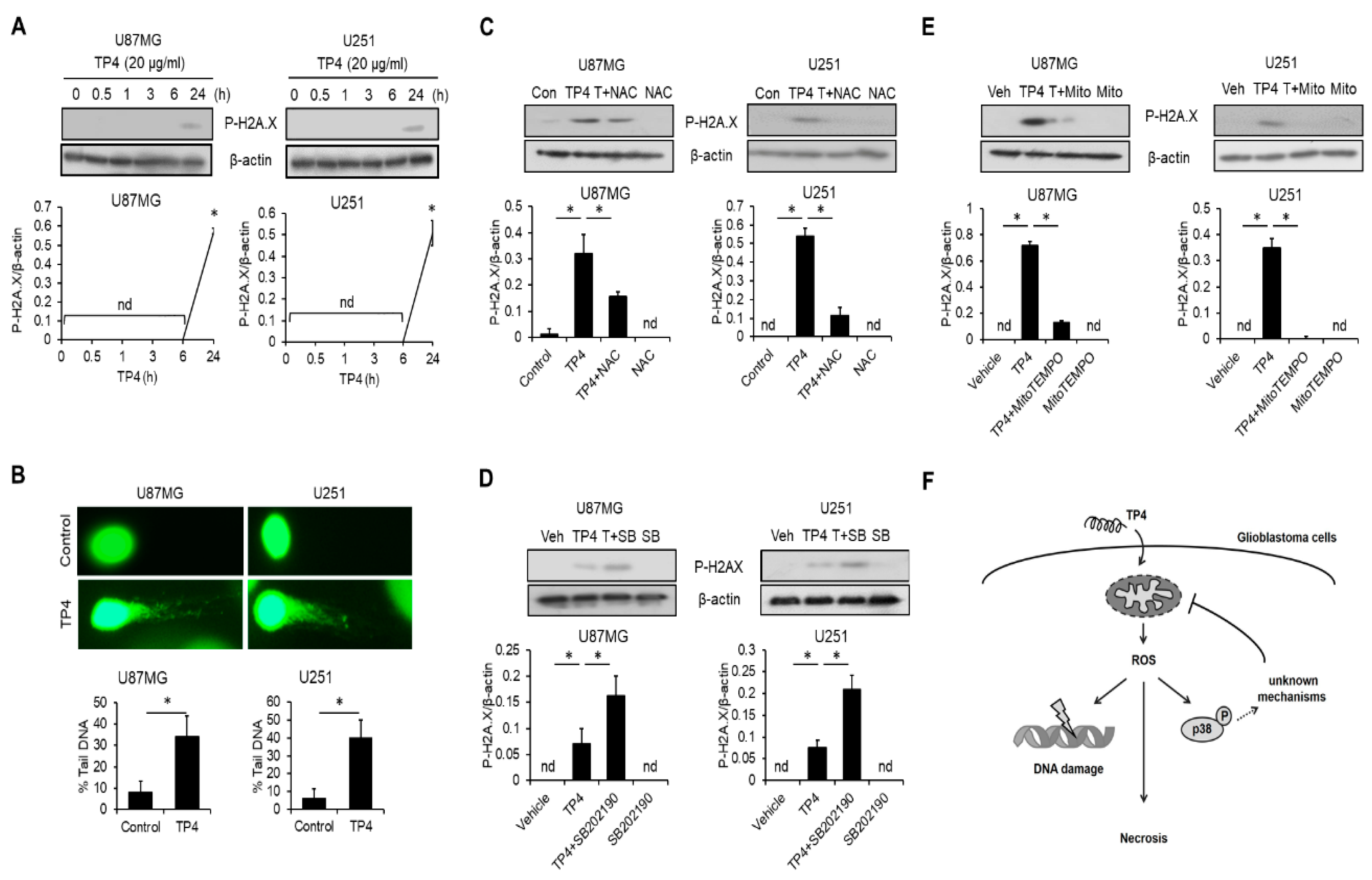
2.8. TP4 Induces ROS-Mediated DNA Damage
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Western Blotting
4.4. Annexin V-FITC Binding Assay
4.5. Cytotoxicity
4.6. ROS and Mitochondrial Functional Analysis
4.7. Comet Assay
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Holland, E.C. Glioblastoma multiforme: The terminator. Proc. Natl. Acad. Sci. USA 2000, 97, 6242–6244. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. Glioblastoma: Overview of disease and treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [PubMed]
- Kleihues, P.; Sobin, L.H. World health organization classification of tumors. Cancer 2000, 88, 2887. [Google Scholar] [CrossRef]
- Arbab, A.S.; Jain, M.; Achyut, B.R. P53 mutation: Critical mediator of therapy resistance against tumor microenvironment. Biochem. Physiol. 2016, 5, e153. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, S.; Tada, K.; Nakamura, H.; Makino, K.; Kochi, M.; Saya, H.; Kuratsu, J.; Ushio, Y. Influence of p53 mutations on prognosis of patients with glioblastoma. Cancer 2002, 95, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Blough, M.D.; Beauchamp, D.C.; Westgate, M.R.; Kelly, J.J.; Cairncross, J.G. Effect of aberrant p53 function on temozolomide sensitivity of glioma cell lines and brain tumor initiating cells from glioblastoma. J. Neurooncol. 2011, 102, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Batista, L.F.; Roos, W.P.; Christmann, M.; Menck, C.F.; Kaina, B. Differential sensitivity of malignant glioma cells to methylating and chloroethylating anticancer drugs: P53 determines the switch by regulating xpc, ddb2, and DNA double-strand breaks. Cancer Res. 2007, 67, 11886–11895. [Google Scholar] [CrossRef]
- Blough, M.D.; Zlatescu, M.C.; Cairncross, J.G. O6-methylguanine-DNA methyltransferase regulation by p53 in astrocytic cells. Cancer Res. 2007, 67, 580–584. [Google Scholar] [CrossRef]
- Wang, X.; Chen, J.X.; Liu, J.P.; You, C.; Liu, Y.H.; Mao, Q. Gain of function of mutant tp53 in glioblastoma: Prognosis and response to temozolomide. Ann. Surg. Oncol. 2014, 21, 1337–1344. [Google Scholar] [CrossRef]
- Peng, K.C.; Lee, S.H.; Hour, A.L.; Pan, C.Y.; Lee, L.H.; Chen, J.Y. Five different piscidins from nile tilapia, oreochromis niloticus: Analysis of their expressions and biological functions. PLoS ONE 2012, 7, e50263. [Google Scholar] [CrossRef]
- Narayana, J.L.; Huang, H.N.; Wu, C.J.; Chen, J.Y. Efficacy of the antimicrobial peptide tp4 against helicobacter pylori infection: In vitro membrane perturbation via micellization and in vivo suppression of host immune responses in a mouse model. Oncotarget 2015, 6, 12936–12954. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.N.; Chan, Y.L.; Wu, C.J.; Chen, J.Y. Tilapia piscidin 4 (tp4) stimulates cell proliferation and wound closure in mrsa-infected wounds in mice. Mar. Drugs 2015, 13, 2813–2833. [Google Scholar] [CrossRef] [PubMed]
- Ting, C.H.; Chen, Y.C.; Wu, C.J.; Chen, J.Y. Targeting fosb with a cationic antimicrobial peptide, tp4, for treatment of triple-negative breast cancer. Oncotarget 2016, 7, 40329–40347. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S. The role of p53 in chemosensitivity and radiosensitivity. Oncogene 2003, 22, 7486–7495. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, S.H.; Earnshaw, W.C. Induction of apoptosis by cancer chemotherapy. Exp. Cell Res. 2000, 256, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Levine, B. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2015, 22, 367–376. [Google Scholar] [CrossRef]
- Christofferson, D.E.; Yuan, J. Cyclophilin a release as a biomarker of necrotic cell death. Cell Death Differ. 2010, 17, 1942–1943. [Google Scholar] [CrossRef]
- Villena, J.; Henriquez, M.; Torres, V.; Moraga, F.; Diaz-Elizondo, J.; Arredondo, C.; Chiong, M.; Olea-Azar, C.; Stutzin, A.; Lavandero, S.; et al. Ceramide-induced formation of ros and atp depletion trigger necrosis in lymphoid cells. Free Radic. Biol. Med. 2008, 44, 1146–1160. [Google Scholar] [CrossRef]
- Debatin, K.M.; Poncet, D.; Kroemer, G. Chemotherapy: Targeting the mitochondrial cell death pathway. Oncogene 2002, 21, 8786–8803. [Google Scholar] [CrossRef]
- Watanabe, T.; Sekine, S.; Naguro, I.; Sekine, Y.; Ichijo, H. Apoptosis signal-regulating kinase 1 (ask1)-p38 pathway-dependent cytoplasmic translocation of the orphan nuclear receptor nr4a2 is required for oxidative stress-induced necrosis. J. Biol. Chem. 2015, 290, 10791–10803. [Google Scholar] [CrossRef]
- Maynard, S.; Schurman, S.H.; Harboe, C.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009, 30, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Singh, K.; Almasan, A. Histone h2ax phosphorylation: A marker for DNA damage. Methods Mol. Biol. 2012, 920, 613–626. [Google Scholar] [PubMed]
- Liu, A.; Sankey, E.W.; Bettegowda, C.; Burger, P.C.; Jallo, G.I.; Groves, M.L. Poor prognosis despite aggressive treatment in adults with intramedullary spinal cord glioblastoma. J. Clin. Neurosci. 2015, 22, 1628–1631. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.P.; Olofsson, T.; Qu, M.; Hesselager, G.; Soussi, T.; Kalimo, H.; Smits, A.; Nister, M. Molecular genetic analysis of p53 intratumoral heterogeneity in human astrocytic brain tumors. J. Neuropathol. Exp. Neurol. 2007, 66, 944–954. [Google Scholar] [CrossRef] [PubMed]
- Safari, M.; Khoshnevisan, A. Cancer stem cells and chemoresistance in glioblastoma multiform: A review article. J. Stem Cells 2015, 10, 271–285. [Google Scholar] [PubMed]
- Valtorta, S.; Lo Dico, A.; Raccagni, I.; Gaglio, D.; Belloli, S.; Politi, L.S.; Martelli, C.; Diceglie, C.; Bonanomi, M.; Ercoli, G.; et al. Metformin and temozolomide, a synergic option to overcome resistance in glioblastoma multiforme models. Oncotarget 2017, 8, 113090–113104. [Google Scholar] [CrossRef]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, R.J.; Maciejewski, J.P.; Wilmink, J.W.; van Noorden, C.J.F. Wild-type and mutated idh1/2 enzymes and therapy responses. Oncogene 2018, 37, 1949–1960. [Google Scholar] [CrossRef]
- Guo, G.; Gong, K.; Ali, S.; Ali, N.; Shallwani, S.; Hatanpaa, K.J.; Pan, E.; Mickey, B.; Burma, S.; Wang, D.H.; et al. A tnf-jnk-axl-erk signaling axis mediates primary resistance to egfr inhibition in glioblastoma. Nat. Neurosci. 2017, 20, 1074–1084. [Google Scholar] [CrossRef]
- Da Ros, M.; De Gregorio, V.; Iorio, A.L.; Giunti, L.; Guidi, M.; de Martino, M.; Genitori, L.; Sardi, I. Glioblastoma chemoresistance: The double play by microenvironment and blood-brain barrier. Int. J. Mol. Sci. 2018, 19, E2879. [Google Scholar] [CrossRef]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and niche concept. Cancers 2018, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Spencer, D.; Pytel, P.; Ahmed, A.U.; Lesniak, M.S. The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert Rev. Neurother. 2015, 15, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Roos, A.; Ding, Z.; Loftus, J.C.; Tran, N.L. Molecular and microenvironmental determinants of glioma stem-like cell survival and invasion. Front. Oncol. 2017, 7, 120. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The role of hypoxia in glioblastoma invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Mei, H.; Li, X.; Ma, Y.; Zeng, A.H.; Wang, Y.; Lu, X.; Chu, F.; Wu, Q.; Zhu, J. Apoptosis-inducing activity of the antimicrobial peptide cecropin of musca domestica in human hepatocellular carcinoma cell line bel-7402 and the possible mechanism. Acta Biochim. Biophys. Sin. 2010, 42, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.R.; Chen, Y.L.; Hsu, H.C.; Chen, W.J. Antimicrobial peptide gw-h1-induced apoptosis of human gastric cancer ags cell line is enhanced by suppression of autophagy. Mol. Cell Biochem. 2015, 400, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Lin, W.J.; Wu, J.L.; Her, G.M.; Hui, C.F. Epinecidin-1 peptide induces apoptosis which enhances antitumor effects in human leukemia u937 cells. Peptides 2009, 30, 2365–2373. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Thompson, C.B. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin. Cancer Res. 2007, 13, 7271–7279. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, X. Intratumoral dendritic cells in the anti-tumor immune response. Cell. Mol. Immunol. 2015, 12, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Zhang, Q.; Liu, Z.; Wang, S.; Li, D.; Liu, C.; Liu, Y.; Shao, Y. Cyclophilin a as a potential genetic adjuvant to improve hiv-1 gag DNA vaccine immunogenicity by eliciting broad and long-term gag-specific cellular immunity in mice. Hum. Vaccines Immunother. 2016, 12, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Walzer, T.; Dalod, M.; Robbins, S.H.; Zitvogel, L.; Vivier, E. Natural-killer cells and dendritic cells: “L’union fait la force”. Blood 2005, 106, 2252–2258. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lostao, L.; Anel, A.; Pardo, J. How do cytotoxic lymphocytes kill cancer cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and mitochondrial ros in cancer: Novel targets for anticancer therapy. J. Cell Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Polson, E.S.; Kuchler, V.B.; Abbosh, C.; Ross, E.M.; Mathew, R.K.; Beard, H.A.; da Silva, B.; Holding, A.N.; Ballereau, S.; Chuntharpursat-Bon, E.; et al. Khs101 disrupts energy metabolism in human glioblastoma cells and reduces tumor growth in mice. Sci. Transl. Med. 2018, 10, eaar2718. [Google Scholar] [CrossRef] [PubMed]
- Arif, T.; Krelin, Y.; Nakdimon, I.; Benharroch, D.; Paul, A.; Dadon-Klein, D.; Shoshan-Barmatz, V. Vdac1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro Oncol. 2017, 19, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Gergely, P., Jr.; Grossman, C.; Niland, B.; Puskas, F.; Neupane, H.; Allam, F.; Banki, K.; Phillips, P.E.; Perl, A. Mitochondrial hyperpolarization and atp depletion in patients with systemic lupus erythematosus. Arthritis Rheumatol. 2002, 46, 175–190. [Google Scholar] [CrossRef]
- Katoh, I.; Sato, S.; Fukunishi, N.; Yoshida, H.; Imai, T.; Kurata, S. Apaf-1-deficient fog mouse cell apoptosis involves hypo-polarization of the mitochondrial inner membrane, atp depletion and citrate accumulation. Cell Res. 2008, 18, 1210–1219. [Google Scholar] [CrossRef]
- Su, B.C.; Mo, F.E. Ccn1 enables fas ligand-induced apoptosis in cardiomyoblast h9c2 cells by disrupting caspase inhibitor xiap. Cell Signal. 2014, 26, 1326–1334. [Google Scholar] [CrossRef]
- Orrenius, S. Reactive oxygen species in mitochondria-mediated cell death. Drug Metab. Rev. 2007, 39, 443–455. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Kohsaka, S.; Takahashi, K.; Wang, L.; Tanino, M.; Kimura, T.; Nishihara, H.; Tanaka, S. Inhibition of gsh synthesis potentiates temozolomide-induced bystander effect in glioblastoma. Cancer Lett. 2013, 331, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y. Apoptosis and necrosis: Intracellular atp level as a determinant for cell death modes. Cell Death Differ. 1997, 4, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Troyano, A.; Sancho, P.; Fernandez, C.; de Blas, E.; Bernardi, P.; Aller, P. The selection between apoptosis and necrosis is differentially regulated in hydrogen peroxide-treated and glutathione-depleted human promonocytic cells. Cell Death Differ. 2003, 10, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Uzquiza, A.; Arechederra, M.; Bragado, P.; Aguirre-Ghiso, J.A.; Porras, A. P38alpha mediates cell survival in response to oxidative stress via induction of antioxidant genes: Effect on the p70s6k pathway. J. Biol. Chem. 2012, 287, 2632–2642. [Google Scholar] [CrossRef]
- Fukui, M.; Zhu, B.T. Mitochondrial superoxide dismutase sod2, but not cytosolic sod1, plays a critical role in protection against glutamate-induced oxidative stress and cell death in ht22 neuronal cells. Free Radic. Biol. Med. 2010, 48, 821–830. [Google Scholar] [CrossRef]
- Ho, Y.S.; Xiong, Y.; Ma, W.; Spector, A.; Ho, D.S. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J. Biol. Chem. 2004, 279, 32804–32812. [Google Scholar] [CrossRef]
- Hirose, Y.; Katayama, M.; Stokoe, D.; Haas-Kogan, D.A.; Berger, M.S.; Pieper, R.O. The p38 mitogen-activated protein kinase pathway links the DNA mismatch repair system to the g2 checkpoint and to resistance to chemotherapeutic DNA-methylating agents. Mol. Cell. Biol. 2003, 23, 8306–8315. [Google Scholar] [CrossRef]
- Deslouches, B.; Di, Y.P. Antimicrobial peptides with selective antitumor mechanisms: Prospect for anticancer applications. Oncotarget 2017, 8, 46635–46651. [Google Scholar] [CrossRef]
- Matsuzaki, K. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta 2009, 1788, 1687–1692. [Google Scholar] [CrossRef]
- Bhowmik, A.; Khan, R.; Ghosh, M.K. Blood brain barrier: A challenge for effectual therapy of brain tumors. Biomed. Res. Int. 2015, 2015, 320941. [Google Scholar] [CrossRef] [PubMed]
- Blakeley, J. Drug delivery to brain tumors. Curr. Neurol. Neurosci. Rep. 2008, 8, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Pfeffer, J.L.; Gururangan, S.; Lester, T.; Lim, D.A.; Shaywitz, A.J.; Westphal, M.; Slavc, I. Intracerebroventricular delivery as a safe, long-term route of drug administration. Pediatr. Neurol. 2017, 67, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Attenello, F.J.; Mukherjee, D.; Datoo, G.; McGirt, M.J.; Bohan, E.; Weingart, J.D.; Olivi, A.; Quinones-Hinojosa, A.; Brem, H. Use of gliadel (bcnu) wafer in the surgical treatment of malignant glioma: A 10-year institutional experience. Ann. Surg. Oncol. 2008, 15, 2887–2893. [Google Scholar] [CrossRef] [PubMed]
- Wait, S.D.; Prabhu, R.S.; Burri, S.H.; Atkins, T.G.; Asher, A.L. Polymeric drug delivery for the treatment of glioblastoma. Neuro Oncol. 2015, 17 (Suppl. 2), ii9–ii23. [Google Scholar] [CrossRef] [PubMed]
- Martyniuk, C.J.; Feswick, A.; Fang, B.; Koomen, J.M.; Barber, D.S.; Gavin, T.; Lopachin, R.M. Protein targets of acrylamide adduct formation in cultured rat dopaminergic cells. Toxicol. Lett. 2013, 219, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Su, B.C.; Huang, H.N.; Lin, T.W.; Hsiao, C.D.; Chen, J.Y. Epinecidin-1 protects mice from lps-induced endotoxemia and cecal ligation and puncture-induced polymicrobial sepsis. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3028–3037. [Google Scholar] [CrossRef]
- Kobayashi, S.; Volden, P.; Timm, D.; Mao, K.; Xu, X.; Liang, Q. Transcription factor gata4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. J. Biol. Chem. 2010, 285, 793–804. [Google Scholar] [CrossRef]
- Wang, X.; Ottosson, A.; Ji, C.; Feng, X.; Nordenskjold, M.; Henter, J.I.; Fadeel, B.; Zheng, C. Proteasome inhibition induces apoptosis in primary human natural killer cells and suppresses nkp46-mediated cytotoxicity. Haematologica 2009, 94, 470–478. [Google Scholar] [CrossRef]
- Lee, K.; Jeong, J.E.; Kim, I.H.; Kim, K.S.; Ju, B.G. Cyclo(phenylalanine-proline) induces DNA damage in mammalian cells via reactive oxygen species. J. Cell. Mol. Med. 2015, 19, 2851–2864. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, B.-C.; Pan, C.-Y.; Chen, J.-Y. Antimicrobial Peptide TP4 Induces ROS-Mediated Necrosis by Triggering Mitochondrial Dysfunction in Wild-Type and Mutant p53 Glioblastoma Cells. Cancers 2019, 11, 171. https://doi.org/10.3390/cancers11020171
Su B-C, Pan C-Y, Chen J-Y. Antimicrobial Peptide TP4 Induces ROS-Mediated Necrosis by Triggering Mitochondrial Dysfunction in Wild-Type and Mutant p53 Glioblastoma Cells. Cancers. 2019; 11(2):171. https://doi.org/10.3390/cancers11020171
Chicago/Turabian StyleSu, Bor-Chyuan, Chieh-Yu Pan, and Jyh-Yih Chen. 2019. "Antimicrobial Peptide TP4 Induces ROS-Mediated Necrosis by Triggering Mitochondrial Dysfunction in Wild-Type and Mutant p53 Glioblastoma Cells" Cancers 11, no. 2: 171. https://doi.org/10.3390/cancers11020171
APA StyleSu, B. -C., Pan, C. -Y., & Chen, J. -Y. (2019). Antimicrobial Peptide TP4 Induces ROS-Mediated Necrosis by Triggering Mitochondrial Dysfunction in Wild-Type and Mutant p53 Glioblastoma Cells. Cancers, 11(2), 171. https://doi.org/10.3390/cancers11020171