Newer-Generation EGFR Inhibitors in Lung Cancer: How Are They Best Used?


Abstract
:1. Introduction
2. Sequencing Targeted Therapies
3. EGFR Inhibitors
4. Osimertinib
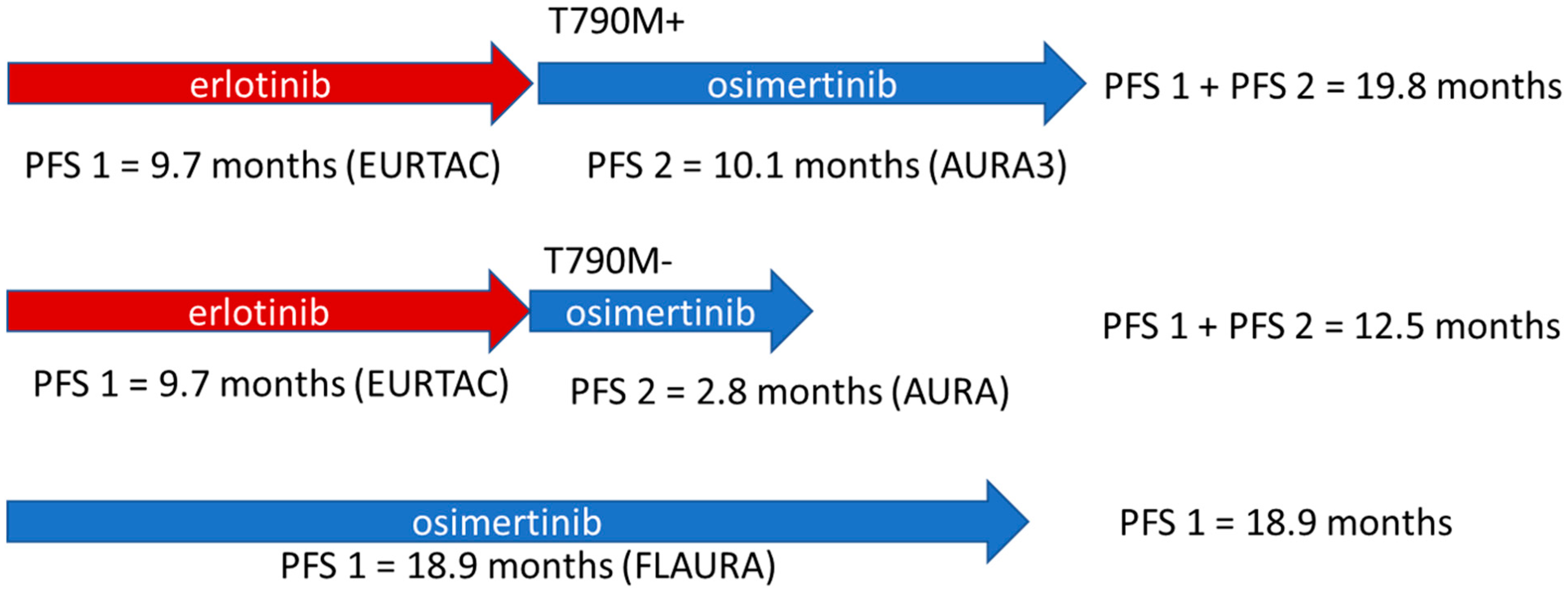
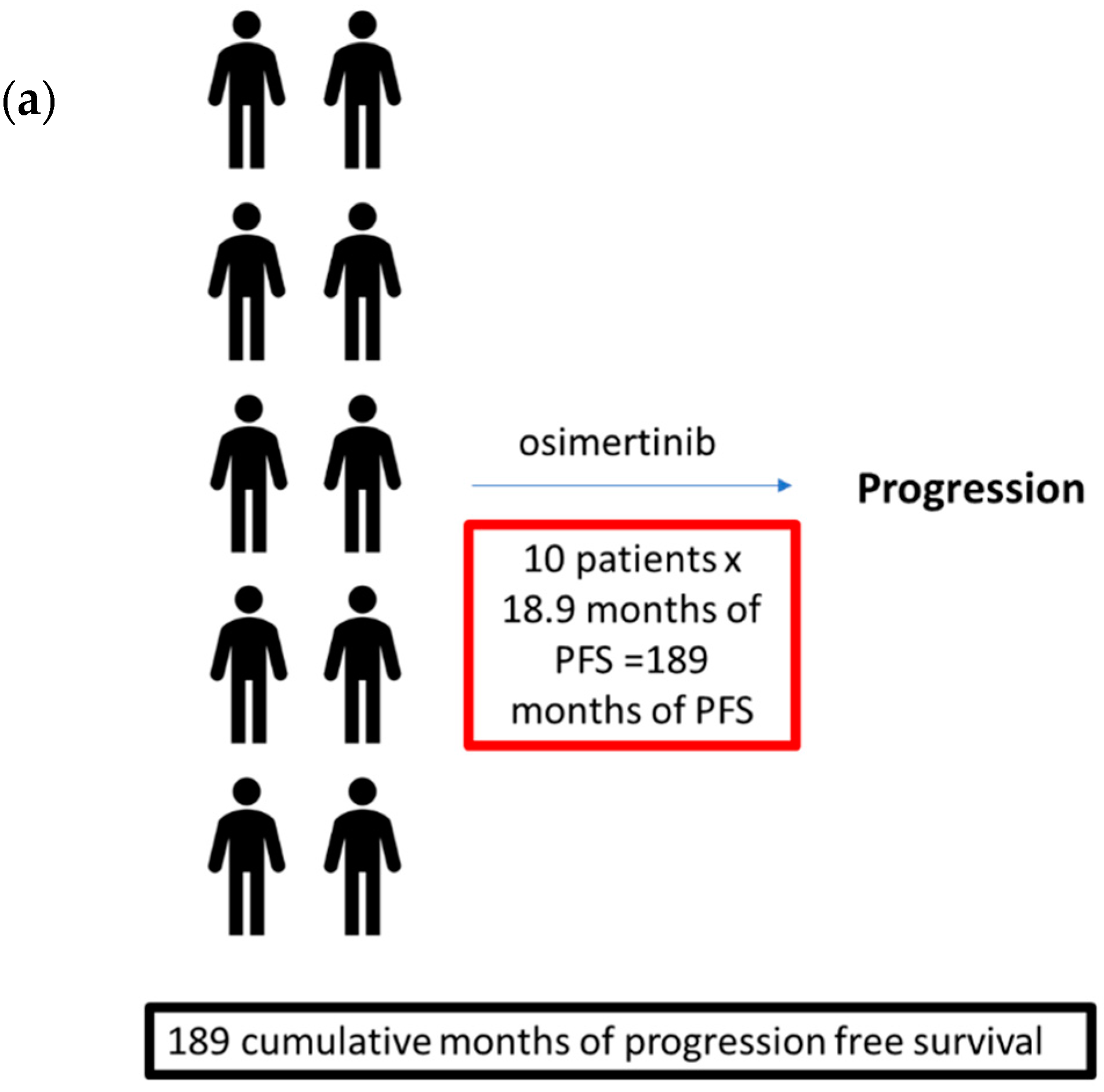
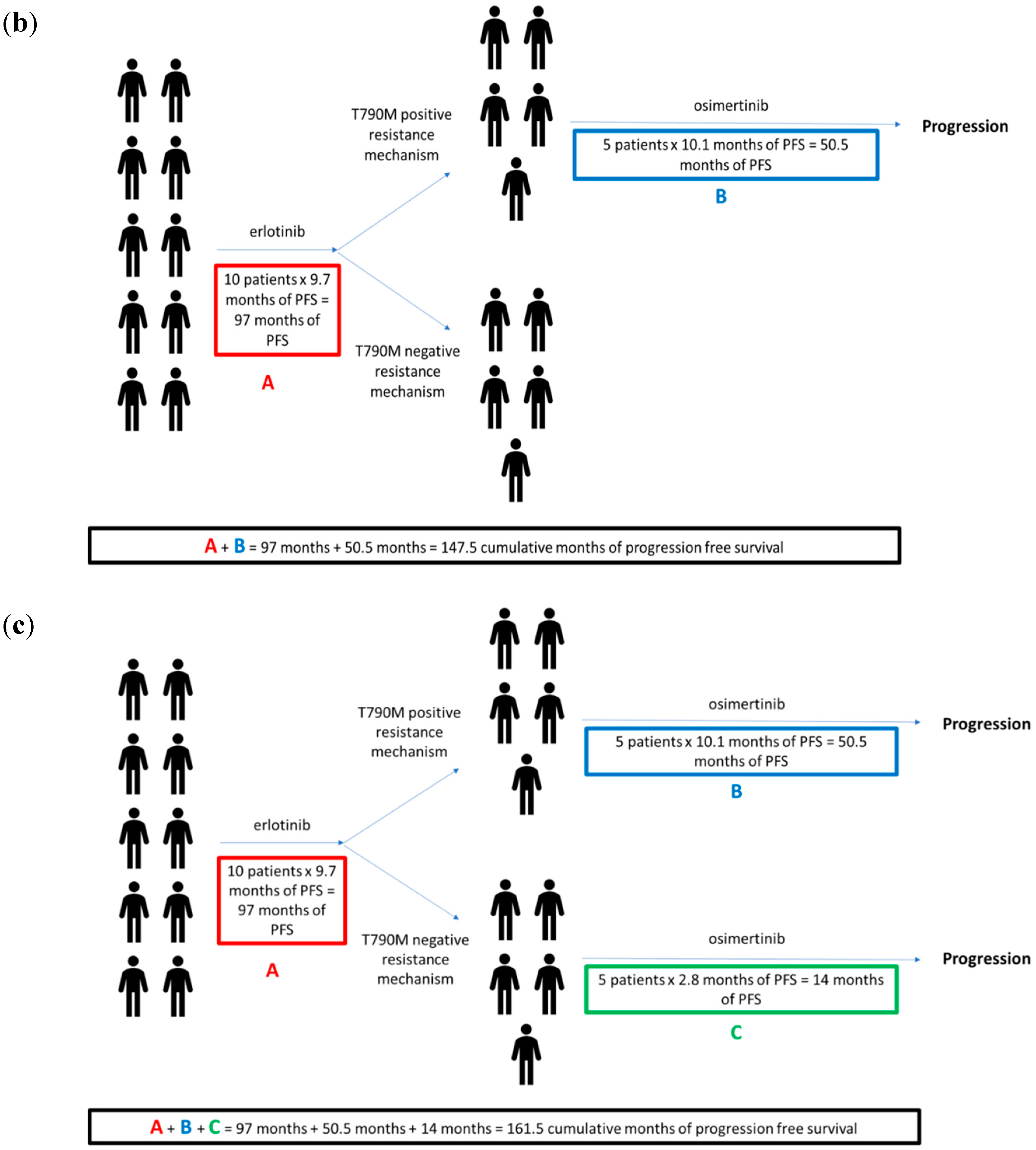
5. The Optimal Use of Osimertinib
6. Acquired Resistance to Osimertinib
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Druker, B.J.; Guilhot, F.; O’Brien, S.G.; Gathmann, I.; Kantarjian, H.; Gattermann, N.; Deininger, M.W.; Silver, R.T.; Goldman, J.M.; Stone, R.M.; et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med. 2006, 355, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Guilhot, F.; Druker, B.; Larson, R.A.; Gathmann, I.; So, C.; Waltzman, R.; O’Brien, S.G. High rates of durable response are achieved with imatinib after treatment with interferon alpha plus cytarabine: Results from the International Randomized Study of Interferon and STI571 (IRIS) trial. Haematologica 2009, 94, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.P.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S.; et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017, 376, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lavallade, H.; Apperley, J.F.; Khorashad, J.S.; Milojkovic, D.; Reid, A.G.; Bua, M.; Szydlo, R.; Olavarria, E.; Kaeda, J.; Goldman, J.M.; et al. Imatinib for newly diagnosed patients with chronic myeloid leukemia: Incidence of sustained responses in an intention-to-treat analysis. J. Clin. Oncol. 2008, 26, 3358–3363. [Google Scholar] [CrossRef] [PubMed]
- Lucas, C.M.; Wang, L.; Austin, G.M.; Knight, K.; Watmough, S.J.; Shwe, K.H.; Dasgupta, R.; Butt, N.M.; Galvani, D.; Hoyle, C.F.; et al. A population study of imatinib in chronic myeloid leukaemia demonstrates lower efficacy than in clinical trials. Leukemia 2008, 22, 1963–1966. [Google Scholar] [CrossRef] [PubMed]
- Giles, F.J.; le Coutre, P.D.; Pinilla-Ibarz, J.; Larson, R.A.; Gattermann, N.; Ottmann, O.G.; Hochhaus, A.; Radich, J.P.; Saglio, G.; Hughes, T.P.; et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia 2013, 27, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Giles, F.J.; Bhalla, K.N.; Pinilla-Ibarz, J.; Larson, R.A.; Gattermann, N.; Ottmann, O.G.; Hochhaus, A.; Radich, J.P.; Saglio, G.; et al. Nilotinib is effective in patients with chronic myeloid leukemia in chronic phase after imatinib resistance or intolerance: 24-month follow-up results. Blood 2011, 117, 1141–1145. [Google Scholar] [CrossRef] [PubMed]
- Le Coutre, P.D.; Giles, F.J.; Hochhaus, A.; Apperley, J.F.; Ossenkoppele, G.J.; Blakesley, R.; Shou, Y.; Gallagher, N.J.; Baccarani, M.; Cortes, J.; et al. Nilotinib in patients with Ph+ chronic myeloid leukemia in accelerated phase following imatinib resistance or intolerance: 24-month follow-up results. Leukemia 2012, 26, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Gandhi, L.; Gadgeel, S.; Riely, G.J.; Cetnar, J.; West, H.; Camidge, D.R.; Socinski, M.A.; Chiappori, A.; Mekhail, T.; et al. Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer: A single-group, multicentre, phase 2 trial. Lancet Oncol. 2016, 17, 234–242. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, D.W.; Mehra, R.; Tan, D.S.; Felip, E.; Chow, L.Q.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; De Pas, T.; et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 370, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Gadgeel, S.M.; Gandhi, L.; Riely, G.J.; Chiappori, A.A.; West, H.L.; Azada, M.C.; Morcos, P.N.; Lee, R.M.; Garcia, L.; Yu, L.; et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): Results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014, 15, 1119–1128. [Google Scholar] [CrossRef]
- Hochhaus, A.; Saglio, G.; Hughes, T.P.; Larson, R.A.; Kim, D.W.; Issaragrisil, S.; le Coutre, P.D.; Etienne, G.; Dorlhiac-Llacer, P.E.; Clark, R.E.; et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia 2016, 30, 1044–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saglio, G.; Kim, D.W.; Issaragrisil, S.; le Coutre, P.; Etienne, G.; Lobo, C.; Pasquini, R.; Clark, R.E.; Hochhaus, A.; Hughes, T.P.; et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N. Engl. J. Med. 2010, 362, 2251–2259. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.W.; Ou, S.I.; Perol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Yang, J.C.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.M.; Boyer, M.; et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Mok, T.; Kim, D.W.; Wu, Y.L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crino, L.; Ahn, M.J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Bang, Y.J.; Kwak, E.L.; Iafrate, A.J.; Varella-Garcia, M.; Fox, S.B.; Riely, G.J.; Solomon, B.; Ou, S.H.; Kim, D.W.; et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: Updated results from a phase 1 study. Lancet Oncol. 2012, 13, 1011–1019. [Google Scholar] [CrossRef]
- Hida, T.; Nokihara, H.; Kondo, M.; Kim, Y.H.; Azuma, K.; Seto, T.; Takiguchi, Y.; Nishio, M.; Yoshioka, H.; Imamura, F.; et al. Alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer (J-ALEX): An open-label, randomised phase 3 trial. Lancet 2017, 390, 29–39. [Google Scholar] [CrossRef]
- Zhou, C.; Lu, Y.; Kim, S.; Reungwetwattana, T.; Zhou, J.; Zhang, Y.; He, J.; Yang, J.; Cheng, Y.; Lee, S.H.; et al. Primary results of ALESIA: A randomised, phase III, open-label study of alectinib vs crizotinib in Asian patients with treatment-naïve ALK+ Advanced Non-Small Cell Lung Cancer. In Proceedings of the European Society for Medical Oncology 2018 Congress, Munich, Germany, 22 October 2018. [Google Scholar]
- Solomon, B.J.; Besse, B.; Bauer, T.M.; Felip, E.; Soo, R.A.; Camidge, D.R.; Chiari, R.; Bearz, A.; Lin, C.C.; Gadgeel, S.M.; et al. Lorlatinib in patients with ALK-positive non-small-cell lung cancer: Results from a global phase 2 study. Lancet Oncol. 2018, 19, 1654–1667. [Google Scholar] [CrossRef]
- Shaw, A.T.; Friboulet, L.; Leshchiner, I.; Gainor, J.F.; Bergqvist, S.; Brooun, A.; Burke, B.J.; Deng, Y.L.; Liu, W.; Dardaei, L.; et al. Resensitization to Crizotinib by the Lorlatinib ALK Resistance Mutation L1198F. N. Engl. J. Med. 2016, 374, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Isozaki, H.; Ichihara, E.; Takigawa, N.; Sendo, T.; Tanimoto, M.; Kiura, K. Crizotinib to overcome alectinib-resistance in non-small cell lung cancer (NSCLC) harboring EML4-ALK. J. Clin. Oncol. 2015, 33, e19140. [Google Scholar]
- Isozaki, H.; Ichihara, E.; Takigawa, N.; Ohashi, K.; Ochi, N.; Yasugi, M.; Ninomiya, T.; Yamane, H.; Hotta, K.; Sakai, K.; et al. Non-Small Cell Lung Cancer Cells Acquire Resistance to the ALK Inhibitor Alectinib by Activating Alternative Receptor Tyrosine Kinases. Cancer Res. 2016, 76, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Kim, D.W.; Tiseo, M.; Langer, C.J.; Ahn, M.J.; Shaw, A.T.; Huber, R.M.; Hochmair, M.J.; Lee, D.H.; Bazhenova, L.A.; et al. Exploratory Analysis of Brigatinib Activity in Patients With Anaplastic Lymphoma Kinase-Positive Non-Small-Cell Lung Cancer and Brain Metastases in Two Clinical Trials. J. Clin. Oncol. 2018, 36, 2693–2701. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.; Reungwetwattana, T.; Chewaskulyong, B.; Dechaphunkul, A.; Lee, K.H.; Imamura, F.; Nogami, N.; Ohe, Y.; Cheng, Y.; Cho, B.C.; et al. Osimertinib vs standard of care (SoC) EGFR-TKI as first-line therapy in patients (pts) with EGFRm advanced NSCLC: FLAURA. In Proceedings of the ESMO 2017 Congress, Madrid, Spain, 9 September 2017. [Google Scholar]
- Riely, G.J.; Politi, K.A.; Miller, V.A.; Pao, W. Update on epidermal growth factor receptor mutations in non-small cell lung cancer. Clin. Cancer Res. 2006, 12, 7232–7241. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.; O’Brien, S.G.; Guilhot, F.; Goldman, J.M.; Hochhaus, A.; Hughes, T.P.; Radich, J.P.; Hatfield, A.K.; Mone, M.; Filian, J.; et al. International Randomized Study of Interferon Vs STI571 (IRIS) 8-Year Follow up: Sustained Survival and Low Risk for Progression or Events in Patients with Newly Diagnosed Chronic Myeloid Leukemia in Chronic Phase (CML-CP) Treated with Imatinib. Blood 2009, 114, 1126. [Google Scholar]
- Camidge, D.R.; Pao, W.; Sequist, L.V. Acquired resistance to TKIs in solid tumours: Learning from lung cancer. Nat. Rev. Clin. Oncol. 2014, 11, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, N.; Iwama, E.; Kubo, N.; Harada, T.; Miyawaki, K.; Tanaka, K.; Okamoto, I.; Baba, E.; Akashi, K.; Sasaki, H.; et al. Most T790M mutations are present on the same EGFR allele as activating mutations in patients with non-small cell lung cancer. Lung Cancer 2017, 108, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Godin-Heymann, N.; Ulkus, L.; Brannigan, B.W.; McDermott, U.; Lamb, J.; Maheswaran, S.; Settleman, J.; Haber, D.A. The T790M “gatekeeper” mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Mol. Cancer Ther. 2008, 7, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sos, M.L.; Rode, H.B.; Heynck, S.; Peifer, M.; Fischer, F.; Kluter, S.; Pawar, V.G.; Reuter, C.; Heuckmann, J.M.; Weiss, J.; et al. Chemogenomic profiling provides insights into the limited activity of irreversible EGFR Inhibitors in tumor cells expressing the T790M EGFR resistance mutation. Cancer Res. 2010, 70, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Westover, D.; Zugazagoitia, J.; Cho, B.C.; Lovly, C.M.; Paz-Ares, L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann. Oncol. 2018, 29, i10–i19. [Google Scholar] [CrossRef] [PubMed]
- Heon, S.; Yeap, B.Y.; Britt, G.J.; Costa, D.B.; Rabin, M.S.; Jackman, D.M.; Johnson, B.E. Development of central nervous system metastases in patients with advanced non-small cell lung cancer and somatic EGFR mutations treated with gefitinib or erlotinib. Clin. Cancer Res. 2010, 16, 5873–5882. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Spicer, J. Epidermal growth factor receptor tyrosine kinase inhibitors in the treatment of epidermal growth factor receptor-mutant non-small cell lung cancer metastatic to the brain. Clin. Cancer Res. 2012, 18, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Wakeling, A.E.; Guy, S.P.; Woodburn, J.R.; Ashton, S.E.; Curry, B.J.; Barker, A.J.; Gibson, K.H. ZD1839 (Iressa): An orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002, 62, 5749–5754. [Google Scholar] [PubMed]
- Weber, B.; Winterdahl, M.; Memon, A.; Sorensen, B.S.; Keiding, S.; Sorensen, L.; Nexo, E.; Meldgaard, P. Erlotinib accumulation in brain metastases from non-small cell lung cancer: Visualization by positron emission tomography in a patient harboring a mutation in the epidermal growth factor receptor. J. Thorac. Oncol. 2011, 6, 1287–1289. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.; Katakami, N.; Yoshioka, H.; Takeshita, J.; Tanaka, K.; Nanjo, S.; Fujita, S.; Kaji, R.; Imai, Y.; Monden, K.; et al. Rebiopsy of non-small cell lung cancer patients with acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitor: Comparison between T790M mutation-positive and mutation-negative populations. Cancer 2013, 119, 4325–4332. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Tan, E.H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.C.; Lee, K.H.; Lu, S.; et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016, 17, 577–589. [Google Scholar] [CrossRef]
- Mok, T.S.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Lee, M.; Linke, R.; Rosell, R.; Corral, J.; et al. Improvement in Overall Survival in a Randomized Study That Compared Dacomitinib With Gefitinib in Patients With Advanced Non-Small-Cell Lung Cancer and EGFR-Activating Mutations. J. Clin. Oncol. 2018, 36, 2244–2250. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Tan, E.H.; O’Byrne, K.; Zhang, L.; Hirsh, V.; Boyer, M.; Yang, J.C.; Mok, T.; Lee, K.H.; Lu, S.; et al. Afatinib versus gefitinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: Overall survival data from the phase IIb LUX-Lung 7 trial. Ann. Oncol. 2017, 28, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Miller, V.A.; Hirsh, V.; Cadranel, J.; Chen, Y.M.; Park, K.; Kim, S.W.; Zhou, C.; Su, W.C.; Wang, M.; Sun, Y.; et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): A phase 2b/3 randomised trial. Lancet Oncol. 2012, 13, 528–538. [Google Scholar] [CrossRef]
- Soria, J.C.; Felip, E.; Cobo, M.; Lu, S.; Syrigos, K.; Lee, K.H.; Goker, E.; Georgoulias, V.; Li, W.; Isla, D.; et al. Afatinib versus erlotinib as second-line treatment of patients with advanced squamous cell carcinoma of the lung (LUX-Lung 8): An open-label randomised controlled phase 3 trial. Lancet Oncol. 2015, 16, 897–907. [Google Scholar] [CrossRef]
- Moyer, J.D.; Barbacci, E.G.; Iwata, K.K.; Arnold, L.; Boman, B.; Cunningham, A.; DiOrio, C.; Doty, J.; Morin, M.J.; Moyer, M.P.; et al. Induction of apoptosis and cell cycle arrest by CP-358,774, an inhibitor of epidermal growth factor receptor tyrosine kinase. Cancer Res. 1997, 57, 4838–4848. [Google Scholar] [PubMed]
- Hirano, T.; Yasuda, H.; Tani, T.; Hamamoto, J.; Oashi, A.; Ishioka, K.; Arai, D.; Nukaga, S.; Miyawaki, M.; Kawada, I.; et al. In vitro modeling to determine mutation specificity of EGFR tyrosine kinase inhibitors against clinically relevant EGFR mutants in non-small-cell lung cancer. Oncotarget 2015, 6, 38789–38803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.R.; Padera, R.F.; Shapiro, G.I.; Baum, A.; Himmelsbach, F.; et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702–4711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Ballard, P.; Yates, J.W.; Yang, Z.; Kim, D.W.; Yang, J.C.; Cantarini, M.; Pickup, K.; Jordan, A.; Hickey, M.; Grist, M.; et al. Preclinical Comparison of Osimertinib with Other EGFR-TKIs in EGFR-Mutant NSCLC Brain Metastases Models, and Early Evidence of Clinical Brain Metastases Activity. Clin. Cancer Res. 2016, 22, 5130–5140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxnard, G.R.; Thress, K.S.; Alden, R.S.; Lawrance, R.; Paweletz, C.P.; Cantarini, M.; Yang, J.C.; Barrett, J.C.; Janne, P.A. Association Between Plasma Genotyping and Outcomes of Treatment With Osimertinib (AZD9291) in Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 3375–3382. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Yang, J.C.; Lee, C.K.; Kurata, T.; Kim, D.W.; John, T.; Nogami, N.; Ohe, Y.; Mann, H.; Rukazenkov, Y.; et al. Osimertinib As First-Line Treatment of EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2017, 36, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Wu, Y.L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.P.; O’Byrne, K.; Feng, J.; et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef]
- Janne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, J.L.; Heideman, D.A.; Thunnissen, E.; Paul, M.A.; van Wijk, A.W.; Postmus, P.E.; Smit, E.F. Incidence of T790M mutation in (sequential) rebiopsies in EGFR-mutated NSCLC-patients. Lung Cancer 2014, 85, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Papadimitrakopoulou, V.A.; Collins, B.; Chmielecki, J.; Barrett, J.C.; Mok, T.S.K.; Wu, Y.-L.; Han, J.-Y.; Ahn, M.-J.; Ramalingam, S.S.; John, T.; et al. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Rukazenkov, Y.; Todd, A.; Markovets, A.; Chmielecki, J.; Barrett, J.C.; Gray, J.; Cheng, Y.; Zhou, C.; Ohe, Y.; et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Arulananda, S.; Do, H.; Musafer, A.; Mitchell, P.; Dobrovic, A.; John, T. Combination Osimertinib and Gefitinib in C797S and T790M EGFR-Mutated Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2017, 12, 1728–1732. [Google Scholar] [CrossRef] [PubMed]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [PubMed]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Tian, S.K.; Drilon, A.E.; Borsu, L.; Riely, G.J.; Arcila, M.E.; Ladanyi, M. Acquired Resistance of EGFR-Mutant Lung Cancer to a T790M-Specific EGFR Inhibitor: Emergence of a Third Mutation (C797S) in the EGFR Tyrosine Kinase Domain. JAMA Oncol. 2015, 1, 982–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersanelli, M.; Minari, R.; Bordi, P.; Gnetti, L.; Bozzetti, C.; Squadrilli, A.; Lagrasta, C.A.; Bottarelli, L.; Osipova, G.; Capelletto, E.; et al. L718Q Mutation as New Mechanism of Acquired Resistance to AZD9291 in EGFR-Mutated NSCLC. J. Thorac. Oncol. 2016, 11, e121–e123. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Azuma, K.; Nagai, H.; Kim, Y.H.; Togashi, Y.; Sesumi, Y.; Chiba, M.; Shimoji, M.; Sato, K.; Tomizawa, K.; et al. Characterization of EGFR T790M, L792F, and C797S Mutations as Mechanisms of Acquired Resistance to Afatinib in Lung Cancer. Mol. Cancer Ther. 2017, 16, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, Y.; Liu, D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017, 385, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Loriot, Y.; Andre, F.; Gobert, A.; Auger, N.; Lacroix, L.; Soria, J.C. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann. Oncol. 2015, 26, 2073–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz-Cuaran, S.; Scheffler, M.; Plenker, D.; Dahmen, L.; Scheel, A.H.; Fernandez-Cuesta, L.; Meder, L.; Lovly, C.M.; Persigehl, T.; Merkelbach-Bruse, S.; et al. Heterogeneous Mechanisms of Primary and Acquired Resistance to Third-Generation EGFR Inhibitors. Clin. Cancer Res. 2016, 22, 4837–4847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piotrowska, Z.; Niederst, M.J.; Karlovich, C.A.; Wakelee, H.A.; Neal, J.W.; Mino-Kenudson, M.; Fulton, L.; Hata, A.N.; Lockerman, E.L.; Kalsy, A.; et al. Heterogeneity Underlies the Emergence of EGFRT790 Wild-Type Clones Following Treatment of T790M-Positive Cancers with a Third-Generation EGFR Inhibitor. Cancer Discov. 2015, 5, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Fu, W.; Zheng, L.; Liu, Z.; Liang, G. Recent progress of small-molecule epidermal growth factor receptor (EGFR) inhibitors against C797S resistance in non-small-cell lung cancer. J. Med. Chem. 2018, 61, 4290–4300. [Google Scholar] [CrossRef] [PubMed]
- Knebel, F.H.; Bettoni, F.; Shimada, A.K.; Cruz, M.; Alessi, J.V.; Negrao, M.V.; Reis, L.F.L.; Katz, A.; Camargo, A.A. Sequential liquid biopsies reveal dynamic alterations of EGFR driver mutations and indicate EGFR amplification as a new mechanism of resistance to osimertinib in NSCLC. Lung Cancer 2017, 108, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.I.; Cui, J.; Schrock, A.B.; Goldberg, M.E.; Zhu, V.W.; Albacker, L.; Stephens, P.J.; Miller, V.A.; Ali, S.M. Emergence of novel and dominant acquired EGFR solvent-front mutations at Gly796 (G796S/R) together with C797S/R and L792F/H mutations in one EGFR (L858R/T790M) NSCLC patient who progressed on osimertinib. Lung Cancer 2017, 108, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Hu, M.; Bai, Y.; Zhu, X.; Lu, X.; Wu, C.; Wang, J.; Liu, L.; Wang, Z.; Ni, J.; et al. EGFR G796D mutation mediates resistance to osimertinib. Oncotarget 2017, 8, 49671–49679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.M.; Song, A.; Kim, D.W.; Kim, S.; Ahn, Y.O.; Keam, B.; Jeon, Y.K.; Lee, S.H.; Chung, D.H.; Heo, D.S. Mechanisms of Acquired Resistance to AZD9291: A Mutation-Selective, Irreversible EGFR Inhibitor. J. Thorac. Oncol. 2015, 10, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Yang, J.J.; Huang, J.; Ye, J.Y.; Zhang, X.C.; Tu, H.Y.; Han-Zhang, H.; Wu, Y.L. Lung Adenocarcinoma Harboring EGFR T790M and In Trans C797S Responds to Combination Therapy of First- and Third-Generation EGFR TKIs and Shifts Allelic Configuration at Resistance. J. Thorac. Oncol. 2017, 12, 1723–1727. [Google Scholar] [CrossRef] [PubMed]
- Babu Koyyala, V.P.; Batra, U.; Jain, P.; Sharma, M.; Goyal, P.; Domadia, K.; Botra, S. Good response to erlotinib in a patient after progression on osimertinib: A rare case of spatiotemporal T790M heterogeneity in a patient with epidermal growth factor receptor-mutant nonsmall cell lung cancer. South Asian J Cancer 2017, 6, 179. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Category | Example TKI | Wt EGFR (IC50 in nM) | Exon 19del (IC50 in nM) | L858R (IC50 in nM) | Exon 19del + T790M EGFRm (IC50 in nM) | L858R + T790M (IC50 in nM) | Dosing Schedule | Common Toxicities (% with Grades 3–4) |
|---|---|---|---|---|---|---|---|---|
| First-generation EGFR TKI | erlotinib | 20 [51] | 23 [52] | 39 [52] | 1600 [52] | >10000 [52] | 150 mg once daily | diarrhea (5), rash (12), paronychia (0), stomatitis (1) [16] |
| Second-generation EGFR TKI | afatinib | 0.5 [53] | 0.2 [52] | 0.2 [52] | 141 [52] | 196 [52] | 40 mg once daily | diarrhea (14), rash (16), paronychia (11), stomatitis (8) [18] |
| Third-generation EGFR TKI | osimertinib | 938 [52] | 12 [52] | 9 [52] | 3 [52] | 13 [52] | 80 mg once daily | diarrhea (2), rash (3), paronychia (1), stomatitis (5) [29] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, T.; Gerber, D.E. Newer-Generation EGFR Inhibitors in Lung Cancer: How Are They Best Used? Cancers 2019, 11, 366. https://doi.org/10.3390/cancers11030366
Le T, Gerber DE. Newer-Generation EGFR Inhibitors in Lung Cancer: How Are They Best Used? Cancers. 2019; 11(3):366. https://doi.org/10.3390/cancers11030366
Chicago/Turabian StyleLe, Tri, and David E. Gerber. 2019. "Newer-Generation EGFR Inhibitors in Lung Cancer: How Are They Best Used?" Cancers 11, no. 3: 366. https://doi.org/10.3390/cancers11030366
APA StyleLe, T., & Gerber, D. E. (2019). Newer-Generation EGFR Inhibitors in Lung Cancer: How Are They Best Used? Cancers, 11(3), 366. https://doi.org/10.3390/cancers11030366