Butoxy Mansonone G Inhibits STAT3 and Akt Signaling Pathways in Non-Small Cell Lung Cancers: Combined Experimental and Theoretical Investigations

 and
and
Abstract
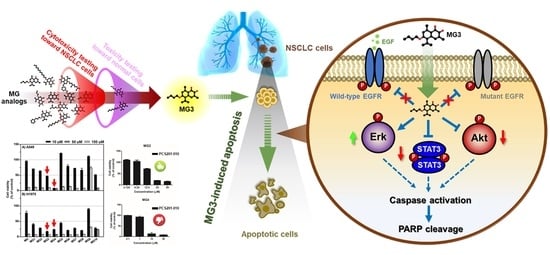
:
1. Introduction
2. Results
2.1. In Vitro Cytotoxicity Screening of MG Derivatives against NSCLC Cell Lines
2.2. Butoxy Mansonone G Exhibits a Potent Cytotoxicity against NSCLC Cells
2.3. Butoxy Mansonone G Induces Apoptosis in A549 and H1975 Cell Lines
2.4. Butoxy Mansonone G Inhibits STAT3 and Akt Signaling Pathways in NSCLC Cell Lines
2.5. Predictive Binding Affinity of Butoxy Mansonone G against STAT3 and Akt Signaling Proteins
2.6. Key Binding Residues
2.7. In Silico Study on Conformational Change of STAT3 and Akt upon Butoxy Mansonone G Binding
3. Discussion
4. Materials and Methods
4.1. Experimental Part
4.1.1. Chemical Reagents and Antibodies
4.1.2. Cell Lines and Culture
4.1.3. Cell Viability Assay
4.1.4. Western Blotting
4.1.5. Flow Cytometric Evaluation of Apoptosis
4.1.6. Statistical Analysis
4.2. Computational Part
4.2.1. Preparation of Initial Structures
4.2.2. Molecular Dynamics (MD) Simulations and Binding Free Energy Calculations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Ahmedin, J. Cancer statistics, 2018. CA A Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.-Y.D.; Cramb, S.M.; Baade, P.D.; Youlden, D.R.; Nwogu, C.; Reid, M.E. The International Epidemiology of Lung Cancer: Latest Trends, Disparities, and Tumor Characteristics. J. Thorac. Oncol. 2016, 11, 1653–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islami, F.; Torre, L.A.; Jemal, A. Global trends of lung cancer mortality and smoking prevalence. Transl. Lung Cancer Res. 2015, 4, 327–338. [Google Scholar]
- Wong, M.C.S.; Lao, X.Q.; Ho, K.-F.; Goggins, W.B.; Tse, S.L.A. Incidence and mortality of lung cancer: Global trends and association with socioeconomic status. Sci. Rep. 2017, 7, 14300. [Google Scholar] [CrossRef]
- Zhang, H. Osimertinib making a breakthrough in lung cancer targeted therapy. Onco Targets Ther. 2016, 9, 5489–5493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar] [PubMed]
- Li, X.; Fan, X.X.; Jiang, Z.B.; Loo, W.T.; Yao, X.J.; Leung, E.L.; Chow, L.W.; Liu, L. Shikonin inhibits gefitinib-resistant non-small cell lung cancer by inhibiting TrxR and activating the EGFR proteasomal degradation pathway. Pharmacol. Res. 2017, 115, 45–55. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Rose, M.C.; Kostyanovskaya, E.; Huang, R.S. Pharmacogenomics of Cisplatin Sensitivity in Non-small Cell Lung Cancer. Genom. Proteom. Bioinform. 2014, 12, 198–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, T.-J.; Ku, W.-H.; Chen, H.-Y.; Hsu, Y.-C.; Hong, Q.-S.; Chang, G.-C.; Yu, S.-L.; Chen, J.J.W. Oncogenic miR-137 contributes to cisplatin resistance via repressing CASP3 in lung adenocarcinoma. Am. J. Cancer Res. 2016, 6, 1317–1330. [Google Scholar] [PubMed]
- Fennell, D.A.; Summers, Y.; Cadranel, J.; Benepal, T.; Christoph, D.C.; Lal, R.; Das, M.; Maxwell, F.; Visseren-Grul, C.; Ferry, D. Cisplatin in the modern era: The backbone of first-line chemotherapy for non-small cell lung cancer. Cancer Treat. Rev. 2016, 44, 42–50. [Google Scholar] [CrossRef]
- Smit, E.; Moro-Sibilot, D.; Carpeño, J.d.C.; Lesniewski-Kmak, K.; Aerts, J.; Villatoro, R.; Kraaij, K.; Nacerddine, K.; Dyachkova, Y.; Smith, K.T.; et al. Cisplatin and carboplatin-based chemotherapy in the first-line treatment of non-small cell lung cancer: Analysis from the European FRAME study. Lung Cancer (Amst. Neth.) 2016, 92, 35–40. [Google Scholar] [CrossRef]
- Ozkaya, S.; Findik, S.; Atici, A.G.; Dirica, A. Cisplatin-based chemotherapy in elderly patients with advanced stage (IIIB and IV) non-small cell lung cancer patients. Neoplasma 2011, 58, 348–351. [Google Scholar] [CrossRef] [Green Version]
- Raimbourg, J.; Joalland, M.P.; Cabart, M.; de Plater, L.; Bouquet, F.; Savina, A.; Decaudin, D.; Bennouna, J.; Vallette, F.M.; Lalier, L. Sensitization of EGFR Wild-Type Non-Small Cell Lung Cancer Cells to EGFR-Tyrosine Kinase Inhibitor Erlotinib. Mol. Cancer Ther. 2017, 16, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Leighl, N.B. Treatment paradigms for patients with metastatic non-small-cell lung cancer: First-, second-, and third-line. Curr. Oncol. (Tor. Ont.) 2012, 19, S52–S58. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Zheng, Y. Cisplatin or Carboplatin for Advanced Non–Small-Cell Lung Cancer? J. Thorac. Oncol. 2014, 9, e70. [Google Scholar] [CrossRef] [Green Version]
- Sarin, N.; Engel, F.; Kalayda, G.V.; Mannewitz, M.; Cinatl, J., Jr.; Rothweiler, F.; Michaelis, M.; Saafan, H.; Ritter, C.A.; Jaehde, U.; et al. Cisplatin resistance in non-small cell lung cancer cells is associated with an abrogation of cisplatin-induced G2/M cell cycle arrest. PLoS ONE 2017, 12, e0181081. [Google Scholar] [CrossRef]
- Rosell, R.; Taron, M.; Barnadas, A.; Scagliotti, G.; Sarries, C.; Roig, B. Nucleotide excision repair pathways involved in Cisplatin resistance in non-small-cell lung cancer. Cancer Control. J. Moffitt Cancer Cent. 2003, 10, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.T.; Chen, H.H.; Song, I.S.; Savaraj, N.; Ishikawa, T. The roles of copper transporters in cisplatin resistance. Cancer Metastasis Rev. 2007, 26, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2011, 31, 1869. [Google Scholar] [CrossRef]
- Weber, B.; Hager, H.; Sorensen, B.S.; McCulloch, T.; Mellemgaard, A.; Khalil, A.A.; Nexo, E.; Meldgaard, P. EGFR mutation frequency and effectiveness of erlotinib: A prospective observational study in Danish patients with non-small cell lung cancer. Lung Cancer (Amst. Neth.) 2014, 83, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, N.; Azuma, K.; Sakai, K.; Hattori, S.; Kawahara, A.; Ishii, H.; Tokito, T.; Kinoshita, T.; Yamada, K.; Nishio, K.; et al. Association of EGFR Exon 19 Deletion and EGFR-TKI Treatment Duration with Frequency of T790M Mutation in EGFR-Mutant Lung Cancer Patients. Sci. Rep. 2016, 6, 36458. [Google Scholar] [CrossRef] [Green Version]
- Wakeling, A.E.; Guy, S.P.; Woodburn, J.R.; Ashton, S.E.; Curry, B.J.; Barker, A.J.; Gibson, K.H. ZD1839 (Iressa). An Orally Active Inhibitor of Epidermal Growth Factor Signaling with Potential for Cancer Therapy. Cancer Res. 2002, 62, 5749–5754. [Google Scholar] [PubMed]
- Wang, S.; Cang, S.; Liu, D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Mayor, S. Osimertinib effective in EGFR T790M-positive lung cancer. Lancet Oncol. 2017, 18, e9. [Google Scholar] [CrossRef]
- Ma, C.; Wei, S.; Song, Y. T790M and acquired resistance of EGFR TKI: A literature review of clinical reports. J. Thorac. Dis. 2011, 3, 10–18. [Google Scholar]
- Ko, B.; Paucar, D.; Halmos, B. EGFR T790M: Revealing the secrets of a gatekeeper. Lung Cancer Targets Ther. 2017, 8, 147–159. [Google Scholar] [CrossRef]
- Hairani, R.; Mongkol, R.; Chavasiri, W. Allyl and prenyl ethers of mansonone G, new potential semisynthetic antibacterial agents. Bioorg. Med. Chem. Lett. 2016, 26, 5300–5303. [Google Scholar] [CrossRef]
- Dai, Y.; Harinantenaina, L.; Brodie, P.J.; Callmander, M.W.; Randrianasolo, S.; Rakotobe, E.; Rasamison, V.E.; Kingston, D.G.I. Isolation and Synthesis of Two Antiproliferative Calamenene-type Sesquiterpenoids from Sterculia tavia from the Madagascar Rain Forest. Bioorg. Med. Chem. 2012, 20, 6940–6944. [Google Scholar] [CrossRef]
- Mongkol, R.; Chavasiri, W. Antimicrobial, herbicidal and antifeedant activities of mansonone E from the heartwoods of Mansonia gagei Drumm. J. Integr. Agric. 2016, 15, 2795–2802. [Google Scholar] [CrossRef]
- El-Halawany, A.M.; Salah El Dine, R.; Hattori, M. Anti-estrogenic activity of mansonone G and mansorin A derivatives. Pharm. Biol. 2013, 51, 948–954. [Google Scholar] [CrossRef] [Green Version]
- Changwong, N.; Sabphon, C.; Ingkaninan, K.; Sawasdee, P. Acetyl- and butyryl-cholinesterase inhibitory activities of mansorins and mansonones. Phytother. Res. PTR 2012, 26, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Hairani, R.; Jeong, H.; Jeong, M.G.; Chavasiri, W.; Hwang, E.S. CBMG, a novel derivative of mansonone G suppresses adipocyte differentiation via suppression of PPARγ activity. Chem. Biol. Interact. 2017, 273, 160–170. [Google Scholar] [CrossRef]
- Cetintas, V.B.; Kucukaslan, A.S.; Kosova, B.; Tetik, A.; Selvi, N.; Cok, G.; Gunduz, C.; Eroglu, Z. Cisplatin resistance induced by decreased apoptotic activity in non-small-cell lung cancer cell lines. Cell Biol. Int. 2012, 36, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Bortner, C.D.; Cidlowski, J.A. A necessary role for cell shrinkage in apoptosis. Biochem. Pharmacol. 1998, 56, 1549–1559. [Google Scholar] [CrossRef]
- Yen, H.-Y.; Liu, Y.-C.; Chen, N.-Y.; Tsai, C.-F.; Wang, Y.-T.; Chen, Y.-J.; Hsu, T.-L.; Yang, P.-C.; Wong, C.-H. Effect of sialylation on EGFR phosphorylation and resistance to tyrosine kinase inhibition. Proc. Natl. Acad. Sci. USA 2015, 112, 6955–6960. [Google Scholar] [CrossRef] [Green Version]
- Lelj-Garolla, B.; Kumano, M.; Beraldi, E.; Nappi, L.; Rocchi, P.; Ionescu, D.N.; Fazli, L.; Zoubeidi, A.; Gleave, M.E. Hsp27 Inhibition with OGX-427 Sensitizes Non–Small Cell Lung Cancer Cells to Erlotinib and Chemotherapy. Mol. Cancer Ther. 2015, 14, 1107–1116. [Google Scholar] [CrossRef]
- Shin, D.-S.; Kim, H.-N.; Shin, K.D.; Yoon, Y.J.; Kim, S.-J.; Han, D.C.; Kwon, B.-M. Cryptotanshinone Inhibits Constitutive Signal Transducer and Activator of Transcription 3 Function through Blocking the Dimerization in DU145 Prostate Cancer Cells. Cancer Res. 2009, 69, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Siddiquee, K.; Zhang, S.; Guida, W.C.; Blaskovich, M.A.; Greedy, B.; Lawrence, H.R.; Yip, M.L.R.; Jove, R.; McLaughlin, M.M.; Lawrence, N.J.; et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7391–7396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pachl, F.; Plattner, P.; Ruprecht, B.; Medard, G.; Sewald, N.; Kuster, B. Characterization of a chemical affinity probe targeting Akt kinases. J. Proteome Res. 2013, 12, 3792–3800. [Google Scholar] [CrossRef] [PubMed]
- Burns, S.; Travers, J.; Collins, I.; Rowlands, M.G.; Newbatt, Y.; Thompson, N.; Garrett, M.D.; Workman, P.; Aherne, W. Identification of small-molecule inhibitors of protein kinase B (PKB/AKT) in an AlphaScreenTM high-throughput screen. J. Biomol. Screen. 2006, 11, 822–827. [Google Scholar] [CrossRef]
- Samadi Moghaddam, M.; Heiny, M.; Shastri, V.P. Enhanced cellular uptake of nanoparticles by increasing the hydrophobicity of poly (lactic acid) through copolymerization with cell-membrane-lipid components. Chem. Commun. 2015, 51, 14605–14608. [Google Scholar] [CrossRef] [PubMed]
- Franks, N.P.; Lieb, W.R. Partitioning of long-chain alcohols into lipid bilayers: Implications for mechanisms of general anesthesia. Proc. Natl. Acad. Sci. USA 1986, 83, 5116–5120. [Google Scholar] [CrossRef]
- Li, Y.; Song, Z.; Jin, Y.; Tang, Z.; Kang, J.; Ma, X. Novel Selective and Potent EGFR Inhibitor that Overcomes T790M-Mediated Resistance in Non-Small Cell Lung Cancer. Molecules (Basel Switz.) 2016, 21, 1462. [Google Scholar] [CrossRef] [PubMed]
- Vazirian, M.; Naser Ostad, S.; Manayi, A.; Hadjiakhoondi, A.; Khanavi, M. Comparison of cytotoxic activity of some Iranian Stachys spp. extracts on different cancer cell lines. Res. J. Pharmacogn. 2014, 1, 23–28. [Google Scholar]
- Patel, R.M.; Patel, S.K. Cytotoxic activity of methanolic extract of artocarpus heterophyllus against a549, hela and mcf-7 cell lines. J. Appl. Pharm. 2011, 1, 167–171. [Google Scholar]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef]
- Suda, K.; Onozato, R.; Yatabe, Y.; Mitsudomi, T. EGFR T790M Mutation: A Double Role in Lung Cancer Cell Survival? J. Thorac. Oncol. 2009, 4, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Henkels, K.M.; Turchi, J.J. Cisplatin-induced apoptosis proceeds by caspase-3-dependent and -independent pathways in cisplatin-resistant and -sensitive human ovarian cancer cell lines. Cancer Res. 1999, 59, 3077–3083. [Google Scholar] [PubMed]
- Cummings, B.S.; Schnellmann, R.G. Cisplatin-induced renal cell apoptosis: Caspase 3-dependent and -independent pathways. J. Pharmacol. Exp. Ther. 2002, 302, 8–17. [Google Scholar] [CrossRef]
- Kaushal, G.P.; Kaushal, V.; Hong, X.; Shah, S.V. Role and regulation of activation of caspases in cisplatin-induced injury to renal tubular epithelial cells. Kidney Int. 2001, 60, 1726–1736. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.H.; Li, J.; Chen, P.; Jiang, H.G.; Wu, M.; Chen, Y.C. RNA interferences targeting the Fanconi anemia/BRCA pathway upstream genes reverse cisplatin resistance in drug-resistant lung cancer cells. J. Biomed. Sci. 2015, 22, 77. [Google Scholar] [CrossRef] [Green Version]
- Aziz, M.H.; Dreckschmidt, N.E.; Verma, A.K. Plumbagin, a Medicinal Plant–Derived Naphthoquinone, Is a Novel Inhibitor of the Growth and Invasion of Hormone-Refractory Prostate Cancer. Cancer Res. 2008, 68, 9024–9032. [Google Scholar] [CrossRef]
- Sandur, S.K.; Pandey, M.K.; Sung, B.; Aggarwal, B.B. 5-hydroxy-2-methyl-1, 4-naphthoquinone, a vitamin K3 analogue, suppresses STAT3 activation pathway through induction of protein tyrosine phosphatase, SHP-1: Potential role in chemosensitization. Mol. Cancer Res. MCR 2010, 8, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Sand, J.M.; Bin Hafeez, B.; Jamal, M.S.; Witkowsky, O.; Siebers, E.M.; Fischer, J.; Verma, A.K. Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone), isolated from Plumbago zeylanica, inhibits ultraviolet radiation-induced development of squamous cell carcinomas. Carcinogenesis 2012, 33, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.Y.; Fu, J.Y.; Yang, M.K.; Han, H.W.; Wang, P.F.; Zhang, Y.H.; Lin, H.Y.; Tang, C.Y.; Qi, J.L.; Yang, R.W.; et al. Identification of new shikonin derivatives as STAT3 inhibitors. Biochem. Pharmacol. 2017, 146, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.-Y.; Zhu, X.; Luo, Y.-L.; Lin, H.-Y.; Tang, C.-Y.; Qi, J.-L.; Pang, Y.-J.; Yang, R.-W.; Lu, G.-H.; Wang, X.-M.; et al. Identification of New Shikonin Derivatives as Antitumor Agents Targeting STAT3 SH2 Domain. Sci. Rep. 2017, 7, 2863. [Google Scholar] [CrossRef]
- Kawiak, A.; Lojkowska, E. Ramentaceone, a Naphthoquinone Derived from Drosera sp., Induces Apoptosis by Suppressing PI3K/Akt Signaling in Breast Cancer Cells. PLoS ONE 2016, 11, e0147718. [Google Scholar] [CrossRef]
- Nishina, A.; Miura, A.; Goto, M.; Terakado, K.; Sato, D.; Kimura, H.; Hirai, Y.; Sato, H.; Phay, N. Mansonone E from Mansonia gagei Inhibited alpha-MSH-Induced Melanogenesis in B16 Cells by Inhibiting CREB Expression and Phosphorylation in the PI3K/Akt Pathway. Biol. Pharm. Bull. 2018, 41, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Singh, J.; Zhang, X.; Yue, P.; Page, B.D.G.; Sharmeen, S.; Shahani, V.M.; Zhao, W.; Schimmer, A.D.; Turkson, J.; et al. Disruption of Transcriptionally Active Stat3 Dimers with Non-phosphorylated, Salicylic Acid-Based Small Molecules: Potent in vitro and Tumor Cell Activities. Chembiochem A Eur. J. Chem. Biol. 2009, 10, 1959–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.J.; Leung, K.H.; Chan, D.S.; Wang, Y.T.; Ma, D.L.; Leung, C.H. Identification of a natural product-like STAT3 dimerization inhibitor by structure-based virtual screening. Cell Death Dis. 2014, 5, e1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, S.; Yu, R.; Yu, Y.; Li, Y. Dual-inhibitors of STAT5 and STAT3: Studies from molecular docking and molecular dynamics simulations. J. Mol. Model. 2014, 20, 2399. [Google Scholar] [CrossRef] [PubMed]
- Sgrignani, J.; Garofalo, M.; Matkovic, M.; Merulla, J.; Catapano, C.V.; Cavalli, A. Structural Biology of STAT3 and Its Implications for Anticancer Therapies Development. Int. J. Mol. Sci. 2018, 19, 1591. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.G.; Verdonk, M.L.; Graham, B.; Saalau-Bethell, S.; Hamlett, C.C.; McHardy, T.; Collins, I.; Garrett, M.D.; Workman, P.; Woodhead, S.J.; et al. A structural comparison of inhibitor binding to PKB, PKA and PKA-PKB chimera. J. Mol. Biol. 2007, 367, 882–894. [Google Scholar] [CrossRef]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Jackson, P.A.; Widen, J.C.; Harki, D.A.; Brummond, K.M. Covalent Modifiers: A Chemical Perspective on the Reactivity of α,β-Unsaturated Carbonyls with Thiols via Hetero-Michael Addition Reactions. J. Med. Chem. 2017, 60, 839–885. [Google Scholar] [CrossRef]
- Yang, J.; Cron, P.; Thompson, V.; Good, V.M.; Hess, D.; Hemmings, B.A.; Barford, D. Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol. Cell 2002, 9, 1227–1240. [Google Scholar] [CrossRef]
- Addie, M.; Ballard, P.; Buttar, D.; Crafter, C.; Currie, G.; Davies, B.R.; Debreczeni, J.; Dry, H.; Dudley, P.; Greenwood, R.; et al. Discovery of 4-amino-N-[(1S)-1-(4-chlorophenyl)-3-hydroxypropyl]-1-(7H-pyrrolo[2,3-d]pyrimidin -4-yl)piperidine-4-carboxamide (AZD5363), an orally bioavailable, potent inhibitor of Akt kinases. J. Med. Chem. 2013, 56, 2059–2073. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2002, 22, 7265. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.J.; Chiu, G.N. Role of oxidative stress, endoplasmic reticulum stress and ERK activation in triptolide-induced apoptosis. Int. J. Oncol. 2013, 42, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, S.; Schnellmann, R.G. A death-promoting role for extracellular signal-regulated kinase. J. Pharmacol. Exp. Ther. 2006, 319, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Persons, D.L.; Yazlovitskaya, E.M.; Pelling, J.C. Effect of extracellular signal-regulated kinase on p53 accumulation in response to cisplatin. J. Biol. Chem. 2000, 275, 35778–35785. [Google Scholar] [CrossRef]
- Greig, M.J.; Niessen, S.; Weinrich, S.L.; Feng, J.L.; Shi, M.; Johnson, T.O. Effects of Activating Mutations on EGFR Cellular Protein Turnover and Amino Acid Recycling Determined Using SILAC Mass Spectrometry. Int. J. Cell Biol. 2015, 2015, 798936. [Google Scholar] [CrossRef]
- Becker, S.; Groner, B.; Muller, C.W. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature 1998, 394, 145–151. [Google Scholar] [CrossRef]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]
- Mahalapbutr, P.; Chusuth, P.; Kungwan, N.; Chavasiri, W.; Wolschann, P.; Rungrotmongkol, T. Molecular recognition of naphthoquinone-containing compounds against human DNA topoisomerase IIα ATPase domain: A molecular modeling study. J. Mol. Liq. 2017, 247, 374–385. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Wu, G.; Robertson, D.H.; Brooks, C.L., 3rd; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- Olsson, M.H.; Sondergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Phanich, J.; Rungrotmongkol, T.; Kungwan, N.; Hannongbua, S. Role of R292K mutation in influenza H7N9 neuraminidase toward oseltamivir susceptibility: MD and MM/PB (GB)SA study. J. Comput.-Aided Mol. Des. 2016, 30, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Mahalapbutr, P.; Nutho, B.; Wolschann, P.; Chavasiri, W.; Kungwan, N.; Rungrotmongkol, T. Molecular insights into inclusion complexes of mansonone E and H enantiomers with various beta-cyclodextrins. J. Mol. Graph. Model. 2018, 79, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Meeprasert, A.; Hannongbua, S.; Rungrotmongkol, T. Key Binding and Susceptibility of NS3/4A Serine Protease Inhibitors against Hepatitis C Virus. J. Chem. Inf. Model. 2014, 54, 1208–1217. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangpheak, W.; Khuntawee, W.; Wolschann, P.; Pongsawasdi, P.; Rungrotmongkol, T. Enhanced stability of a naringenin/2, 6-dimethyl β-cyclodextrin inclusion complex: Molecular dynamics and free energy calculations based on MM- and QM-PBSA/GBSA. J. Mol. Graph. Model. 2014, 50, 10–15. [Google Scholar] [CrossRef]
- Mahalapbutr, P.; Thitinanthavet, K.; Kedkham, T.; Nguyen, H.; Theu, L.T.H.; Dokmaisrijan, S.; Huynh, L.; Kungwan, N.; Rungrotmongkol, T. A theoretical study on the molecular encapsulation of luteolin and pinocembrin with various derivatized beta-cyclodextrins. J. Mol. Struct. 2019, 1180, 480–490. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- York, D.M.; Darden, T.A.; Pedersen, L.G. The effect of long-range electrostatic interactions in simulations of macromolecular crystals: A comparison of the Ewald and truncated list methods. J. Chem. Phys. 1993, 99, 8345–8348. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MG Analogs | A549 | H1975 | ||
|---|---|---|---|---|
| %CV10 µM | %CV100 µM | %CV10 µM | %CV100 µM | |
| MG | 95.31 ± 2.35 | 10.71 ± 0.95 | 77.28 ± 5.80 | 8.26 ± 0.79 |
| MG1 | 70.55 ± 0.27 | 9.59 ± 1.48 | 40.46 ± 2.83 | 7.04 ± 0.22 |
| MG2 | 63.57 ± 0.91 | 8.51 ± 0.66 | 28.33 ± 0.82 | 7.29 ± 0.57 |
| MG3 | 46.90 ± 1.21 | 8.94 ± 0.67 | 17.04 ± 1.42 | 6.50 ± 0.28 |
| MG4 | 7.93 ± 0.43 | 7.10 ± 0.22 a | 23.52 ± 1.44 | 7.42 ± 0.55 a |
| MG5 | 121.54 ± 0.36 | 12.12 ± 1.71 | 44.35 ± 2.98 | 7.79 ± 0.12 |
| MG6 | 79.56 ± 4.53 | 12.54 ± 1.33 | 20.64 ± 1.17 | 6.73 ± 0.15 |
| MG7 | 71.84 ± 1.15 | 7.45 ± 0.04 | 27.53 ± 1.54 | 6.57 ± 0.25 |
| MG8 | 66.59 ± 5.98 | 7.93 ± 0.06 | 20.24 ± 0.92 | 8.25 ± 0.32 |
| MG9 | 110.92 ± 1.72 | 72.86 ± 3.75 | 89.84 ± 2.39 | 29.50 ± 2.80 |
| MG10 | 51.00 ± 0.99 | 8.74 ± 0.30 a | 27.85 ± 2.35 | 23.19 ± 1.38 a |
| STAT3 | Akt | |||||
|---|---|---|---|---|---|---|
| CTS | S3I201 | MG3 | Uprosertib | H8 | MG3 | |
| ΔEele | −6.80 ± 0.44 | −110.36 ± 3.35 | −1.56 ± 0.30 | −146.76 ± 1.43 | −168.13 ± 1.16 | −9.43 ± 0.17 |
| ΔEvdW | −35.61 ± 0.21 | −37.47 ± 0.23 | −35.77 ±0.22 | −44.62 ± 0.20 | −33.92 ± 0.18 | −39.14 ± 0.17 |
| ΔEMM | −42.41 ± 0.48 | −147.83 ± 3.36 | −37.33 ±0.37 | −191.39 ± 1.48 | −202.05 ± 1.18 | −48.57 ± 0.24 |
| ΔGsolv, non-polar | −4.46 ± 0.02 | −5.71 ± 0.02 | −4.89 ± 0.02 | −6.04 ± 0.02 | −4.95 ± 0.01 | −4.85 ± 0.01 |
| ΔGsolv, polar | 23.99 ± 0.39 | 124.43 ± 3.15 | 15.67 ± 0.27 | 162.16 ± 1.22 | 176.02 ± 1.12 | 24.55 ± 0.15 |
| ΔGsolv | 19.53 ± 0.39 | 118.72 ± 3.15 | 10.78 ± 0.27 | 156.11 ± 1.22 | 171.07 ± 1.12 | 19.70 ± 0.15 |
| ΔEele+ΔGsolv, polar | 17.19 ± 0.58 | 14.07 ± 4.59 | 14.11 ± 0.40 | 15.40 ± 1.87 | 7.89 ± 1.61 | 15.12 ± 0.22 |
| ΔEvdW+ΔGsolv, non-polar | −40.07 ± 0.21 | −43.18 ± 0.23 | −40.66 ±0.22 | −50.66 ± 0.20 | −38.87 ± 0.18 | −43.99 ± 0.17 |
| −TΔS | 17.78 ± 1.69 | 25.38 ± 2.16 | 18.02 ± 1.93 | 24.82 ± 0.90 | 21.28 ± 0.66 | 19.67 ± 1.54 |
| ΔGbind | −5.09 ± 0.42 | −3.73 ± 0.58 | −8.54 ± 0.48 | −10.45 ± 0.38 | −9.68 ± 0.29 | −9.19 ± 0.39 |
| ΔGbind, exp | −7.26 | −5.54 | n/a | −9.19 | −6.46 | n/a |
| IC50 (μM) | 4.6 [40] | 86 [41] | n/a | 0.18 [42] | 18 [43] | n/a |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahalapbutr, P.; Wonganan, P.; Chavasiri, W.; Rungrotmongkol, T. Butoxy Mansonone G Inhibits STAT3 and Akt Signaling Pathways in Non-Small Cell Lung Cancers: Combined Experimental and Theoretical Investigations. Cancers 2019, 11, 437. https://doi.org/10.3390/cancers11040437
Mahalapbutr P, Wonganan P, Chavasiri W, Rungrotmongkol T. Butoxy Mansonone G Inhibits STAT3 and Akt Signaling Pathways in Non-Small Cell Lung Cancers: Combined Experimental and Theoretical Investigations. Cancers. 2019; 11(4):437. https://doi.org/10.3390/cancers11040437
Chicago/Turabian StyleMahalapbutr, Panupong, Piyanuch Wonganan, Warinthorn Chavasiri, and Thanyada Rungrotmongkol. 2019. "Butoxy Mansonone G Inhibits STAT3 and Akt Signaling Pathways in Non-Small Cell Lung Cancers: Combined Experimental and Theoretical Investigations" Cancers 11, no. 4: 437. https://doi.org/10.3390/cancers11040437
APA StyleMahalapbutr, P., Wonganan, P., Chavasiri, W., & Rungrotmongkol, T. (2019). Butoxy Mansonone G Inhibits STAT3 and Akt Signaling Pathways in Non-Small Cell Lung Cancers: Combined Experimental and Theoretical Investigations. Cancers, 11(4), 437. https://doi.org/10.3390/cancers11040437