Pheochromocytomas and Paragangliomas: From Genetic Diversity to Targeted Therapies




{kind=link}
Abstract
:1. Introduction
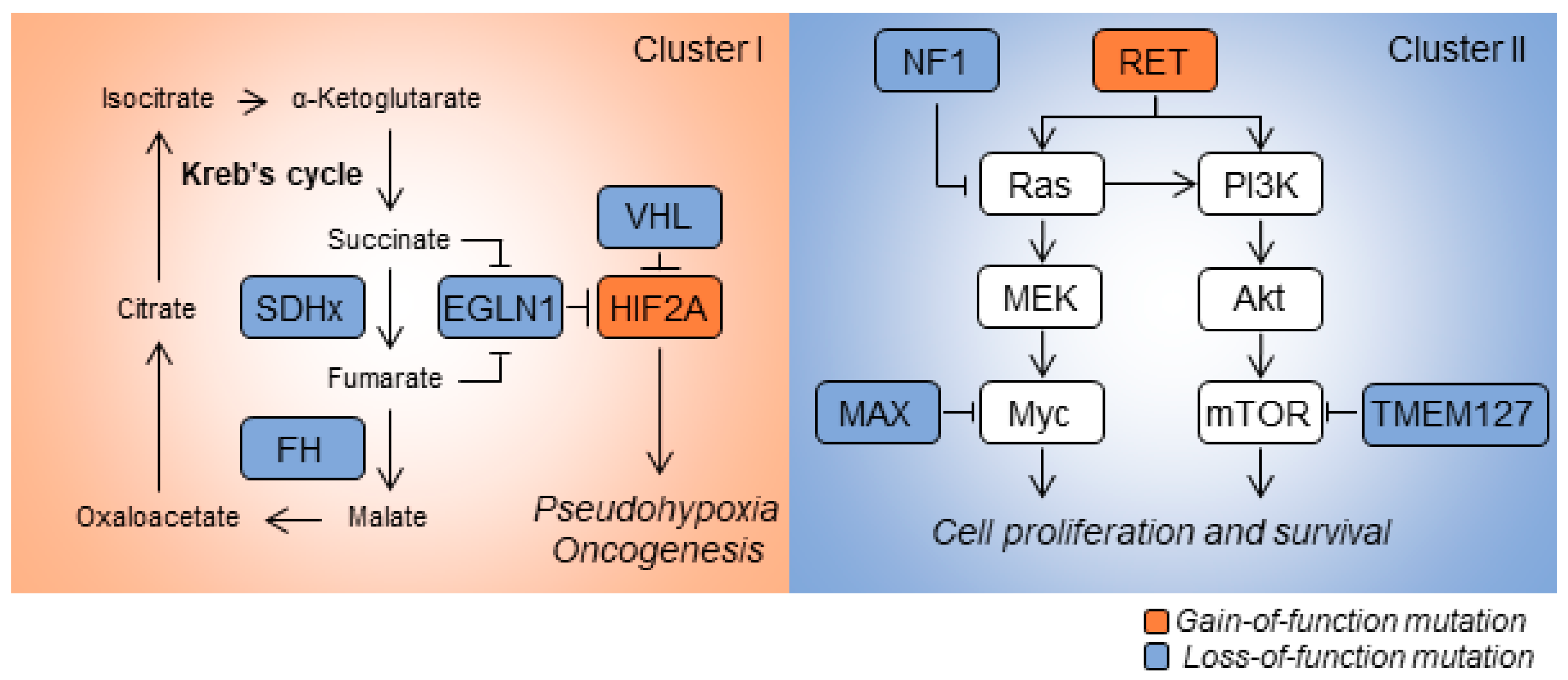
2. Genetics of PCPGs
2.1. SDHx
2.2. Von Hippel-Lindau (VHL)
2.3. HIF2A
2.4. Neurofibromin 1 (NF1)
2.5. RET
2.6. MAX
2.7. Harvey Rat Sarcoma Viral Oncogene Homologue (HRAS)
3. Current Therapies and Limitations
4. Targeted Molecular Therapies
4.1. Antiangiogenic Therapies
4.2. Hypoxia-Inducible Factor (HIF) Inhibitors
4.3. mTOR Inhibitors
4.4. DNA Demethylation
4.5. DNA-Alkylating Agents
4.6. PARP Inhibitors
4.7. Histone Deacetylase Inhibitors
4.8. Immunotherapy
4.9. Other Potential Therapies
5. Future Directions
6. Conclusions
Funding
Conflicts of Interest
References
- Crona, J.; Delgado Verdugo, A.; Maharjan, R.; Stalberg, P.; Granberg, D.; Hellman, P.; Bjorklund, P. Somatic mutations in H-RAS in sporadic pheochromocytoma and paraganglioma identified by exome sequencing. J. Clin. Endocrinol. Metab. 2013, 98, E1266–E1271. [Google Scholar] [PubMed]
- Luchetti, A.; Walsh, D.; Rodger, F.; Clark, G.; Martin, T.; Irving, R.; Sanna, M.; Yao, M.; Robledo, M.; Neumann, H.P.; et al. Profiling of somatic mutations in phaeochromocytoma and paraganglioma by targeted next generation sequencing analysis. Int. J. Endocrinol. 2015, 2015, 138573. [Google Scholar] [CrossRef] [PubMed]
- King, K.S.; Prodanov, T.; Kantorovich, V.; Fojo, T.; Hewitt, J.K.; Zacharin, M.; Wesley, R.; Lodish, M.; Raygada, M.; Gimenez-Roqueplo, A.P.; et al. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: Significant link to SDHB mutations. J. Clin. Oncol. 2011, 29, 4137–4142. [Google Scholar] [CrossRef] [PubMed]
- Erlic, Z.; Neumann, H.P. Familial pheochromocytoma. Hormones (Athens) 2009, 8, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnichon, N.; Vescovo, L.; Amar, L.; Libe, R.; de Reynies, A.; Venisse, A.; Jouanno, E.; Laurendeau, I.; Parfait, B.; Bertherat, J.; et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum. Mol. Genet. 2011, 20, 3974–3985. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Abraham, J.; Hung, E.; Averbuch, S.; Merino, M.; Steinberg, S.M.; Pacak, K.; Fojo, T. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: Recommendation from a 22-year follow-up of 18 patients. Cancer 2008, 113, 2020–2028. [Google Scholar] [CrossRef]
- Jochmanova, I.; Pacak, K. Genomic Landscape of Pheochromocytoma and Paraganglioma. Trends Cancer 2018, 4, 6–9. [Google Scholar] [CrossRef]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef]
- Castro-Vega, L.J.; Letouze, E.; Burnichon, N.; Buffet, A.; Disderot, P.H.; Khalifa, E.; Loriot, C.; Elarouci, N.; Morin, A.; Menara, M.; et al. Multi-omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat. Commun. 2015, 6, 6044. [Google Scholar] [CrossRef]
- Gill, A.J.; Benn, D.E.; Chou, A.; Clarkson, A.; Muljono, A.; Meyer-Rochow, G.Y.; Richardson, A.L.; Sidhu, S.B.; Robinson, B.G.; Clifton-Bligh, R.J. Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Hum. Pathol. 2010, 41, 805–814. [Google Scholar] [CrossRef]
- Gottlieb, E.; Tomlinson, I.P. Mitochondrial tumour suppressors: A genetic and biochemical update. Nat. Rev. Cancer 2005, 5, 857–866. [Google Scholar] [CrossRef]
- Vicha, A.; Taieb, D.; Pacak, K. Current views on cell metabolism in SDHx-related pheochromocytoma and paraganglioma. Endocr. Relat. Cancer 2014, 21, R261–277. [Google Scholar] [CrossRef]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der Mey, A.; Taschner, P.E.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef]
- Niemann, S.; Muller, U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat. Genet. 2000, 26, 268–270. [Google Scholar] [CrossRef]
- Jafri, M.; Maher, E.R. The genetics of phaeochromocytoma: Using clinical features to guide genetic testing. Eur. J. Endocrinol. 2012, 166, 151–158. [Google Scholar] [CrossRef]
- Pasini, B.; Stratakis, C.A. SDH mutations in tumorigenesis and inherited endocrine tumours: Lesson from the phaeochromocytoma-paraganglioma syndromes. J. Intern. Med. 2009, 266, 19–42. [Google Scholar] [CrossRef]
- Bayley, J.P.; Oldenburg, R.A.; Nuk, J.; Hoekstra, A.S.; van der Meer, C.A.; Korpershoek, E.; McGillivray, B.; Corssmit, E.P.; Dinjens, W.N.; de Krijger, R.R.; et al. Paraganglioma and pheochromocytoma upon maternal transmission of SDHD mutations. BMC Med. Genet. 2014, 15, 111. [Google Scholar] [CrossRef]
- Van der Mey, A.G.; Maaswinkel-Mooy, P.D.; Cornelisse, C.J.; Schmidt, P.H.; van de Kamp, J.J. Genomic imprinting in hereditary glomus tumours: Evidence for new genetic theory. Lancet 1989, 2, 1291–1294. [Google Scholar] [CrossRef]
- Hao, H.X.; Khalimonchuk, O.; Schraders, M.; Dephoure, N.; Bayley, J.P.; Kunst, H.; Devilee, P.; Cremers, C.W.; Schiffman, J.D.; Bentz, B.G.; et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 2009, 325, 1139–1142. [Google Scholar] [CrossRef]
- Burnichon, N.; Briere, J.J.; Libe, R.; Vescovo, L.; Riviere, J.; Tissier, F.; Jouanno, E.; Jeunemaitre, X.; Benit, P.; Tzagoloff, A.; et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum. Mol. Genet. 2010, 19, 3011–3020. [Google Scholar] [CrossRef] [Green Version]
- Comino-Mendez, I.; Gracia-Aznarez, F.J.; Schiavi, F.; Landa, I.; Leandro-Garcia, L.J.; Leton, R.; Honrado, E.; Ramos-Medina, R.; Caronia, D.; Pita, G.; et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat. Genet. 2011, 43, 663–667. [Google Scholar] [CrossRef]
- Astuti, D.; Latif, F.; Dallol, A.; Dahia, P.L.; Douglas, F.; George, E.; Skoldberg, F.; Husebye, E.S.; Eng, C.; Maher, E.R. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 2001, 69, 49–54. [Google Scholar] [CrossRef]
- Timmers, H.J.; Kozupa, A.; Eisenhofer, G.; Raygada, M.; Adams, K.T.; Solis, D.; Lenders, J.W.; Pacak, K. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. J. Clin. Endocrinol. Metab. 2007, 92, 779–786. [Google Scholar] [CrossRef]
- Kunst, H.P.; Rutten, M.H.; de Monnink, J.P.; Hoefsloot, L.H.; Timmers, H.J.; Marres, H.A.; Jansen, J.C.; Kremer, H.; Bayley, J.P.; Cremers, C.W. SDHAF2 (PGL2-SDH5) and hereditary head and neck paraganglioma. Clin. Cancer Res. 2011, 17, 247–254. [Google Scholar] [CrossRef]
- Korpershoek, E.; Favier, J.; Gaal, J.; Burnichon, N.; van Gessel, B.; Oudijk, L.; Badoual, C.; Gadessaud, N.; Venisse, A.; Bayley, J.P.; et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J. Clin. Endocrinol. Metab. 2011, 96, E1472–1476. [Google Scholar] [CrossRef]
- Saxena, N.; Maio, N.; Crooks, D.R.; Ricketts, C.J.; Yang, Y.; Wei, M.H.; Fan, T.W.; Lane, A.N.; Sourbier, C.; Singh, A.; et al. SDHB-Deficient Cancers: The Role of Mutations That Impair Iron Sulfur Cluster Delivery. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef]
- de Cubas, A.A.; Korpershoek, E.; Inglada-Perez, L.; Letouze, E.; Curras-Freixes, M.; Fernandez, A.F.; Comino-Mendez, I.; Schiavi, F.; Mancikova, V.; Eisenhofer, G.; et al. DNA Methylation Profiling in Pheochromocytoma and Paraganglioma Reveals Diagnostic and Prognostic Markers. Clin. Cancer Res. 2015, 21, 3020–3030. [Google Scholar] [CrossRef] [Green Version]
- Lonser, R.R.; Glenn, G.M.; Walther, M.; Chew, E.Y.; Libutti, S.K.; Linehan, W.M.; Oldfield, E.H. von Hippel-Lindau disease. Lancet 2003, 361, 2059–2067. [Google Scholar] [CrossRef]
- Friedrich, C.A. Genotype-phenotype correlation in von Hippel-Lindau syndrome. Hum. Mol. Genet. 2001, 10, 763–767. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis. Blood 2009, 114, 2015–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capodimonti, S.; Teofili, L.; Martini, M.; Cenci, T.; Iachininoto, M.G.; Nuzzolo, E.R.; Bianchi, M.; Murdolo, M.; Leone, G.; Larocca, L.M. Von hippel-lindau disease and erythrocytosis. J. Clin. Oncol. 2012, 30, e137–e139. [Google Scholar] [CrossRef]
- Koch, C.A.; Huang, S.C.; Zhuang, Z.; Stolle, C.; Stolle, C.; Azumi, N.; Chrousos, G.P.; Vortmeyer, A.O.; Pacak, K. Somatic VHL gene deletion and point mutation in MEN 2A-associated pheochromocytoma. Oncogene 2002, 17, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Merlo, A.; de Quiros, S.B.; de Santa-Maria, I.S.; Pitiot, A.S.; Balbin, M.; Astudillo, A.; Scola, B.; Aristegui, M.; Quer, M.; Suarez, C.; et al. Identification of somatic VHL gene mutations in sporadic head and neck paragangliomas in association with activation of the HIF-1alpha/miR-210 signaling pathway. J. Clin. Endocrinol. Metab. 2013, 98, E1661–E1666. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shepard, M.J.; Zhang, C.; Dong, L.; Walker, D.; Guedez, L.; Park, S.; Wang, Y.; Chen, S.; Pang, Y.; et al. Deletion of the von Hippel-Lindau Gene in Hemangioblasts Causes Hemangioblastoma-like Lesions in Murine Retina. Cancer Res. 2018, 78, 1266–1274. [Google Scholar] [CrossRef]
- Roe, J.S.; Kim, H.; Lee, S.M.; Kim, S.T.; Cho, E.J.; Youn, H.D. p53 stabilization and transactivation by a von Hippel-Lindau protein. Mol. Cell 2006, 22, 395–405. [Google Scholar] [CrossRef]
- Semenza, G.L.; Rue, E.A.; Iyer, N.V.; Pang, M.G.; Kearns, W.G. Assignment of the hypoxia-inducible factor 1alpha gene to a region of conserved synteny on mouse chromosome 12 and human chromosome 14q. Genomics 1996, 34, 437–439. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci. STKE 2007, 2007, cm8. [Google Scholar] [CrossRef]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar]
- Amar, L.; Baudin, E.; Burnichon, N.; Peyrard, S.; Silvera, S.; Bertherat, J.; Bertagna, X.; Schlumberger, M.; Jeunemaitre, X.; Gimenez-Roqueplo, A.P.; et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J. Clin. Endocrinol. Metab. 2007, 92, 3822–3828. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1alpha and HIF2alpha: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef]
- Percy, M.J.; Furlow, P.W.; Lucas, G.S.; Li, X.; Lappin, T.R.; McMullin, M.F.; Lee, F.S. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N. Engl. J. Med. 2008, 358, 162–168. [Google Scholar] [CrossRef]
- Zhuang, Z.; Yang, C.; Lorenzo, F.; Merino, M.; Fojo, T.; Kebebew, E.; Popovic, V.; Stratakis, C.A.; Prchal, J.T.; Pacak, K. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N. Engl. J. Med. 2012, 367, 922–930. [Google Scholar] [CrossRef]
- Yang, C.; Hong, C.S.; Prchal, J.T.; Balint, M.T.; Pacak, K.; Zhuang, Z. Somatic mosaicism of EPAS1 mutations in the syndrome of paraganglioma and somatostatinoma associated with polycythemia. Hum. Genome. Var. 2015, 2, 15053. [Google Scholar] [CrossRef] [PubMed]
- Ratner, N.; Miller, S.J. A RASopathy gene commonly mutated in cancer: The neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer 2015, 15, 290–301. [Google Scholar] [CrossRef]
- Bausch, B.; Borozdin, W.; Mautner, V.F.; Hoffmann, M.M.; Boehm, D.; Robledo, M.; Cascon, A.; Harenberg, T.; Schiavi, F.; Pawlu, C.; et al. Germline NF1 mutational spectra and loss-of-heterozygosity analyses in patients with pheochromocytoma and neurofibromatosis type 1. J. Clin. Endocrinol. Metab. 2007, 92, 2784–2792. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Primers 2017, 3, 17004. [Google Scholar] [CrossRef]
- Welander, J.; Larsson, C.; Backdahl, M.; Hareni, N.; Sivler, T.; Sivler, T.; Brauckhoff, M.; Soderkvist, P.; Gimm, O. Integrative genomics reveals frequent somatic NF1 mutations in sporadic pheochromocytomas. Hum. Mol. Genet. 2012, 21, 5406–5416. [Google Scholar] [CrossRef] [Green Version]
- Correia, M.J.; Lopes, L.O.; Bugalho, M.J.; Cristina, L.; Santos, A.I.; Bordalo, A.D.; Pinho, B.; da Silva, H.L.; Goncalves, M.D.; Ribeiro, C.; et al. Multiple endocrine neoplasia type 2A. Study of a family. Rev. Port. Cardiol. 2000, 19, 11–31. [Google Scholar]
- Bergsland, E.K. The evolving landscape of neuroendocrine tumors. Semin. Oncol. 2013, 40, 4–22. [Google Scholar] [CrossRef]
- Machens, A.; Brauckhoff, M.; Holzhausen, H.J.; Thanh, P.N.; Lehnert, H.; Dralle, H. Codon-specific development of pheochromocytoma in multiple endocrine neoplasia type 2. J. Clin. Endocrinol. Metab. 2005, 90, 3999–4003. [Google Scholar] [CrossRef]
- Boedeker, C.C.; Erlic, Z.; Richard, S.; Kontny, U.; Gimenez-Roqueplo, A.P.; Cascon, A.; Robledo, M.; de Campos, J.M.; van Nederveen, F.H.; de Krijger, R.R.; et al. Head and neck paragangliomas in von Hippel-Lindau disease and multiple endocrine neoplasia type 2. J. Clin. Endocrinol. Metab. 2009, 94, 1938–1944. [Google Scholar] [CrossRef]
- Fishbein, L.; Nathanson, K.L. Pheochromocytoma and paraganglioma: Understanding the complexities of the genetic background. Cancer Genet. 2012, 205, 1–11. [Google Scholar] [CrossRef]
- Plaza-Menacho, I.; Barnouin, K.; Goodman, K.; Martinez-Torres, R.J.; Borg, A.; Murray-Rust, J.; Mouilleron, S.; Knowles, P.; McDonald, N.Q. Oncogenic RET kinase domain mutations perturb the autophosphorylation trajectory by enhancing substrate presentation in trans. Mol. Cell 2014, 53, 738–751. [Google Scholar] [CrossRef]
- Burnichon, N.; Cascon, A.; Schiavi, F.; Morales, N.P.; Comino-Mendez, I.; Abermil, N.; Inglada-Perez, L.; de Cubas, A.A.; Amar, L.; Barontini, M.; et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin. Cancer Res. 2012, 18, 2828–2837. [Google Scholar] [CrossRef]
- Blackwood, E.M.; Eisenman, R.N. Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef]
- Bousset, K.; Henriksson, M.; Luscher-Firzlaff, J.M.; Litchfield, D.W.; Luscher, B. Identification of casein kinase II phosphorylation sites in Max: Effects on DNA-binding kinetics of Max homo- and Myc/Max heterodimers. Oncogene 1993, 8, 3211–3220. [Google Scholar]
- Yoshimoto, K.; Iwahana, H.; Fukuda, A.; Sano, T.; Katsuragi, K.; Kinoshita, M.; Saito, S.; Itakura, M. ras mutations in endocrine tumors: Mutation detection by polymerase chain reaction-single strand conformation polymorphism. Jpn. J. Cancer Res. 1992, 83, 1057–1062. [Google Scholar] [CrossRef]
- Safford, S.D.; Coleman, R.E.; Gockerman, J.P.; Moore, J.; Feldman, J.M.; Leight, G.S., Jr.; Tyler, D.S.; Olson, J.A., Jr. Iodine -131 metaiodobenzylguanidine is an effective treatment for malignant pheochromocytoma and paraganglioma. Surgery 2003, 134, 956–962. [Google Scholar] [CrossRef]
- Fitzgerald, P.A.; Goldsby, R.E.; Huberty, J.P.; Price, D.C.; Hawkins, R.A.; Veatch, J.J.; Dela Cruz, F.; Jahan, T.M.; Linker, C.A.; Damon, L.; et al. Malignant pheochromocytomas and paragangliomas: A phase II study of therapy with high-dose 131I-metaiodobenzylguanidine (131I-MIBG). Ann. NY Acad. Sci. 2006, 1073, 465–490. [Google Scholar] [CrossRef]
- van Hulsteijn, L.T.; Niemeijer, N.D.; Dekkers, O.M.; Corssmit, E.P. (131)I-MIBG therapy for malignant paraganglioma and phaeochromocytoma: Systematic review and meta-analysis. Clin. Endocrinol. (Oxf) 2014, 80, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Otte, A.; Jermann, E.; Behe, M.; Goetze, M.; Bucher, H.C.; Roser, H.W.; Heppeler, A.; Mueller-Brand, J.; Maecke, H.R. DOTATOC: A powerful new tool for receptor-mediated radionuclide therapy. Eur. J. Nucl. Med. 1997, 24, 792–795. [Google Scholar] [PubMed]
- Kwekkeboom, D.J.; Bakker, W.H.; Kam, B.L.; Teunissen, J.J.; Kooij, P.P.; de Herder, W.W.; Feelders, R.A.; van Eijck, C.H.; de Jong, M.; Srinivasan, A.; et al. Treatment of patients with gastro-entero-pancreatic (GEP) tumours with the novel radiolabelled somatostatin analogue [177Lu-DOTA(0),Tyr3]octreotate. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Bodei, L.; Mueller-Brand, J.; Baum, R.P.; Pavel, M.E.; Horsch, D.; O–Dorisio, M.S.; O–Dorisio, T.M.; Howe, J.R.; Cremonesi, M.; Kwekkeboom, D.J.; et al. The joint IAEA, EANM, and SNMMI practical guidance on peptide receptor radionuclide therapy (PRRNT) in neuroendocrine tumours. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 800–816. [Google Scholar] [CrossRef]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 Trial of (177)Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Favier, J.; Igaz, P.; Burnichon, N.; Amar, L.; Libe, R.; Badoual, C.; Tissier, F.; Bertherat, J.; Plouin, P.F.; Jeunemaitre, X.; et al. Rationale for anti-angiogenic therapy in pheochromocytoma and paraganglioma. Endocr. Pathol. 2012, 23, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Janeway, K.A.; Kim, S.Y.; Lodish, M.; Nose, V.; Rustin, P.; Gaal, J.; Dahia, P.L.; Liegl, B.; Ball, E.R.; Raygada, M.; et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc. Natl. Acad. Sci. USA 2011, 108, 314–318. [Google Scholar] [CrossRef]
- Ricketts, C.; Woodward, E.R.; Killick, P.; Morris, M.R.; Astuti, D.; Latif, F.; Maher, E.R. Germline SDHB mutations and familial renal cell carcinoma. J. Natl. Cancer Inst. 2008, 100, 1260–1262. [Google Scholar] [CrossRef]
- Tuthill, M.; Barod, R.; Pyle, L.; Cook, T.; Chew, S.; Gore, M.; Maxwell, P.; Eisen, T. A report of succinate dehydrogenase B deficiency associated with metastatic papillary renal cell carcinoma: Successful treatment with the multi-targeted tyrosine kinase inhibitor sunitinib. BMJ Case Rep. 2009, 2009. [Google Scholar] [CrossRef]
- Joshua, A.M.; Ezzat, S.; Asa, S.L.; Evans, A.; Broom, R.; Freeman, M.; Knox, J.J. Rationale and evidence for sunitinib in the treatment of malignant paraganglioma/pheochromocytoma. J. Clin. Endocrinol. Metab. 2009, 94, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Hahn, N.M.; Reckova, M.; Cheng, L.; Baldridge, L.A.; Cummings, O.W.; Sweeney, C.J. Patient with malignant paraganglioma responding to the multikinase inhibitor sunitinib malate. J. Clin. Oncol. 2009, 27, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, C.; Cabanillas, M.E.; Santarpia, L.; Jonasch, E.; Kyle, K.L.; Lano, E.A.; Matin, S.F.; Nunez, R.F.; Perrier, N.D.; Phan, A.; et al. Use of the tyrosine kinase inhibitor sunitinib in a patient with von Hippel-Lindau disease: Targeting angiogenic factors in pheochromocytoma and other von Hippel-Lindau disease-related tumors. J. Clin. Endocrinol. Metab. 2009, 94, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, K.; Miura, T.; Shioji, G.; Tsuboi, N. Sunitinib treatment for refractory malignant pheochromocytoma. Neuro. Endocrinol. Lett. 2012, 33, 260–264. [Google Scholar] [PubMed]
- Ayala-Ramirez, M.; Chougnet, C.N.; Habra, M.A.; Palmer, J.L.; Leboulleux, S.; Cabanillas, M.E.; Caramella, C.; Anderson, P.; Al Ghuzlan, A.; Waguespack, S.G.; et al. Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas. J. Clin. Endocrinol. Metab. 2012, 97, 4040–4050. [Google Scholar] [CrossRef]
- Welsh, S.J.; Williams, R.R.; Birmingham, A.; Newman, D.J.; Kirkpatrick, D.L.; Powis, G. The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1alpha and vascular endothelial growth factor formation. Mol. Cancer Ther. 2003, 2, 235–243. [Google Scholar] [PubMed]
- Welsh, S.; Williams, R.; Kirkpatrick, L.; Paine-Murrieta, G.; Powis, G. Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1alpha. Mol. Cancer Ther. 2004, 3, 233–244. [Google Scholar]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Courtney, K.D.; Infante, J.R.; Lam, E.T.; Figlin, R.A.; Rini, B.I.; Brugarolas, J.; Zojwalla, N.J.; Lowe, A.M.; Wang, K.; Wallace, E.M.; et al. Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2alpha Antagonist in Patients With Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J. Clin. Oncol. 2018, 36, 867–874. [Google Scholar] [CrossRef]
- Pang, Y.; Yang, C.; Schovanek, J.; Wang, H.; Bullova, P.; Caisova, V.; Gupta, G.; Wolf, K.I.; Semenza, G.L.; Zhuang, Z.; et al. Anthracyclines suppress pheochromocytoma cell characteristics, including metastasis, through inhibition of the hypoxia signaling pathway. Oncotarget 2017, 8, 22313–22324. [Google Scholar] [CrossRef] [Green Version]
- Burnichon, N.; Lepoutre-Lussey, C.; Laffaire, J.; Gadessaud, N.; Molinie, V.; Hernigou, A.; Plouin, P.F.; Jeunemaitre, X.; Favier, J.; Gimenez-Roqueplo, A.P. A novel TMEM127 mutation in a patient with familial bilateral pheochromocytoma. Eur. J. Endocrinol. 2011, 164, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Attie, T.; Pelet, A.; Edery, P.; Eng, C.; Mulligan, L.M.; Amiel, J.; Boutrand, L.; Beldjord, C.; Nihoul-Fekete, C.; Munnich, A.; et al. Diversity of RET proto-oncogene mutations in familial and sporadic Hirschsprung disease. Hum. Mol. Genet. 1995, 4, 1381–1386. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell. Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Druce, M.R.; Kaltsas, G.A.; Fraenkel, M.; Gross, D.J.; Grossman, A.B. Novel and evolving therapies in the treatment of malignant phaeochromocytoma: Experience with the mTOR inhibitor everolimus (RAD001). Horm. Metab. Res. 2009, 41, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Kim, T.W.; Park, Y.S.; Shin, S.J.; Shin, S.H.; Song, E.K.; Lee, H.J.; Lee, K.W.; Bang, Y.J. Phase 2 study of everolimus monotherapy in patients with nonfunctioning neuroendocrine tumors or pheochromocytomas/paragangliomas. Cancer 2012, 118, 6162–6170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giubellino, A.; Bullova, P.; Nolting, S.; Turkova, H.; Powers, J.F.; Liu, Q.; Guichard, S.; Tischler, A.S.; Grossman, A.B.; Pacak, K. Combined inhibition of mTORC1 and mTORC2 signaling pathways is a promising therapeutic option in inhibiting pheochromocytoma tumor growth: In vitro and in vivo studies in female athymic nude mice. Endocrinology 2013, 154, 646–655. [Google Scholar] [CrossRef]
- Matro, J.; Giubellino, A.; Pacak, K. Current and future therapeutic approaches for metastatic pheochromocytoma and paraganglioma: Focus on SDHB tumors. Horm. Metab. Res. 2013, 45, 147–153. [Google Scholar] [CrossRef]
- Letouze, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef]
- Killian, J.K.; Kim, S.Y.; Miettinen, M.; Smith, C.; Merino, M.; Tsokos, M.; Quezado, M.; Smith, W.I., Jr.; Jahromi, M.S.; Xekouki, P.; et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013, 3, 648–657. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- SongTao, Q.; Lei, Y.; Si, G.; YanQing, D.; HuiXia, H.; XueLin, Z.; LanXiao, W.; Fei, Y. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci. 2012, 103, 269–273. [Google Scholar] [CrossRef]
- Lu, Y.; Kwintkiewicz, J.; Liu, Y.; Tech, K.; Frady, L.N.; Su, Y.T.; Bautista, W.; Moon, S.I.; MacDonald, J.; Ewend, M.G.; et al. Chemosensitivity of IDH1-Mutated Gliomas Due to an Impairment in PARP1-Mediated DNA Repair. Cancer Res. 2017, 77, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Hadoux, J.; Favier, J.; Scoazec, J.Y.; Leboulleux, S.; Al Ghuzlan, A.; Caramella, C.; Deandreis, D.; Borget, I.; Loriot, C.; Chougnet, C.; et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int. J. Cancer 2014, 135, 2711–2720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulke, M.H.; Stuart, K.; Enzinger, P.C.; Ryan, D.P.; Clark, J.W.; Muzikansky, A.; Vincitore, M.; Michelini, A.; Fuchs, C.S. Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J. Clin. Oncol. 2006, 24, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, P.L.; Sundaram, R.K.; Oeck, S.; Corso, C.D.; Liu, Y.; Noorbakhsh, S.; Niger, M.; Boeke, M.; Ueno, D.; Kalathil, A.N.; et al. Krebs-cycle-deficient hereditary cancer syndromes are defined by defects in homologous-recombination DNA repair. Nat. Genet. 2018, 50, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Lu, Y.; Caisova, V.; Liu, Y.; Bullova, P.; Huynh, T.T.; Zhou, Y.; Yu, D.; Frysak, Z.; Hartmann, I.; et al. Targeting NAD(+)/PARP DNA Repair Pathway as a Novel Therapeutic Approach to SDHB-Mutated Cluster I Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2018, 24, 3423–3432. [Google Scholar] [CrossRef]
- Zhang, Z.; Guo, Z.; Zhan, Y.; Li, H.; Wu, S. Role of histone acetylation in activation of nuclear factor erythroid 2-related factor 2/heme oxygenase 1 pathway by manganese chloride. Toxicol. Appl. Pharmacol. 2017, 336, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Li, Q.Z.; Chen, L.; Chen, B.D.; Zhang, C.; Wang, X.; Li, W.P. HPOB, an HDAC6 inhibitor, attenuates corticosterone-induced injury in rat adrenal pheochromocytoma PC12 cells by inhibiting mitochondrial GR translocation and the intrinsic apoptosis pathway. Neurochem. Int. 2016, 99, 239–251. [Google Scholar] [CrossRef]
- Cayo, M.A.; Cayo, A.K.; Jarjour, S.M.; Chen, H. Sodium butyrate activates Notch1 signaling, reduces tumor markers, and induces cell cycle arrest and apoptosis in pheochromocytoma. Am. J. Transl. Res. 2009, 1, 178–183. [Google Scholar]
- Adler, J.T.; Hottinger, D.G.; Kunnimalaiyaan, M.; Chen, H. Histone deacetylase inhibitors upregulate Notch-1 and inhibit growth in pheochromocytoma cells. Surgery 2008, 144, 956–961. [Google Scholar] [CrossRef]
- Yang, C.; Matro, J.C.; Huntoon, K.M.; Ye, D.Y.; Huynh, T.T.; Fliedner, S.M.; Breza, J.; Zhuang, Z.; Pacak, K. Missense mutations in the human SDHB gene increase protein degradation without altering intrinsic enzymatic function. FASEB J. 2012, 26, 4506–4516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatfield, S.M.; Sitkovsky, M. A2A adenosine receptor antagonists to weaken the hypoxia-HIF-1alpha driven immunosuppression and improve immunotherapies of cancer. Curr. Opin. Pharmacol. 2016, 29, 90–96. [Google Scholar] [CrossRef]
- Labiano, S.; Palazon, A.; Bolanos, E.; Azpilikueta, A.; Sanchez-Paulete, A.R.; Morales-Kastresana, A.; Quetglas, J.I.; Perez-Gracia, J.L.; Gurpide, A.; Rodriguez-Ruiz, M.; et al. Hypoxia-induced soluble CD137 in malignant cells blocks CD137L-costimulation as an immune escape mechanism. Oncoimmunology 2016, 5, e1062967. [Google Scholar] [CrossRef] [PubMed]
- Chouaib, S.; Noman, M.Z.; Kosmatopoulos, K.; Curran, M.A. Hypoxic stress: Obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 2017, 36, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Hadoux, J.; Terroir, M.; Leboulleux, S.; Deschamps, F.; Al Ghuzlan, A.; Hescot, S.; Tselikas, L.; Borget, I.; Caramella, C.; Deandreis, D.; et al. Interferon-alpha Treatment for Disease Control in Metastatic Pheochromocytoma/Paraganglioma Patients. Horm. Cancer 2017, 8, 330–337. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, Y.; Celiku, O.; Li, A.; Wu, Q.; Zhou, Y.; Yang, C. Targeting IDH1-Mutated Malignancies with NRF2 Blockade. J. Natl. Cancer Inst. 2019. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Ghayee, H.K.; Bhagwandin, V.J.; Stastny, V.; Click, A.; Ding, L.H.; Mizrachi, D.; Zou, Y.S.; Chari, R.; Lam, W.L.; Bachoo, R.M.; et al. Progenitor cell line (hPheo1) derived from a human pheochromocytoma tumor. PLoS ONE 2013, 8, e65624. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, Y.; Liu, Y.; Pacak, K.; Yang, C. Pheochromocytomas and Paragangliomas: From Genetic Diversity to Targeted Therapies. Cancers 2019, 11, 436. https://doi.org/10.3390/cancers11040436
Pang Y, Liu Y, Pacak K, Yang C. Pheochromocytomas and Paragangliomas: From Genetic Diversity to Targeted Therapies. Cancers. 2019; 11(4):436. https://doi.org/10.3390/cancers11040436
Chicago/Turabian StylePang, Ying, Yang Liu, Karel Pacak, and Chunzhang Yang. 2019. "Pheochromocytomas and Paragangliomas: From Genetic Diversity to Targeted Therapies" Cancers 11, no. 4: 436. https://doi.org/10.3390/cancers11040436
APA StylePang, Y., Liu, Y., Pacak, K., & Yang, C. (2019). Pheochromocytomas and Paragangliomas: From Genetic Diversity to Targeted Therapies. Cancers, 11(4), 436. https://doi.org/10.3390/cancers11040436