A Novel Role for Helicobacter pylori Gamma-Glutamyltranspeptidase in Regulating Autophagy and Bacterial Internalization in Human Gastric Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Helicobacter Pylori Gamma-Glutamyltranspeptidase Inhibits Autophagy in Human Gastric Cancer Cells
2.2. HpGGT Activity in the Supernatants of H. pylori Cultures Was Sufficient to Inhibit Autophagy of Gastric Cells
2.3. HpGGT Did Not Affect the Early Steps of Autophagy
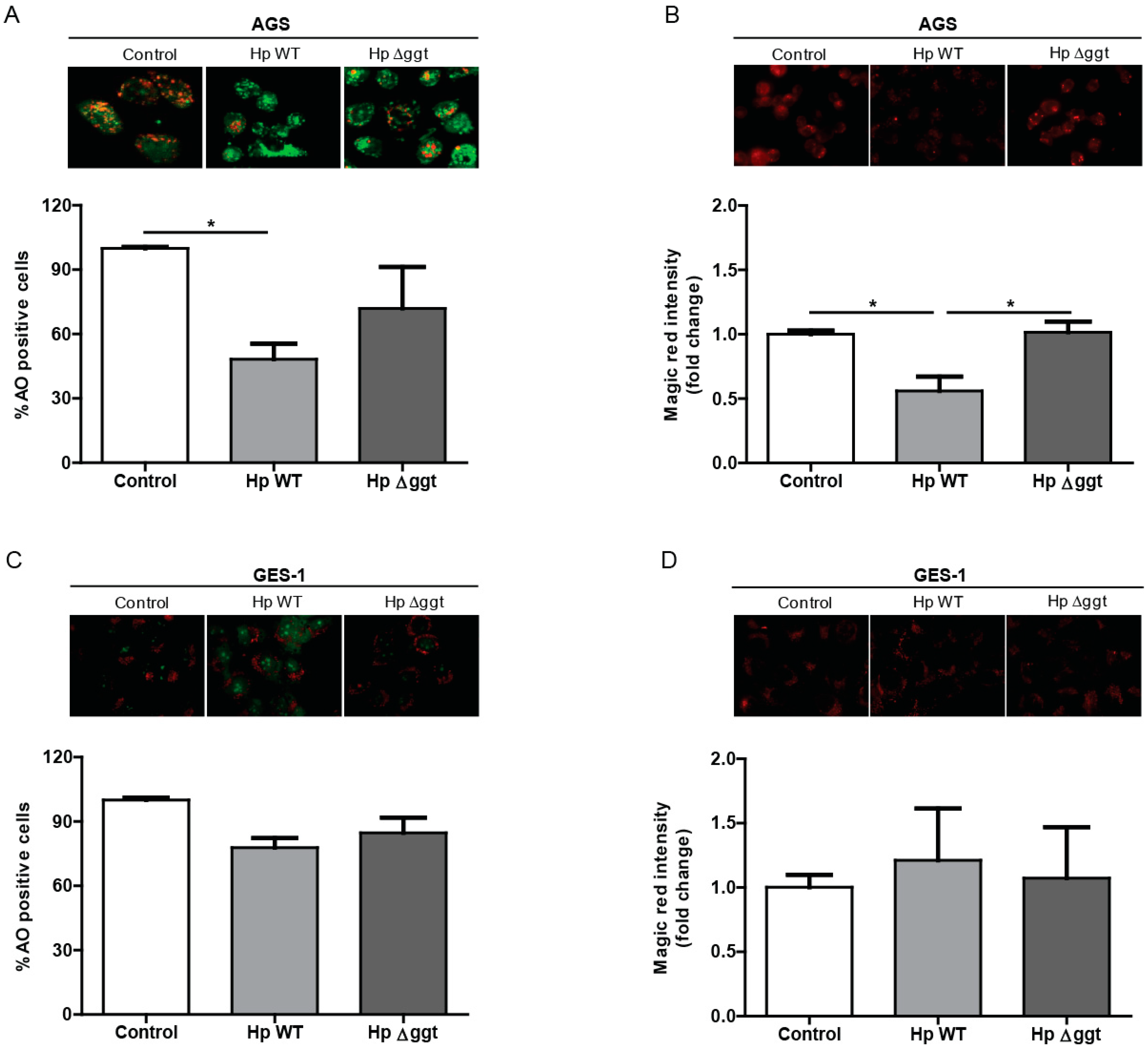
2.4. HpGGT Contributes to Autophagy Inhibition without Modifying Lysosome Acidification
2.5. HpGGT Inhibits Autophagy of Gastric Cells by Decreasing Cathepsin B Activity in the Lysosome
2.6. Loss of HpGGT Decreases H. pylori Internalization in Gastric Cells
3. Discussion
4. Material and Methods
4.1. Cell lines, Strains, and Culture Conditions
4.2. Infection of Gastric Cells with H. pylori
4.3. Concentrated Culture Supernatants
4.4. Autophagosome Analysis
4.5. Immunofluorescence (IF) and Confocal Microscopy
4.6. LysoTracker Red Staining
4.7. Flow Cytometry
4.8. Measurement of Lysosomal Membrane Stability
4.9. Cathepsin B Activity (Magic Red)
4.10. Adherence and Gentamicin Internalization Assays
4.11. Western Blot Analysis
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.; Forman, D.; Bray, F. Cancer incidence and mortality—Major patterns in GLOBOCAN 2012, worldwide and Georgia. Int. J. Cancer 2015, 136, 359–386. [Google Scholar] [CrossRef] [PubMed]
- Pormohammad, A.; Ghotaslo, R.; Leylabadlo, H.E.; Nasiri, M.J.; Dabiri, H.; Hashemi, A. Risk of gastric cancer in association with Helicobacter pylori different virulence factors: A systematic review and meta-analysis. Microb. Pathog. 2018, 118, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Matos, J.I.; De Sousa, H.A.C.; Marcos-Pinto, R.; Dinis-Ribeiro, M. Helicobacter pylori CagA and VacA genotypes and gastric phenotype: A meta-analysis. Eur. J. Gastroenterol. Hepatol. 2013, 25, 1431–1441. [Google Scholar] [CrossRef]
- Ricci, V.; Giannouli, M.; Romano, M.; Zarrilli, R. Helicobacter pylori gamma-glutamyl transpeptidase and its pathogenic role. World J. Gastroenterol. 2014, 20, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Hanigan, M.H. gamma-Glutamyl transpeptidase, a glutathionase: Its expression and function in carcinogenesis. Chem. Biol. Interact. 1998, 111–112, 333–342. [Google Scholar] [CrossRef]
- Chevalier, C.; Thiberge, J.M.; Ferrero, R.L.; Labigne, A. Essential role of Helicobacter pylori γ-glutamyltranspeptidase for the colonization of the gastric mucosa of mice. Mol. Microbiol. 1999, 31, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Lee, S.G.; Park, M.G.; Song, J.Y.; Kang, H.L.; Lee, W.K.; Cho, M.J.; Rhee, K.H.; Youn, H.S.; Baik, S.C. γ-Glutamyltranspeptidase of Helicobacter pylori induces mitochondria-mediated apoptosis in AGS cells. Biochem. Biophys. Res. Commun. 2007, 355, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, M.; Bravo, D.; Canales, J.; Sanhueza, C.; Díaz, N.; Almarza, O.; Toledo, H.; Quest, A.F.G. Helicobacter pylori-induced loss of survivin and gastric cell viability is attributable to secreted bacterial gamma-glutamyl transpeptidase activity. J. Infect. Dis. 2013, 208, 1131–1141. [Google Scholar] [CrossRef]
- Kim, K.M.; Lee, S.G.; Kim, J.M.; Kim, D.S.; Song, J.Y.; Kang, H.L.; Lee, W.K.; Cho, M.J.; Rhee, K.H.; Youn, H.S.; et al. Helicobacter pylori γ-glutamyltranspeptidase induces cell cycle arrest at the G1-S phase transition. J. Microbiol. 2010, 48, 372–377. [Google Scholar] [CrossRef]
- Gong, M.; Ling, S.S.M.; Lui, S.Y.; Yeoh, K.G.; Ho, B. Helicobacter pylori γ-glutamyl transpeptidase is a pathogenic factor in the development of peptic ulcer disease. Gastroenterology 2010, 139, 564–573. [Google Scholar] [CrossRef]
- Colombo, M.I. Pathogens and autophagy: Subverting to survive. Cell Death Differ. 2005, 12, 1481–1483. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Siqueira, M.; de Moraes Ribeiro, R.; Travassos, L.H. Autophagy and its interaction with intracellular bacterial pathogens. Front. Immunol. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Terebiznik, M.R.; Raju, D.; Vázquez, C.L.; Torbricki, K.; Kulkarni, R.; Blanke, S.R.; Yoshimori, T.; Colombo, M.I.; Jones, N.L. Effect of Helicobacter pylori’s vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy 2009, 5, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Raju, D.; Hussey, S.; Ang, M.; Terebiznik, M.R.; Sibony, M.; Galindo-Mata, E.; Gupta, V.; Blanke, S.R.; Delgado, A.; Romero-Gallo, J.; et al. Vaculolating Cytotoxin and Variants in Atg16L1 that Disrupt Autophagy Promote Helicobacter Pylori Infection in Humans. Gastroenterology 2012, 142, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Björkholm, B.; Zhukhovitsky, V.; Löfman, C.; Hultén, K.; Enroth, H.; Block, M.; Rigo, R.; Falk, P.; Engstrand, L. Helicobacter pylori Entry into Human Gastric Epithelial Cells: A Potential Determinant of Virulence, Persistence, and Treatment Failures. Helicobacter 2000, 5, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Amieva, M.R.; Salama, N.R.; Tompkins, L.S.; Falkow, S. Helicobacter pylori enter and survive within multivesicular vacuoles of epithelial cells. Cell. Microbiol. 2002, 4, 677–690. [Google Scholar] [CrossRef]
- Terebiznik, M.R.; Vazquez, C.L.; Torbicki, K.; Banks, D.; Wang, T.; Hong, W.; Blanke, S.R.; Colombo, M.I.; Jones, N.L. Helicobacter pylori VacA toxin promotes bacterial intracellular survival in gastric epithelial cells. Infect. Immun. 2006, 74, 6599–6614. [Google Scholar] [CrossRef]
- Tang, B.; Li, N.; Gu, J.; Li, Q.; Zhuang, Y.; Fang, Y.; Yu, B.; Wang, H.; Zhang, J.; Xie, Q.; et al. Compromised autophagy by MIR30B benefits the intracellular survival of Helicobacter pylori. Autophagy 2012, 8, 1045–1057. [Google Scholar] [CrossRef]
- Yang, X.J.; Si, R.H.; Liang, Y.H.; Ma, B.Q.; Jiang, Z.B.; Wang, B.; Gao, P. Mir-30d increases intracellular survival of Helicobacter pylori through inhibition of autophagy pathway. World J. Gastroenterol. 2016, 22, 3978–3991. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, W.; Cho, C.H.; Chan, F.K.L.; Yu, J.; Ross Fitzgerald, J.; Cheung, C.K.Y.; Xiao, Z.G.; Shen, J.; Li, L.F.; et al. Reduced lysosomal clearance of autophagosomes promotes survival and colonization of Helicobacter pylori. J. Pathol. 2018, 244, 432–444. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, L.K.; Jones, N.L. Modulation of autophagy by Helicobacter pylori and its role in gastric carcinogenesis. Trends Microbiol. 2013, 21, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Luo, Y.; Zou, J.; Ouyang, J.; Cai, Z.; Zeng, X.; Ling, H.; Zeng, T. Autophagy and its role in gastric cancer. Clin. Chim. Acta 2018, 489, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Yang, Y. Functional role of autophagy in gastric cancer. Oncotarget 2016, 7, 17641–17651. [Google Scholar] [CrossRef] [PubMed]
- Boquet, P.; Ricci, V. Intoxication strategy of Helicobacter pylori VacA toxin. Trends Microbiol. 2012, 20, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.S.M.; Khoo, L.H.B.; Hwang, L.A.; Yeoh, K.G.; Ho, B. Instrumental role of Helicobacter pylori γ-glutamyl transpeptidase in VacA-dependent vacuolation in gastric epithelial cells. PLoS ONE 2015, 10, 1–17. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation Mechanisms and Signaling Pathways of Autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [Green Version]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef]
- Kawai, A.; Uchiyama, H.; Takano, S.; Nakamura, N.; Ohkuma, S. Autophagosome-lysosome fusion depends on the pH in acidic compartments in CHO cells. Autophagy 2007, 3, 154–157. [Google Scholar] [CrossRef]
- Boya, P.; Kroemer, G. Lysosomal membrane permeabilization in cell death. Oncogene 2008, 27, 6434–6451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenzuela, M.; Pérez-Pérez, G.; Corvalán, A.H.; Carrasco, G.; Urra, H.; Bravo, D.; Toledo, H.; Quest, A.F.G. Helicobacter pylori –Induced Loss of the Inhibitor-of-Apoptosis Protein Survivin Is Linked to Gastritis and Death of Human Gastric Cells. J. Infect. Dis. 2010, 202, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Ollinger, K.; Brunk, U.L.F.T. Cellular Injury Induced By Oxidative Stress Is Mediated. Science 1995, 19, 565–574. [Google Scholar]
- Brunk, U.T.; Neuzil, J.; Eaton, J.W. Lysosomal involvement in apoptosis. Redox Rep. 2004, 6, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.W.; Luo, R.H.; Zhao, Q.; Shen, Z.Z.; Huang, L.L.; An, X.Y.; Zhao, L.J.; Wang, J.; Huang, Y.Z. Helicobacter pylori induces mitochondrial DNA mutation and reactive oxygen species level in AGS cells. Int. J. Med. Sci. 2011, 8, 56–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azad, M.B.; Chen, Y.; Gibson, S.B. Regulation of Autophagy by Reactive Oxygen Species (ROS): Implications for Cancer Progression and Treatment. Antioxid. Redox Signal. 2009, 11, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Nardone, G.; Compare, D.; De Colibus, P.; De Nucci, G.; Rocco, A. Helicobacter pylori and epigenetic mechanisms underlying gastric carcinogenesis. Dig. Dis. 2007, 25, 225–229. [Google Scholar] [CrossRef]
- Angrisano, T.; Lembo, F.; Peluso, S.; Keller, S.; Chiariotti, L.; Pero, R. Helicobacter pylori regulates iNOS promoter by histone modifications in human gastric epithelial cells. Med. Microbiol. Immunol. 2012, 201, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Chiariotti, L.; Angrisano, T.; Keller, S.; Florio, E.; Affinito, O.; Pallante, P.; Perrino, C.; Pero, R.; Lembo, F. Epigenetic modifications induced by Helicobacter pylori infection through a direct microbe-gastric epithelial cells cross-talk. Med. Microbiol. Immunol. 2013, 202, 327–337. [Google Scholar] [CrossRef]
- Muhammad, J.; Eladl, M.; Khoder, G. Helicobacter pylori-induced DNA Methylation as an Epigenetic Modulator of Gastric Cancer: Recent Outcomes and Future Direction. Pathogens 2019, 8, 23. [Google Scholar] [CrossRef]
- Muhammad, J.S.; Nanjo, S.; Ando, T.; Yamashita, S.; Maekita, T.; Ushijima, T.; Tabuchi, Y.; Sugiyama, T. Autophagy impairment by Helicobacter pylori-induced methylation silencing of MAP1LC3Av1 promotes gastric carcinogenesis. Int. J. Cancer 2017, 140, 2272–2283. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Nagashima, H.; Uotani, T.; Graham, D.Y.; Yamaoka, Y. Autophagy-related genes in Helicobacter pylori infection. Helicobacter 2017, 22, 1–20. [Google Scholar] [CrossRef]
- Dubois, A.; Borén, T. Helicobacter pylori is invasive and it may be a facultative intracellular organism. Cell. Microbiol. 2007, 9, 1108–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.H.; Lv, Z.F.; Zhong, Y.; Liu, D.S.; Chen, S.P.; Xie, Y. The internalization of Helicobacter pylori plays a role in the failure of H. pylori eradication. Helicobacter 2017, 22, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Wüstner, S.; Anderl, F.; Wanisch, A.; Sachs, C.; Steiger, K.; Nerlich, A.; Vieth, M.; Mejías-Luque, R.; Gerhard, M. Helicobacter pylori γ-glutamyl transferase contributes to colonization and differential recruitment of T cells during persistence. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Terman, A.; Kurz, T.; Gustafsson, B.; Brunk, U.T. Lysosomal labilization. IUBMB Life 2006, 58, 531–539. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bravo, J.; Díaz, P.; Corvalán, A.H.; Quest, A.F.G. A Novel Role for Helicobacter pylori Gamma-Glutamyltranspeptidase in Regulating Autophagy and Bacterial Internalization in Human Gastric Cells. Cancers 2019, 11, 801. https://doi.org/10.3390/cancers11060801
Bravo J, Díaz P, Corvalán AH, Quest AFG. A Novel Role for Helicobacter pylori Gamma-Glutamyltranspeptidase in Regulating Autophagy and Bacterial Internalization in Human Gastric Cells. Cancers. 2019; 11(6):801. https://doi.org/10.3390/cancers11060801
Chicago/Turabian StyleBravo, Jimena, Paula Díaz, Alejandro H. Corvalán, and Andrew F.G. Quest. 2019. "A Novel Role for Helicobacter pylori Gamma-Glutamyltranspeptidase in Regulating Autophagy and Bacterial Internalization in Human Gastric Cells" Cancers 11, no. 6: 801. https://doi.org/10.3390/cancers11060801
APA StyleBravo, J., Díaz, P., Corvalán, A. H., & Quest, A. F. G. (2019). A Novel Role for Helicobacter pylori Gamma-Glutamyltranspeptidase in Regulating Autophagy and Bacterial Internalization in Human Gastric Cells. Cancers, 11(6), 801. https://doi.org/10.3390/cancers11060801