Uncovering Potential Therapeutic Targets in Colorectal Cancer by Deciphering Mutational Status and Expression of Druggable Oncogenes

 ,
, 


Abstract
:1. Introduction
2. Methods
2.1. Sequencing and Expression Database
2.2. Gene Selecting Algorithm
2.3. Survival Analysis
2.4. CRC Specimens for the Validation Cohort
2.5. Nucleic Acid Isolation and Gene Expression Analysis
2.6. Availability of Data and Material
3. Results
3.1. Database Setup
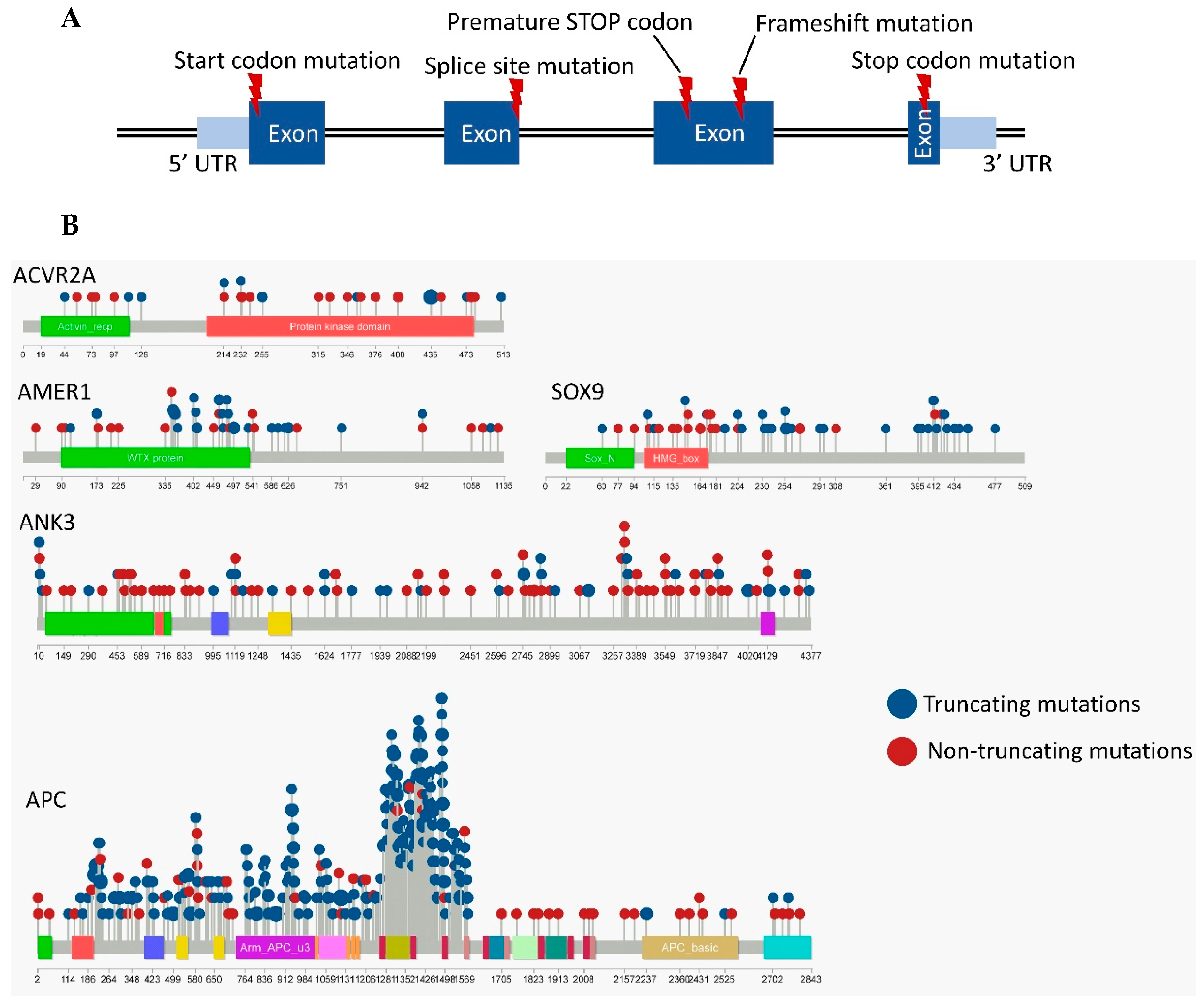
3.2. Frequently Mutated Genes
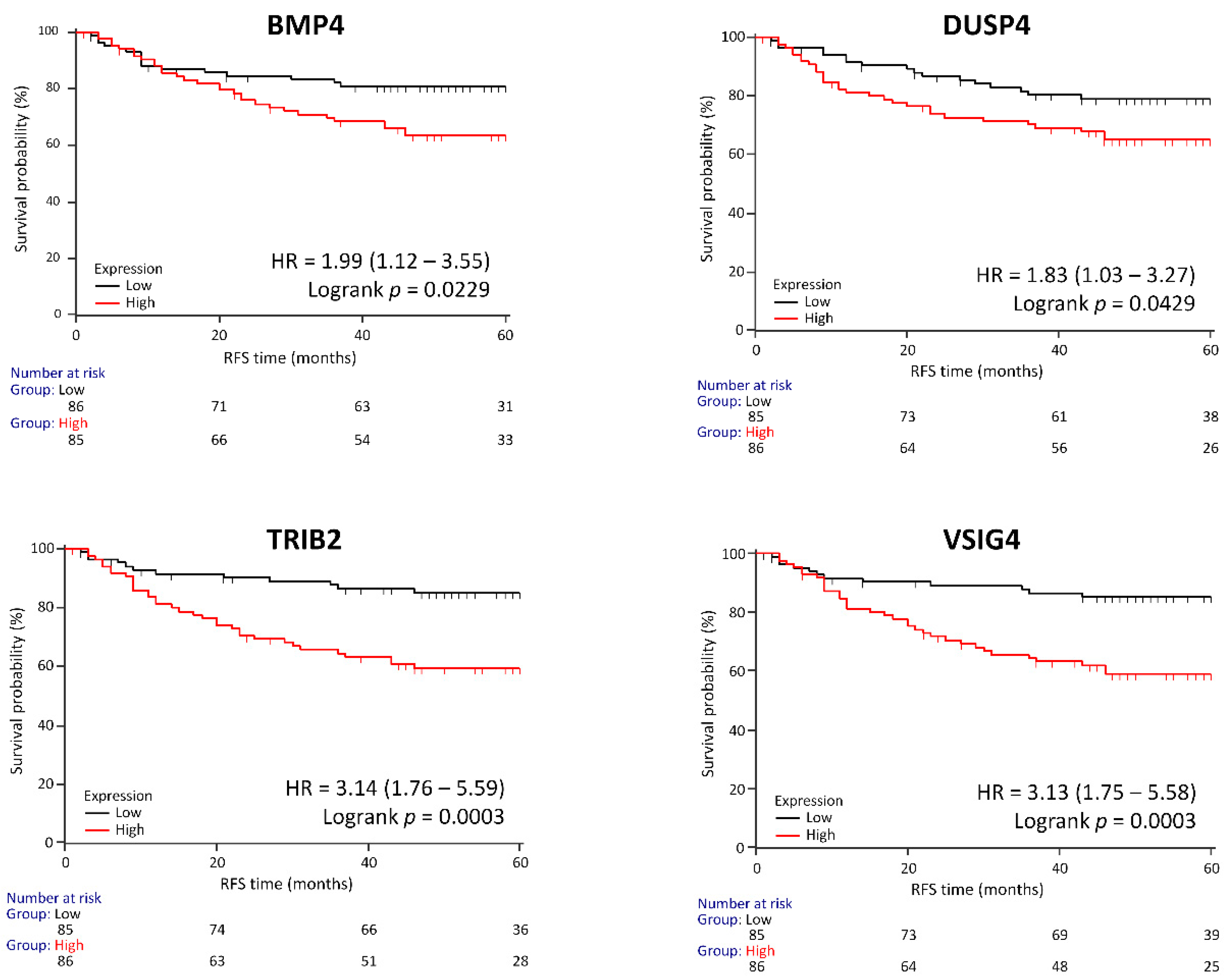
3.3. Survival Analysis
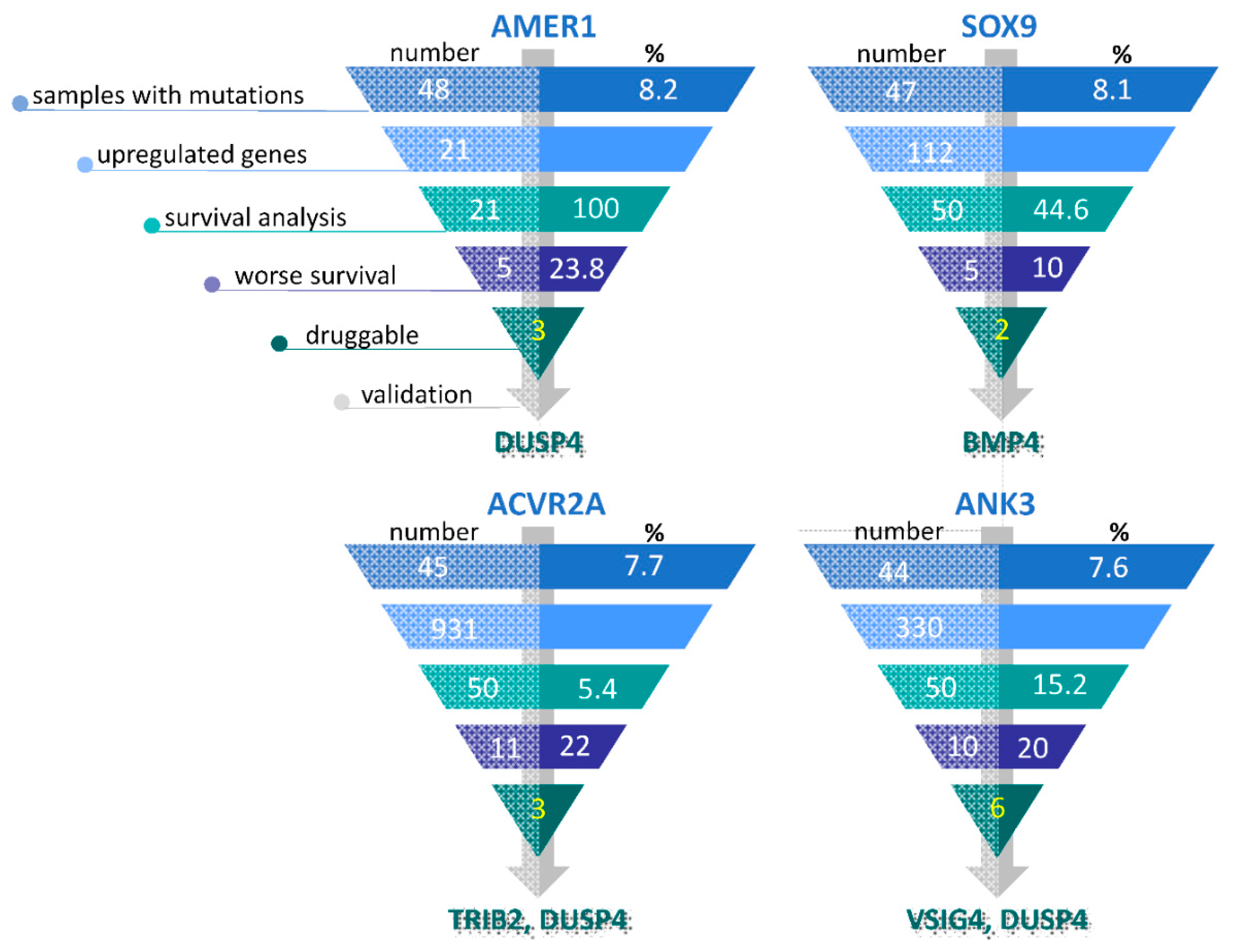
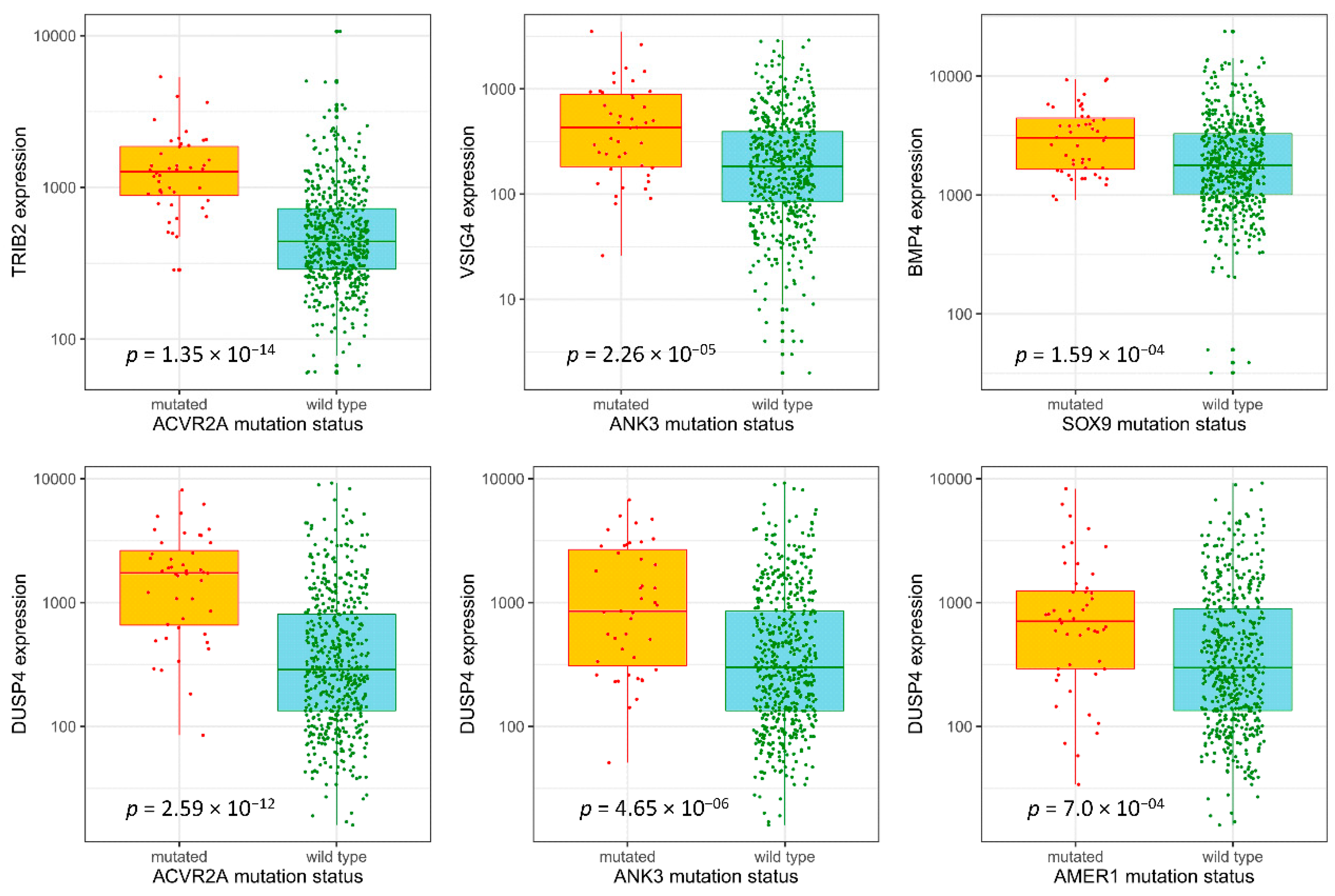
3.4. Selection of Potentially Actionable Genes
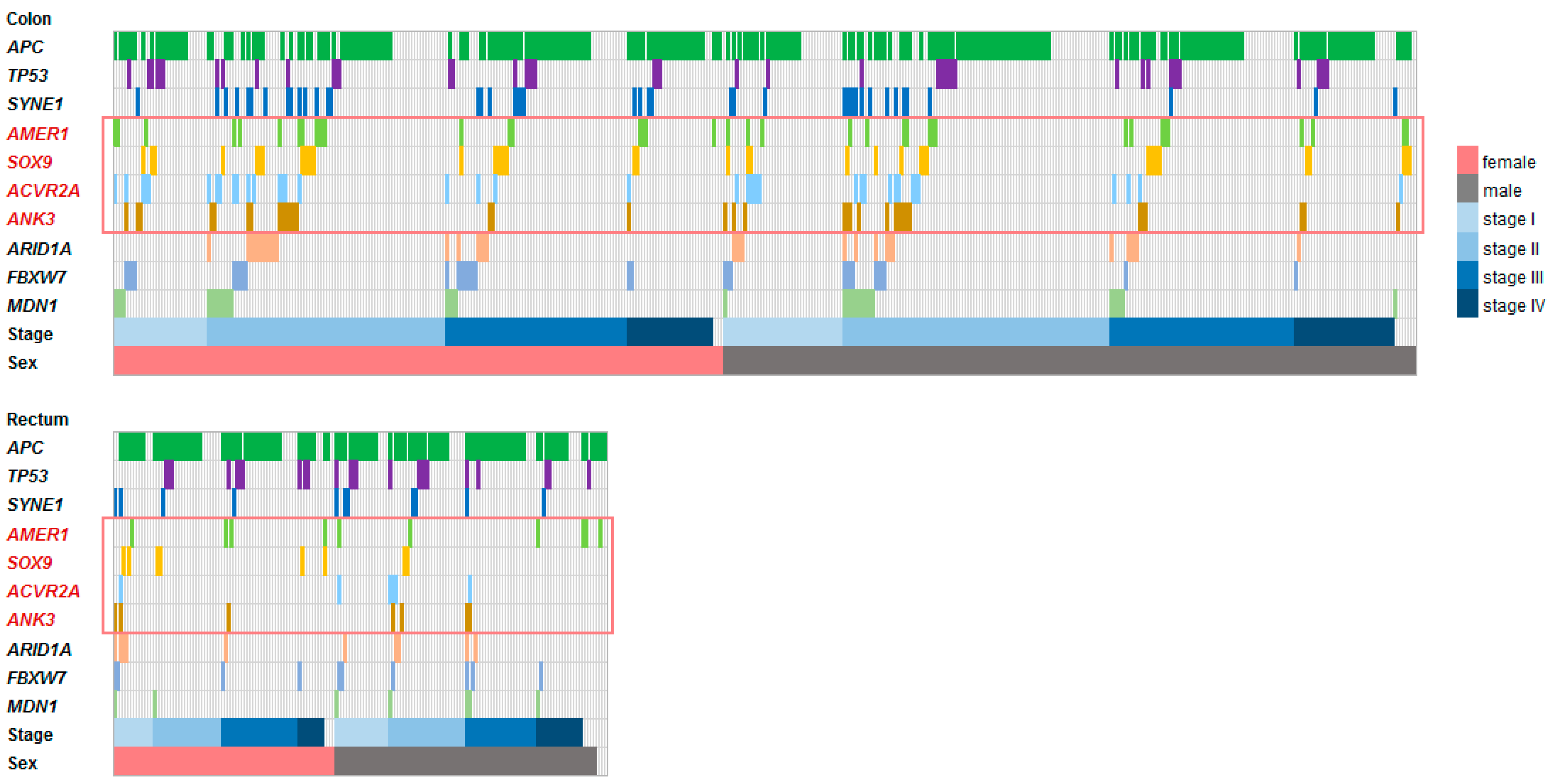
3.5. Validation Sample Set
3.6. Genes Associated with RFS in a Clinical Validation Set
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CRC | colorectal carcinoma |
| MSI | microsatellite instability |
| MMR | DNA Mismatch Repair |
| FFPE | formalin-fixed paraffin-embedded |
| TNM | tumor-node-metastasis |
| AJCC | American Joint Committee on Cancer |
| COAD | colon adenocarcinomas |
| READ | rectal adenocarcinomas |
| RFS | recurrence free survival |
| OS | overall survival |
| AML | acute myeloid leukemia |
| ALL | acute lymphoblastic leukemia |
| BMP | bone morphogenetic protein |
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144. [Google Scholar] [CrossRef] [PubMed]
- Menyhart, O.; Fekete, J.T.; Gyorffy, B. Demographic shift disproportionately increases cancer burden in an aging nation: current and expected incidence and mortality in Hungary up to 2030. Clin. Epidemiol. 2018, 10, 1093–1108. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fedewa, S.A.; Ahnen, D.J.; Meester, R.G.S.; Barzi, A.; Jemal, A. Colorectal cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 177–193. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Siena, S.; Cassidy, J.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J. Clin. Oncol. 2010, 28, 4697–4705. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Tabernero, J.; Lakomy, R.; Prenen, H.; Prausova, J.; Macarulla, T.; Ruff, P.; van Hazel, G.A.; Moiseyenko, V.; Ferry, D.; et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J. Clin. Oncol. 2012, 30, 3499–3506. [Google Scholar] [CrossRef] [PubMed]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouche, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Tabernero, J.; Yoshino, T.; Cohn, A.L.; Obermannova, R.; Bodoky, G.; Garcia-Carbonero, R.; Ciuleanu, T.E.; Portnoy, D.C.; Van Cutsem, E.; Grothey, A.; et al. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): A randomised, double-blind, multicentre, phase 3 study. Lancet Oncol. 2015, 16, 499–508. [Google Scholar] [CrossRef]
- Sztupinszki, Z.; Győrffy, B. Colon cancer subtypes: concordance, effect on survival and selection of the most representative preclinical models. Sci. Rep. 2016, 6, 37169. [Google Scholar] [CrossRef] [Green Version]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Marchionni, L.; Nowak, M.A.; Parmigiani, G.; Vogelstein, B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Cutsem, E.V.; McDermott, R.; Hill, A.; et al. Durable Clinical benefit with nivolumab plus ipilimumab in dna mismatch repair–deficient/microsatellite instability–high metastatic colorectal cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; de Reynies, A.; Giraldo, N.A.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautes-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and Stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clin. Cancer Res. 2016, 22, 4057–4066. [Google Scholar] [CrossRef] [PubMed]
- De Roock, W.; Claes, B.; Bernasconi, D.; De Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Calabrese, P.; Mecklin, J.P.; Järvinen, H.J.; Aaltonen, L.A.; Tavaré, S.; Shibata, D. Numbers of mutations to different types of colorectal cancer. BMC Cancer 2005, 5, 126. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 2011, 6, 479–507. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Á.; Pongor, L.S.; Szabó, A.; Santarpia, M.; Győrffy, B. KRAS driven expression signature has prognostic power superior to mutation status in non-small cell lung cancer. Int. J. Cancer 2017, 140, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Jay, J.J.; Brouwer, C. Lollipops in the clinic: Information Dense mutation plots for precision medicine. PLoS ONE 2016, 11, e0160519. [Google Scholar] [CrossRef]
- Varet, H.; Brillet-Gueguen, L.; Coppee, J.Y.; Dillies, M.A. SARTools: A DESeq2- and EdgeR-Based R Pipeline for comprehensive differential analysis of RNA-Seq Data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Birkbak, N.J.; Gyorffy, B.; Szallasi, Z.; Eklund, A.C. Jetset: selecting the optimal microarray probe set to represent a gene. BMC Bioinform. 2011, 12, 474. [Google Scholar] [CrossRef] [PubMed]
- Cotto, K.C.; Wagner, A.H.; Feng, Y.Y.; Kiwala, S.; Coffman, A.C.; Spies, G.; Wollam, A.; Spies, N.C.; Griffith, O.L.; Griffith, M. DGIdb 3.0: A redesign and expansion of the drug-gene interaction database. Nucleic Acids Res. 2018, 46, d1068–d1073. [Google Scholar] [CrossRef] [PubMed]
- Keeshan, K.; He, Y.; Wouters, B.J.; Shestova, O.; Xu, L.; Sai, H.; Rodriguez, C.G.; Maillard, I.; Tobias, J.W.; Valk, P.; et al. Tribbles homolog 2 inactivates C/EBPalpha and causes acute myelogenous leukemia. Cancer Cell 2006, 10, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.; Kalathur, R.K.; Colaco, L.; Brandao, R.; Ugurel, S.; Futschik, M.; Link, W. TRIB2 as a biomarker for diagnosis and progression of melanoma. Carcinogenesis 2015, 36, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Guo, K.; Sun, X.; Hu, F.; Chen, Q.; Luo, X.; Wang, G.; Hu, J.; Sun, L. TRIB2 functions as novel oncogene in colorectal cancer by blocking cellular senescence through AP4/p21 signaling. Mol. Cancer 2018, 17, 172. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.; Madureira, P.A.; Ferreira, B.; Baptista, I.; Machado, S.; Colaco, L.; Dos Santos, M.; Liu, N.; Dopazo, A.; Ugurel, S.; et al. TRIB2 confers resistance to anti-cancer therapy by activating the serine/threonine protein kinase AKT. Nat. Commun. 2017, 8, 14687. [Google Scholar] [CrossRef]
- Liang, K.L.; Rishi, L.; Keeshan, K. Tribbles in acute leukemia. Blood 2013, 121, 4265–4270. [Google Scholar] [CrossRef] [Green Version]
- Foulkes, D.M.; Byrne, D.P.; Yeung, W.; Shrestha, S.; Bailey, F.P.; Ferries, S.; Eyers, C.E.; Keeshan, K.; Wells, C.; Drewry, D.H.; et al. Covalent inhibitors of EGFR family protein kinases induce degradation of human Tribbles 2 (TRIB2) pseudokinase in cancer cells. Sci. Signal 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- De Pauw, I.; Wouters, A.; Van den Bossche, J.; Peeters, M.; Pauwels, P.; Deschoolmeester, V.; Vermorken, J.B.; Lardon, F. Preclinical and clinical studies on afatinib in monotherapy and in combination regimens: Potential impact in colorectal cancer. Pharmacol. Ther. 2016, 166, 71–83. [Google Scholar] [CrossRef]
- Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [CrossRef] [PubMed] [Green Version]
- Bauer, J.; Ozden, O.; Akagi, N.; Carroll, T.; Principe, D.R.; Staudacher, J.J.; Spehlmann, M.E.; Eckmann, L.; Grippo, P.J.; Jung, B. Activin and TGFbeta use diverging mitogenic signaling in advanced colon cancer. Mol. Cancer 2015, 14, 182. [Google Scholar] [CrossRef] [PubMed]
- Vogt, L.; Schmitz, N.; Kurrer, M.O.; Bauer, M.; Hinton, H.I.; Behnke, S.; Gatto, D.; Sebbel, P.; Beerli, R.R.; Sonderegger, I.; et al. VSIG4, a B7 family-related protein, is a negative regulator of T cell activation. J. Clin. Investig. 2006, 116, 2817–2826. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Guo, S.; Chen, Y.; Cao, D.; Xu, H.; Yang, C.; Fei, L.; Ni, B.; Ruan, Z. VSIG4 expression on macrophages facilitates lung cancer development. Lab. Investig. 2014, 94, 706. [Google Scholar] [CrossRef] [PubMed]
- Glinsky, G.V.; Berezovska, O.; Glinskii, A.B. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J. Clin. Investig. 2005, 115, 1503–1521. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Abou-Ouf, H.; Hegazy, S.A.; Alshalalfa, M.; Stoletov, K.; Lewis, J.; Donnelly, B.; Bismar, T.A. Ankyrin G expression is associated with androgen receptor stability, invasiveness, and lethal outcome in prostate cancer patients. J. Mol. Med. 2016, 94, 1411–1422. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.H.; Park, H.J.; Lee, S.K. The dual role of bone morphogenetic proteins in cancer. Mol. Ther. Oncol. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Bragdon, B.; Moseychuk, O.; Saldanha, S.; King, D.; Julian, J.; Nohe, A. Bone morphogenetic proteins: A critical review. Cell. Signal. 2011, 23, 609–620. [Google Scholar] [CrossRef]
- Irshad, S.; Bansal, M.; Guarnieri, P.; Davis, H.; Al Haj Zen, A.; Baran, B.; Pinna, C.M.A.; Rahman, H.; Biswas, S.; Bardella, C.; et al. Bone morphogenetic protein and notch signalling crosstalk in poor-prognosis, mesenchymal-subtype colorectal cancer. J. Pathol. 2017, 242, 178–192. [Google Scholar] [CrossRef]
- Deng, H.; Makizumi, R.; Ravikumar, T.S.; Dong, H.; Yang, W.; Yang, W.L. Bone morphogenetic protein-4 is overexpressed in colonic adenocarcinomas and promotes migration and invasion of HCT116 cells. Exp. Cell Res. 2007, 313, 1033–1044. [Google Scholar] [CrossRef]
- Wegner, M. All purpose Sox: The many roles of Sox proteins in gene expression. Int. J. Biochem. Cell Biol. 2010, 42, 381–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blache, P.; van de Wetering, M.; Duluc, I.; Domon, C.; Berta, P.; Freund, J.N.; Clevers, H.; Jay, P. SOX9 is an intestine crypt transcription factor, is regulated by the Wnt pathway, and represses the CDX2 and MUC2 genes. J. Cell Biol. 2004, 166, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Javier, B.M.; Yaeger, R.; Wang, L.; Sanchez-Vega, F.; Zehir, A.; Middha, S.; Sadowska, J.; Vakiani, E.; Shia, J.; Klimstra, D.; et al. Recurrent, truncating SOX9 mutations are associated with SOX9 overexpression, KRAS mutation, and TP53 wild type status in colorectal carcinoma. Oncotarget 2016, 7, 50875–50882. [Google Scholar] [CrossRef]
- Matheu, A.; Collado, M.; Wise, C.; Manterola, L.; Cekaite, L.; Tye, A.J.; Canamero, M.; Bujanda, L.; Schedl, A.; Cheah, K.S.; et al. Oncogenicity of the developmental transcription factor Sox9. Cancer Res. 2012, 72, 1301–1315. [Google Scholar] [CrossRef]
- Lu, B.; Fang, Y.; Xu, J.; Wang, L.; Xu, F.; Xu, E.; Huang, Q.; Lai, M. Analysis of SOX9 expression in colorectal cancer. Am. J. Clin. Pathol. 2008, 130, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Pritchett, J.; Athwal, V.; Roberts, N.; Hanley, N.A.; Hanley, K.P. Understanding the role of SOX9 in acquired diseases: lessons from development. Trends Mol. Med. 2011, 17, 166–174. [Google Scholar] [CrossRef]
- Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008, 27, 253–261. [Google Scholar] [CrossRef]
- Groschl, B.; Bettstetter, M.; Giedl, C.; Woenckhaus, M.; Edmonston, T.; Hofstadter, F.; Dietmaier, W. Expression of the MAP kinase phosphatase DUSP4 is associated with microsatellite instability in colorectal cancer (CRC) and causes increased cell proliferation. Int. J. Cancer 2013, 132, 1537–1546. [Google Scholar] [CrossRef]
- Yeon, S.Y.; Jo, Y.S.; Choi, E.J.; Kim, M.S.; Yoo, N.J.; Lee, S.H. Frameshift Mutations in Repeat sequences of ANK3, HACD4, TCP10L, TP53BP1, MFN1, LCMT2, RNMT, TRMT6, METTL8 and METTL16 genes in colon cancers. Pathol. Oncol. Res. 2018, 24, 617–622. [Google Scholar] [CrossRef]
- Sanz-Pamplona, R.; Lopez-Doriga, A.; Pare-Brunet, L.; Lazaro, K.; Bellido, F.; Alonso, M.H.; Ausso, S.; Guino, E.; Beltran, S.; Castro-Giner, F.; et al. Exome sequencing reveals AMER1 as a frequently mutated gene in colorectal cancer. Clin. Cancer Res. 2015, 21, 4709–4718. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | (%) |
|---|---|
| Sex | |
| Male | 55.6 |
| Female | 44.4 |
| Age/Years | |
| <60 | 24.6 |
| ≥60 | 75.4 |
| Stage | |
| I | 9.9 |
| II | 41.5 |
| III | 41.5 |
| IV | 7.0 |
| Venous Invasion | |
| Present | 86.0 |
| Absent | 13.5 |
| N/A | 0.6 |
| Lymphatic Invasion | |
| Present | 57.9 |
| Absent | 41.5 |
| N/A | 0.6 |
| Microsatellite Instability | |
| Yes | 6.4 |
| No | 42.7 |
| N/A | 50.9 |
| Recurrence (Median Follow up 50 Months) | 26.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menyhart, O.; Kakisaka, T.; Pongor, L.S.; Uetake, H.; Goel, A.; Győrffy, B. Uncovering Potential Therapeutic Targets in Colorectal Cancer by Deciphering Mutational Status and Expression of Druggable Oncogenes. Cancers 2019, 11, 983. https://doi.org/10.3390/cancers11070983
Menyhart O, Kakisaka T, Pongor LS, Uetake H, Goel A, Győrffy B. Uncovering Potential Therapeutic Targets in Colorectal Cancer by Deciphering Mutational Status and Expression of Druggable Oncogenes. Cancers. 2019; 11(7):983. https://doi.org/10.3390/cancers11070983
Chicago/Turabian StyleMenyhart, Otília, Tatsuhiko Kakisaka, Lőrinc Sándor Pongor, Hiroyuki Uetake, Ajay Goel, and Balázs Győrffy. 2019. "Uncovering Potential Therapeutic Targets in Colorectal Cancer by Deciphering Mutational Status and Expression of Druggable Oncogenes" Cancers 11, no. 7: 983. https://doi.org/10.3390/cancers11070983
APA StyleMenyhart, O., Kakisaka, T., Pongor, L. S., Uetake, H., Goel, A., & Győrffy, B. (2019). Uncovering Potential Therapeutic Targets in Colorectal Cancer by Deciphering Mutational Status and Expression of Druggable Oncogenes. Cancers, 11(7), 983. https://doi.org/10.3390/cancers11070983