Targeted Treatment Options of Recurrent Radioactive Iodine Refractory Hürthle Cell Cancer

 ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. Treatment Options of Recurrent HCC
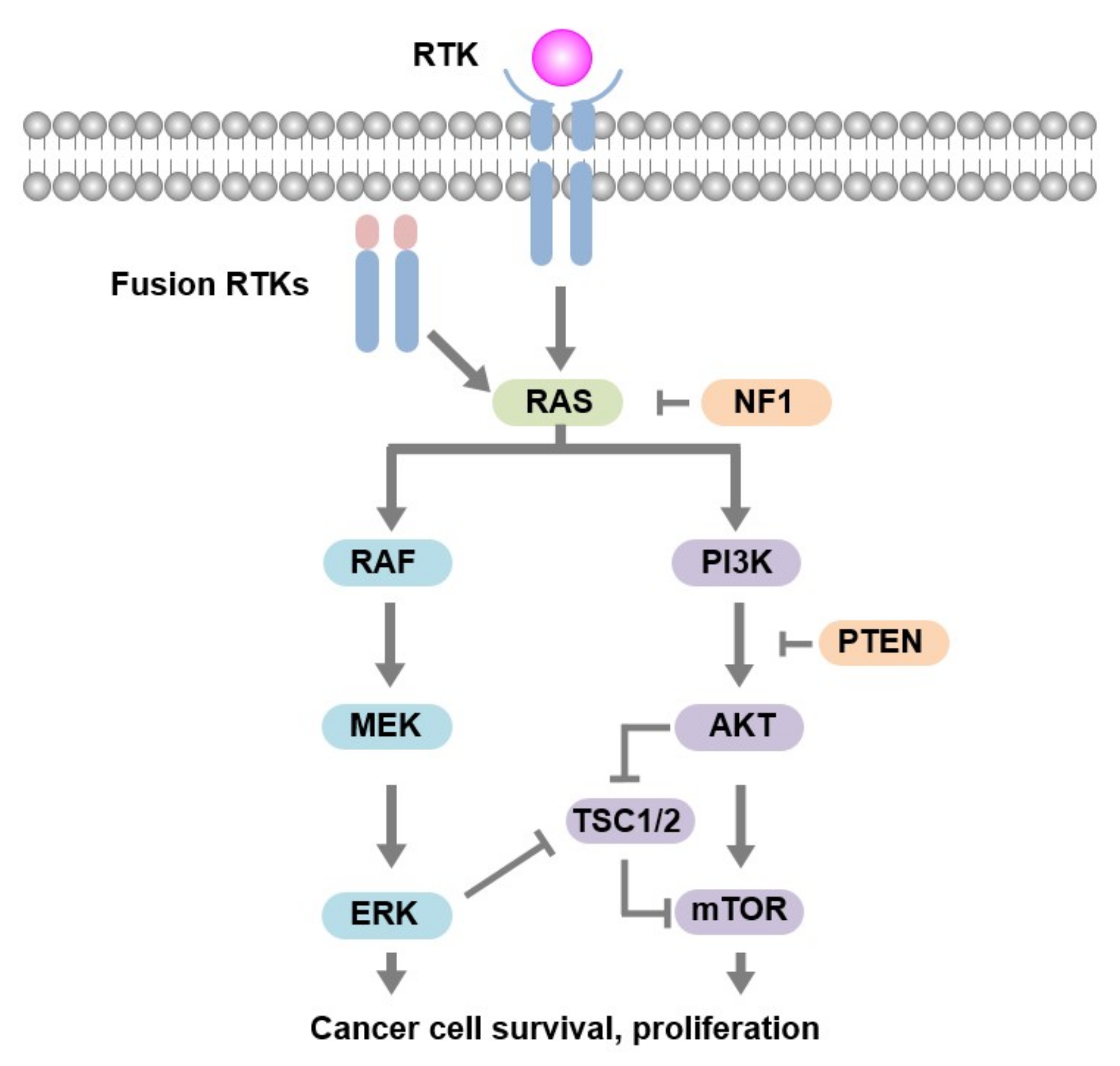
2.2. Rationale for Non-Responsiveness to Panitumumab
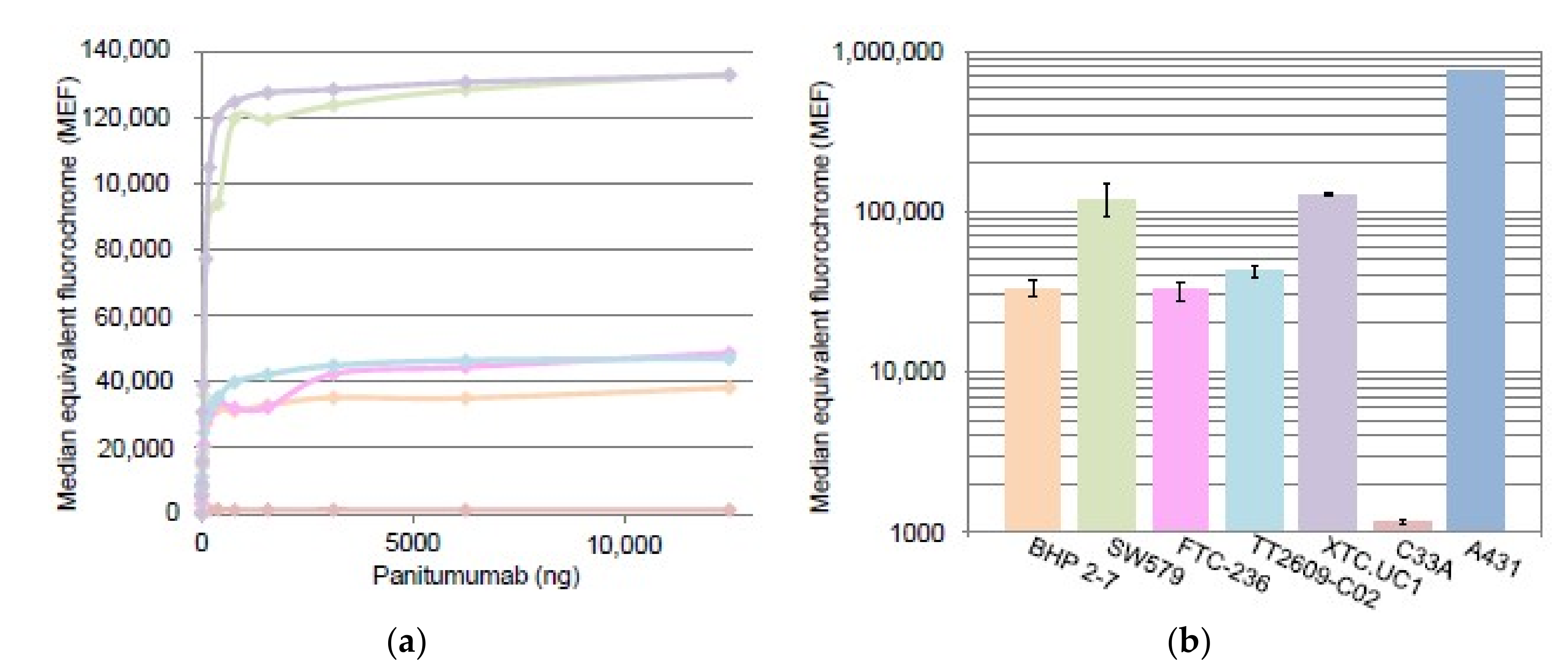
2.2.1. EGFR Quantification by Flow Cytometry
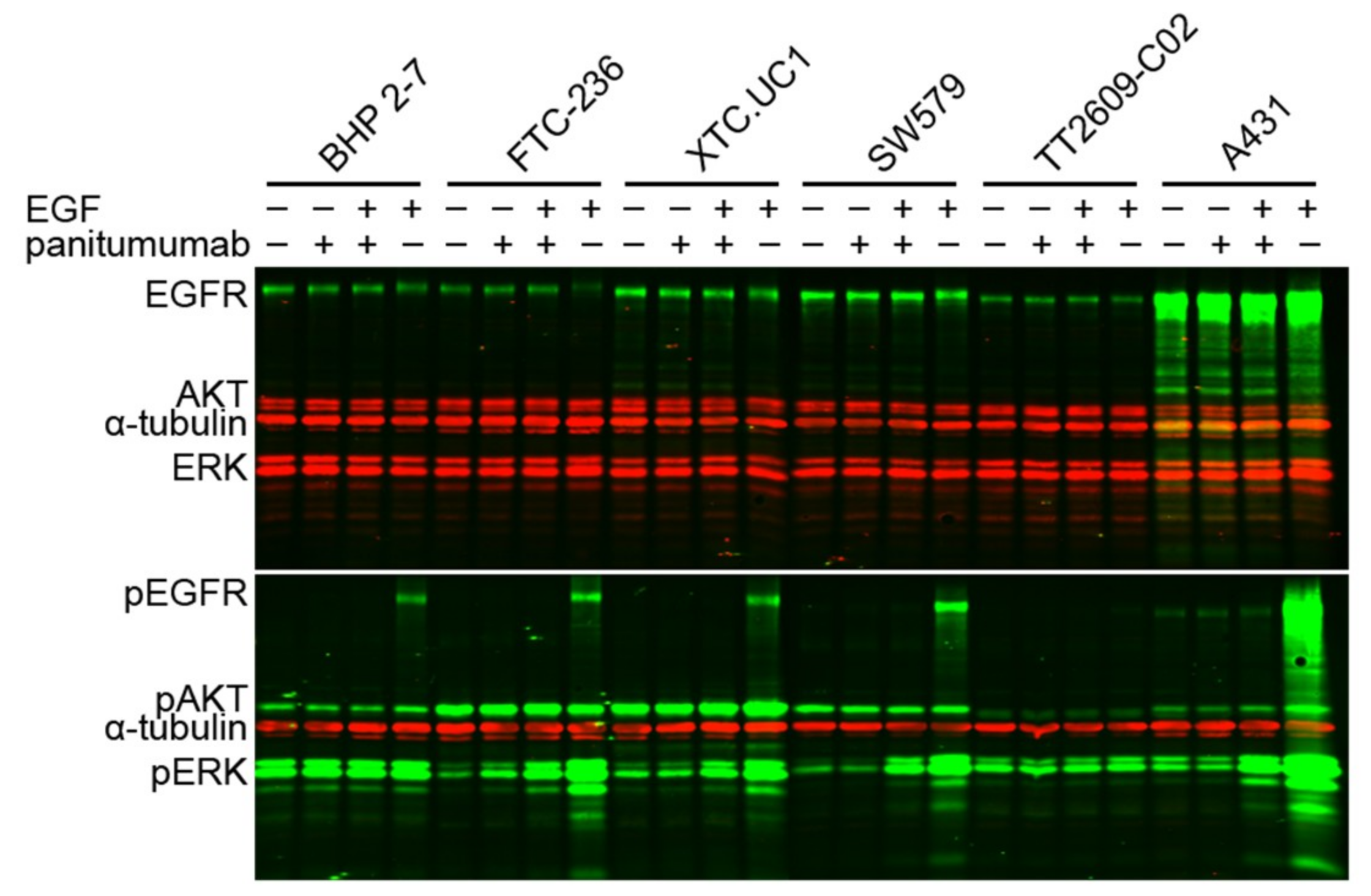
2.2.2. Immunofluorescent Western Blotting Analysis
3. Discussion
4. Materials and Methods
4.1. Patient
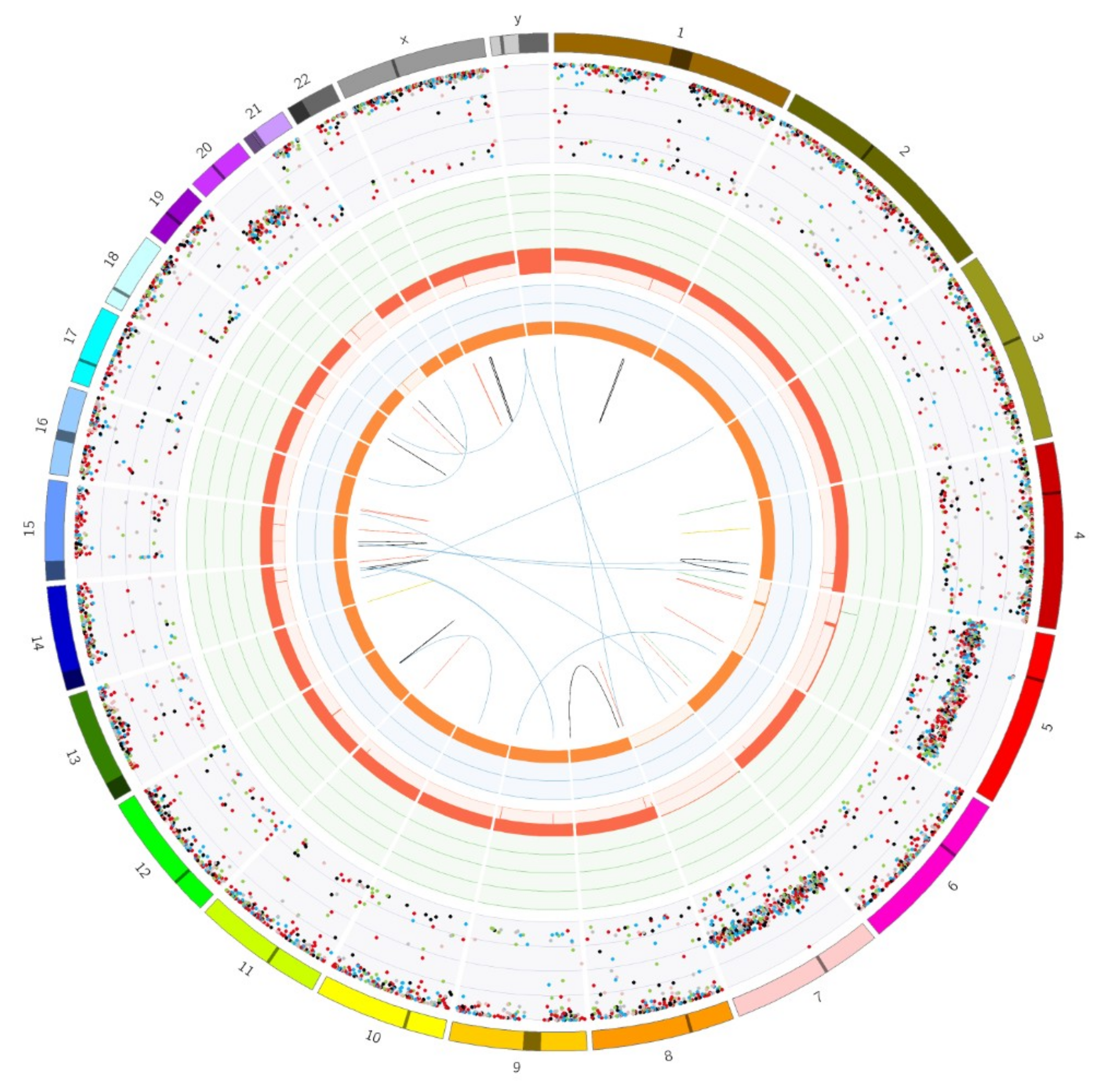
4.2. Somatic Gene Variant Spectrum Screening
4.3. Cell Lines, Cell Culture, DNA Isolation, and CELL Count
4.4. Compounds, Western Blotting
4.5. Antibodies
4.6. EGFR Expression Analysis by Flow Cytometry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lim, H.; Devesa, S.; Sosa, J.; Check, D.; Kitahara, C. Trends in thyroid cancer incidence and mortality in the United States, 1974–2013. JAMA 2017, 317, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Classification of Tumours of Endocrine Organs, 4th ed.; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American thyroid association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: The American thyroid association guidelines task force on thyroid nodules and differentiated thyroid cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M.; Brose, M.; Elisei, R.; Leboulleux, S.; Luster, M.; Pitoia, F.; Pacini, F. Definition and management of radioactive iodine-refractory differentiated thyroid cancer. Lancet Diabetes Endocrinol. 2014, 2, 356–358. [Google Scholar] [CrossRef]
- Durante, C.; Haddy, N.; Baudin, E.; Leboulleux, S.; Hartl, D.; Travagli, J.P.; Caillou, B.; Ricard, M.; Lumbroso, J.D.; Vathaire, F.D.; et al. Long-Term outcome of 444 patients with distant metastases from papillary and follicular thyroid carcinoma: benefits and limits of radioiodine therapy. J. Clin. Endocrinol. Metab. 2006, 91, 2892–2899. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Smit, J.; Capdevila, J.; Elisei, R.; Nutting, C.; Pitoia, F.; Robinson, B.; Schlumberger, M.; Shong, Y.K.; Takami, H. Regional approaches to the management of patients with advanced, radioactive iodine-refractory differentiated thyroid carcinoma. Expert Rev. Anticancer Ther. 2012, 12, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M.; Tahara, M.; Wirth, L.; Robinson, B.; Brose, M.; Elisei, R.; Habra, M.; Newbold, K.; Shah, M.; Hoff, A.; et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N. Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Wada, N.; Duh, Q.; Miura, D.; Brunaud, L.; Wong, M.; Clark, O. Chromosomal aberrations by comparative genomic hybridization in hürthle cell thyroid carcinomas are associated with tumor recurrence. J. Clin. Endocrinol. Metab. 2002, 87, 4595–4601. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Abdulrahman, R.; Corssmit, E.; Morreau, H.; Smit, J.; Kapiteijn, E. Long-term analysis of the efficacy and tolerability of sorafenib in advanced radio-iodine refractory differentiated thyroid carcinoma: Final results of a phase II trial. Eur. J. Endocrinol. 2012, 167, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.; Nutting, C.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; Fouchardiere, C.d.l.; Pacini, F.; Paschke, R.; Shong, Y.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef]
- Goffredo, P.; Roman, S.; Sosa, J. Hurthle cell carcinoma: A population-level analysis of 3311 patients. Cancer 2013, 119, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Oluic, B.; Paunovic, I.; Loncar, Z.; Djukic, V.; Diklic, A.; Jovanovic, M.; Garabinovic, Z.; Slijepcevic, N.; Rovcanin, B.; Micic, D.; et al. Survival and prognostic factors for survival, cancer specific survival and disease free interval in 239 patients with Hurthle cell carcinoma: A single center experience. BMC Cancer 2017, 17, 371. [Google Scholar] [CrossRef] [PubMed]
- Petric, R.; Gazic, B.; Besic, N. Prognostic factors for disease-specific survival in 108 patients with Hürthle cell thyroid carcinoma: A single-institution experience. BMC Cancer 2014, 14, 777. [Google Scholar] [CrossRef] [PubMed]
- Mills, S.; Haq, M.; Smellie, W.; Harmer, C. Hürthle cell carcinoma of the thyroid: Retrospective review of 62 patients treated at the Royal Marsden Hospital between 1946 and 2003. Eur. J. Surg. Oncol. 2009, 35, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Tallini, G. Oncocytic tumours. Virchows Arch. 1998, 433, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Maximo, V.; Lima, J.; Prazeres, H.; Soares, P.; Sobrinho-Simoes, M. The biology and the genetics of Hurthle cell tumors of the thyroid. Endocr. Relat. Cancer 2012, 19, R131–R147. [Google Scholar] [CrossRef] [PubMed]
- Gasparre, G.; Porcelli, A.M.; Bonora, E.; Pennisi, L.F.; Toller, M.; Iommarini, L.; Ghelli, A.; Moretti, M.; Betts, C.M.; Martinelli, G.N.; et al. Disruptive mitochondrial DNA mutations in complex I subunits are markers of oncocytic phenotype in thyroid tumors. Proc. Natl. Acad. Sci. USA 2007, 104, 9001–9006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corver, W.E.; Ruano, D.; Weijers, K.; den Hartog, W.C.; van Nieuwenhuizen, M.P.; de Miranda, N.; van Eijk, R.; Middeldorp, A.; Jordanova, E.S.; Oosting, J.; et al. Genome haploidisation with chromosome 7 retention in oncocytic follicular thyroid carcinoma. PLoS ONE 2012, 7, e38287. [Google Scholar] [CrossRef]
- Corver, W.E.; van Wezel, T.; Molenaar, K.; Schrumpf, M.; van den Akker, B.; van Eijk, R.; Ruano Neto, D.; Oosting, J.; Morreau, H. Near-haploidization significantly associates with oncocytic adrenocortical, thyroid, and parathyroid tumors but not with mitochondrial DNA mutations. Genes Chromosomes Cancer 2014, 53, 833–844. [Google Scholar] [CrossRef]
- Ganly, I.; Makarov, V.; Deraje, S.; Dong, Y.; Reznik, E.; Seshan, V.; Nanjangud, G.; Eng, S.; Bose, P.; Kuo, F.; et al. Integrated genomic analysis of hurthle cell cancer reveals oncogenic drivers, recurrent mitochondrial mutations, and unique chromosomal landscapes. Cancer Cell 2018, 34, 256–270.e5. [Google Scholar] [CrossRef]
- Gopal, R.K.; Kubler, K.; Calvo, S.E.; Polak, P.; Livitz, D.; Rosebrock, D.; Sadow, P.M.; Campbell, B.; Donovan, S.E.; Amin, S.; et al. Widespread chromosomal losses and mitochondrial dna alterations as genetic drivers in hurthle cell carcinoma. Cancer Cell 2018, 34, 242–255.e5. [Google Scholar] [CrossRef]
- Corver, W.; Morreau, H. Unique landscape of widespread chromosomal losses in Hurthle cell carcinoma. Endocr. Relat. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Corver, W.E.; Demmers, J.; Oosting, J.; Sahraeian, S.; Boot, A.; Ruano, D.; Wezel, T.V.; Morreau, H. ROS-induced near-homozygous genomes in thyroid cancer. Endocr. Relat. Cancer 2018, 25, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Ricarte-Filho, J.C.; Ryder, M.; Chitale, D.A.; Rivera, M.; Heguy, A.; Ladanyi, M.; Janakiraman, M.; Solit, D.; Knauf, J.A.; Tuttle, R.M.; et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009, 69, 4885–4893. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Ma, C.; Troxel, A.B.; Stopenski, S.J.; Tang, W.; Cohen, A.B.; Pappas-Paxinos, M.; Johnson, B.A., 3rd; Chen, E.Y.; Feldman, M.D.; et al. pAKT expression and response to sorafenib in differentiated thyroid cancer. Horm. Cancer 2016, 7, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y. Molecular analysis of thyroid tumors. Mod. Pathol. 2011, 24, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Xing, M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Rev. Cancer 2013, 13, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Robbins, H.; Hague, A. The Pi3K/Akt pathway in tumors of endocrine tissues. Front. Endocrinol. 2016, 6, 188. [Google Scholar] [CrossRef]
- Abubaker, J.; Jehan, Z.; Bavi, P.; Sultana, M.; Al-Harbi, S.; Ibrahim, M.; Al-Nuaim, A.; Ahmed, M.; Amin, T.; Al-Fehaily, M.; et al. Clinicopathological analysis of papillary thyroid cancer with PIK3CA alterations in a Middle Eastern population. J. Clin. Endocrinol. Metab. 2008, 93, 611–618. [Google Scholar] [CrossRef]
- Liu, Z.; Hou, P.; Ji, M.; Guan, H.; Studeman, K.; Jensen, K.; Vasko, V.; El-Naggar, A.; Xing, M. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J. Clin. Endocrinol. Metab. 2008, 93, 3106–3116. [Google Scholar] [CrossRef]
- Ganly, I.; Filho, J.R.; Eng, S.; Ghossein, R.; Morris, L.; Liang, Y.; Socci, N.; Kasthuri, K.; Mo, Q.; Fagin, J.; et al. Genomic dissection of Hurthle cell carcinoma reveals a unique class of thyroid malignancy. J. Clin. Endocrinol. Metab. 2013, 98, 962–972. [Google Scholar] [CrossRef]
- Pozdeyev, N.; Gay, L.M.; Sokol, E.S.; Hartmaier, R.; Deaver, K.E.; Davis, S.; French, J.D.; Borre, P.V.; LaBarbera, D.V.; Tan, A.-C.; et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin. Cancer Res. 2018, 24, 3059–3068. [Google Scholar] [CrossRef] [PubMed]
- Ganly, I.; McFadden, D.G. Short review: Genomic alterations in Hurthle cell carcinoma. Thyroid 2019, 29. [Google Scholar] [CrossRef] [PubMed]
- Berdelou, A.; Lamartina, L.; Klain, M.; Leboulleux, S.; Schlumberger, M. Treatment of refractory thyroid cancer. Endocr. Relat. Cancer 2018, 25, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Tumino, D.; Frasca, F.; Newbold, K. Updates on the management of advanced, metastatic, and radioiodine refractory differentiated thyroid cancer. Front. Endocrinol. (Lausanne) 2017, 20, 312. [Google Scholar] [CrossRef] [PubMed]
- Grani, G.; Lamartina, L.; Durante, C.; Filetti, S.; Cooper, D.S. Follicular thyroid cancer and Hurthle cell carcinoma: challenges in diagnosis, treatment, and clinical management. Lancet Diabetes Endocrinol. 2018, 6, 500–514. [Google Scholar] [CrossRef]
- Shackelford, R.E.; Whitling, N.A.; McNab, P.; Japa, S.; Coppola, D. KRAS testing: A tool for the implementation of personalized medicine. Genes Cancer 2012, 3, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Allegra, C.J.; Jessup, J.M.; Somerfield, M.R.; Hamilton, S.R.; Hammond, E.H.; Hayes, D.F.; McAllister, P.K.; Morton, R.F.; Schilsky, R.L. American Society of Clinical Oncology provisional clinical opinion: Testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J. Clin. Oncol. 2009, 27, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Arteaga, C.L. Critical update and emerging trends in epidermal growth factor receptor targeting in cancer. J. Clin. Oncol. 2005, 23, 2445–2459. [Google Scholar] [CrossRef] [PubMed]
- Acquaviva, G.; Visani, M.; Repaci, A.; Rhoden, K.; Biase, D.d.; Pession, A.; Giovanni, T. Molecular pathology of thyroid tumours of follicular cells: A review of genetic alterations and their clinicopathological relevance. Histopathology 2018, 72, 6–31. [Google Scholar] [CrossRef]
- Nikiforova, M.; Lynch, R.; Biddinger, P.; Alexander, E.K.; Gerald, W.D., 2nd; Tallini, G.; Kroll, T.; Nikiforov, Y. RAS point mutations and PAX8-PPAR gamma rearrangement in thyroid tumors: Evidence for distinct molecular pathways in thyroid follicular carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 2318–2326. [Google Scholar] [CrossRef]
- Lim, S.M.; Chang, H.; Yoon, M.J.; Hong, Y.K.; Kim, H.; Chung, W.Y.; Park, C.S.; Nam, K.H.; Kang, S.W.; Kim, M.K.; et al. A multicenter, phase II trial of everolimus in locally advanced or metastatic thyroid cancer of all histologic subtypes. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2013, 24, 3089–3094. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.C.; de Wit, D.; Links, T.P.; van Erp, N.P.; van der Hoeven, J.J.; Gelderblom, H.; Roozen, I.C.; Bos, M.; Corver, W.E.; van Wezel, T.; et al. Everolimus in patients with advanced follicular-derived thyroid cancer: Results of a phase II clinical trial. J. Clin. Endocrinol. Metab. 2017, 102, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Resteghini, C.; Cavalieri, S.; Galbiati, D.; Granata, R.; Alfieri, S.; Bergamini, C.; Bossi, P.; Licitra, L.; Locati, L.D. Management of tyrosine kinase inhibitors (TKI) side effects in differentiated and medullary thyroid cancer patients. Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M.; Newbold, K.; Hasan, B.; Marreaud, S.; Assele, S.; Licitra, L.; Schoffski, P.; Leboulleux, S.; Locati, L.; Godbert, Y.; et al. A randomized doubled blind phase II study exploring the safety and efficacy of nintedanib (BIBF1120) as second line therapy for patients (pts) with differentiated thyroid carcinoma (DTC) progressing after first line therapy: EORTC 1209. J. Clin. Oncol. 2018, 36 (Suppl. abstr 6021). Available online: https://meetinglibrary.asco.org/record/160248/abstract (accessed on 11 March 2019). [CrossRef]
- Gianoukakis, A.G.; Dutcus, C.E.; Batty, N.; Guo, M.; Baig, M. Prolonged duration of response in lenvatinib responders with thyroid cancer. Endocr. Relat. Cancer 2018, 25, 699–704. [Google Scholar] [CrossRef]
- Bissler, J.J.; McCormack, F.X.; Young, L.R.; Elwing, J.M.; Chuck, G.; Leonard, J.M.; Schmithorst, V.J.; Laor, T.; Brody, A.S.; Bean, J.; et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N. Engl. J. Med. 2008, 358, 140–151. [Google Scholar] [CrossRef]
- Davies, D.M.; de Vries, P.J.; Johnson, S.R.; McCartney, D.L.; Cox, J.A.; Serra, A.L.; Watson, P.C.; Howe, C.J.; Doyle, T.; Pointon, K.; et al. Sirolimus therapy for angiomyolipoma in tuberous sclerosis and sporadic lymphangioleiomyomatosis: A phase 2 trial. Clin. Cancer Res. 2011, 17, 4071–4081. [Google Scholar] [CrossRef]
- Klumpen, H.J.; Queiroz, K.C.; Spek, C.A.; van Noesel, C.J.; Brink, H.C.; de Leng, W.W.; de Wilde, R.F.; Mathus-Vliegen, E.M.; Offerhaus, G.J.; Alleman, M.A.; et al. mTOR inhibitor treatment of pancreatic cancer in a patient With Peutz-Jeghers syndrome. J. Clin. Oncol. 2011, 29, e150–e153. [Google Scholar] [CrossRef]
- Wagner, A.J.; Malinowska-Kolodziej, I.; Morgan, J.A.; Qin, W.; Fletcher, C.D.; Vena, N.; Ligon, A.H.; Antonescu, C.R.; Ramaiya, N.H.; Demetri, G.D.; et al. Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: Targeting the pathogenic activation of mTORC1 in tumors. J. Clin. Oncol. 2010, 28, 835–840. [Google Scholar] [CrossRef]
- Iyer, G.; Hanrahan, A.J.; Milowsky, M.I.; Al-Ahmadie, H.; Scott, S.N.; Janakiraman, M.; Pirun, M.; Sander, C.; Socci, N.D.; Ostrovnaya, I.; et al. Genome sequencing identifies a basis for everolimus sensitivity. Science 2012, 338, 221. [Google Scholar] [CrossRef]
- Wagle, N.; Grabiner, B.C.; Van Allen, E.M.; Amin-Mansour, A.; Taylor-Weiner, A.; Rosenberg, M.; Gray, N.; Barletta, J.A.; Guo, Y.; Swanson, S.J.; et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N. Engl. J. Med. 2014, 371, 1426–1433. [Google Scholar] [CrossRef] [PubMed]
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014, 4, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Park, H.S.; Kim, S.; Kim, S.; Ali, S.M.; Greenbowe, J.R.; Yang, I.S.; Kwon, N.J.; Lee, J.L.; Ryu, M.H.; et al. Next-generation sequencing reveals somatic mutations that confer exceptional response to everolimus. Oncotarget 2016, 7, 10547–10556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, A.; Petnga, W.; Macaulay, V.M.; Weyer-Czernilofsky, U.; Bogenrieder, T. Insulin-like growth factor (IGF) pathway targeting in cancer: Role of the IGF axis and opportunities for future combination studies. Target Oncol. 2017, 12, 571–597. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Montalto, G.; Cervello, M.; Nicoletti, F.; Fagone, P.; Malaponte, G.; Mazzarino, M.C.; et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget 2012, 3, 954–987. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- DrugBank. Available online: https://www.drugbank.ca/ (accessed on 11 March 2019).
- Priestley, P.; Baber, J.; Lolkema, M.; Steeghs, N.; de Bruijn, E.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; Roepman, P.; et al. Pan-cancer whole genome analyses of metastatic solid tumors. bioRxiv 2019. Available online: https://www.biorxiv.org/content/10.1101/415133v2.full (accessed on 11 March 2019). [CrossRef]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E. HGVS recommendations for the description of sequence variants—2016 update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Melillo, R.M.; Castellone, M.D.; Guarino, V.; De Falco, V.; Cirafici, A.M.; Salvatore, G.; Caiazzo, F.; Basolo, F.; Giannini, R.; Kruhoffer, M.; et al. The RET/PTC-RAS-BRAF linear signaling cascade mediates the motile and mitogenic phenotype of thyroid cancer cells. J. Clin. Investig. 2005, 115, 1068–1081. [Google Scholar] [CrossRef] [Green Version]
- Schweppe, R.E.; Klopper, J.P.; Korch, C.; Pugazhenthi, U.; Benezra, M.; Knauf, J.A.; Fagin, J.A.; Marlow, L.A.; Copland, J.A.; Smallridge, R.C.; et al. Deoxyribonucleic acid profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. J. Clin. Endocrinol. Metab. 2008, 93, 4331–4341. [Google Scholar] [CrossRef]
- Ohta, K.; Pang, X.; Berg, L.; Hershman, J. Growth inhibition of new human thyroid carcinoma cell lines by activation of adenylate cyclase through the beta-adrenergic receptor. J. Clin. Endocrinol. Metab. 1997, 82, 2633–2638. [Google Scholar]
- Yoshimoto, K.; Iwahana, H.; Fukuda, A.; Sano, T.; Saito, S.; Itakura, M. Role of p53 mutations in endocrine tumorigenesis: Mutation detection by polymerase chain reaction-single strand conformation polymorphism. Cancer Res. 1992, 52, 5061–5064. [Google Scholar] [PubMed]
- Zielke, A.; Tezelman, S.; Jossart, G.; Wong, M.; Siperstein, A.; Duh, Q.; Clark, O. Establishment of a highly differentiated thyroid cancer cell line of Hurthle cell origin. Thyroid 1998, 8, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Hölting, T.; Zielke, A.; Siperstein, A.; Clark, O.; Duh, Q. Transforming growth factor-beta 1 is a negative regulator for differentiated thyroid cancer: Studies of growth, migration, invasion, and adhesion of cultured follicular and papillary thyroid cancer cell lines. J. Clin. Endocrinol. Metab. 1994, 79, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Landa, I.; Ganly, I.; Chan, T.A.; Mitsutake, N.; Matsuse, M.; Ibrahimpasic, T.; Ghossein, R.A.; Fagin, J.A. Frequent somatic TERT promoter mutations in thyroid cancer: Higher prevalence in advanced forms of the disease. J. Clin. Endocrinol. Metab. 2013, 98, E1562–E1566. [Google Scholar] [CrossRef] [PubMed]
- Geldof, A.; Versteegh, L.; Mourik, J.V.; Rooimans, M.; Arwert, F.; Hermsen, M.; Schadee-Eestermans, I.; Dongen, G.V.; Valk, P.V.D.; Clement, E.V.D.; et al. Clonally related but phenotypically divergent human cancer cell lines derived from a single follicular thyroid cancer recurrence (TT2609). Thyroid 2001, 11, 909–917. [Google Scholar] [CrossRef]
- Van de Vijver, M.J.; Kumar, R.; Mendelsohn, J. Ligand-induced activation of A431 cell epidermal growth factor receptors occurs primarily by an autocrine pathway that acts upon receptors on the surface rather than intracellularly. J. Biol. Chem. 1991, 266, 7503–7508. [Google Scholar] [PubMed]
- Giard, D.J.; Aaronson, S.A.; Todaro, G.J.; Arnstein, P.; Kersey, J.H.; Dosik, H.; Parks, W.P. In vitro cultivation of human tumors: Establishment of cell lines derived from a series of solid tumors. J. Natl. Cancer Inst. 1973, 51, 1417–1423. [Google Scholar] [CrossRef]
- Miller, W.E.; Earp, H.S.; Raab-Traub, N. The Epstein-Barr virus latent membrane protein 1 induces expression of the epidermal growth factor receptor. J. Virol. 1995, 69, 4390–4398. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Sex | Age | Origin | Localization | NHG | Variants | Ref. |
|---|---|---|---|---|---|---|---|
| BHP 2–7 | f | uk | PTC | primary | N | RET/PTC1 rearrangement [61,62] | [63] |
| SW579 | m | 59 | PDTC | primary | N | TP53 c.827T > G, p. (Ile255Ser) | [64] 1 |
| XTC.UC1 | f | 63 | HCC | metas. in breast | Y [23] | TP53 c.451C > A, NP_000537.3: p. (Pro151Thr) 3 | [65] 2 |
| FTC-236 | m | 42 | FTC | neck LN metas. | Y [23] | [66] | |
| TT2609-C02 | m | 57 | FTC | primary | N | NRAS c.182A > G, p. (Gln61Arg) [67] | [68] 1 |
| A431 | f | 85 | EC | uk | uk | EGFR overexpression [69] | [70] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aydemirli, M.D.; Corver, W.; Beuk, R.; Roepman, P.; Solleveld-Westerink, N.; van Wezel, T.; Kapiteijn, E.; Morreau, H. Targeted Treatment Options of Recurrent Radioactive Iodine Refractory Hürthle Cell Cancer. Cancers 2019, 11, 1185. https://doi.org/10.3390/cancers11081185
Aydemirli MD, Corver W, Beuk R, Roepman P, Solleveld-Westerink N, van Wezel T, Kapiteijn E, Morreau H. Targeted Treatment Options of Recurrent Radioactive Iodine Refractory Hürthle Cell Cancer. Cancers. 2019; 11(8):1185. https://doi.org/10.3390/cancers11081185
Chicago/Turabian StyleAydemirli, Mehtap Derya, Willem Corver, Ruben Beuk, Paul Roepman, Nienke Solleveld-Westerink, Tom van Wezel, Ellen Kapiteijn, and Hans Morreau. 2019. "Targeted Treatment Options of Recurrent Radioactive Iodine Refractory Hürthle Cell Cancer" Cancers 11, no. 8: 1185. https://doi.org/10.3390/cancers11081185
APA StyleAydemirli, M. D., Corver, W., Beuk, R., Roepman, P., Solleveld-Westerink, N., van Wezel, T., Kapiteijn, E., & Morreau, H. (2019). Targeted Treatment Options of Recurrent Radioactive Iodine Refractory Hürthle Cell Cancer. Cancers, 11(8), 1185. https://doi.org/10.3390/cancers11081185